

Abstract
Objective
C-reactive protein (CRP) is an inflammatory marker that contributes to the prediction of cardiovascular disease. We investigated the influences of CRP polymorphisms on baseline CRP levels and fenofibrate-induced CRP changes in subjects with the metabolic syndrome.
Research Design and Methods
We examined the association of CRP single nucleotide polymorphisms (SNPs) (m772A>G, m301G>A >T, i178T>A, 3u1273C>T, and 3u2131C>T) with baseline plasma CRP levels among 1,123 white U.S. participants in the Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) Study and the modulating effect of these SNPs on CRP response to a 3-week fenofibrate treatment among 290 participants with the metabolic syndrome.
Results
There were strong associations of m301G>A>T (rs3091244; P = 0.003), i178T>A (rs1417938; P = 0.001), 3u1273C>T (rs1130864; P = 0.001), and 3u2131C>T (rs1205; P < 0.001) with baseline CRP levels. Moreover, among subjects with the metabolic syndrome, fenofibrate induced the greatest reduction in CRP levels for TT subjects of the i178T>A compared with TA and AA subjects (−30 for TT, −19 for TA, and −11% for AA; P = 0.004). Similarly, for the m301G>A>T, major allele carriers displayed maximal reduction of CRP over noncarriers (−20 for GG, −15 for GA and GT, and −0.3% for TA and AA; P = 0.020).
Conclusions
Our results demonstrate that common genetic variants within the CRP gene affect baseline CRP levels and further modulate CRP response in subjects with the metabolic syndrome treated with fenofibrate. This knowledge could contribute to a better prediction of therapeutic success.
Creactive protein (CRP) is an important downstream inflammatory marker that integrates the action of several activated cytokines. Current evidence suggests that plasma CRP levels may be an independent predictor of clinical cardiovascular events in both healthy individuals and patients with cardiovascular disease (CVD) (1). In addition, elevated CRP has been shown to associate with multiple risk factors for CVD including obesity, insulin resistance, and hypertension and to predict risk of the metabolic syndrome and diabetes (2).
Plasma CRP levels display extensive interindividual variability. Although plasma CRP levels are influenced by sociodemographic, behavioral, and lifestyle factors and obesity and type 2 diabetes, twin and family studies have estimated that genetic factors could contribute up to 35–50% of the variation of CRP (3,4). Several population-based association studies have shown that common genetic variants at the CRP locus are significantly associated with plasma CRP levels (3–5). Furthermore, the CRP locus has been shown to associate with CVD risk (4), providing additional support for a causal role of CRP in CVD. In line with these results, in vitro studies reveal that the CRP upstream region harbors functional polymorphisms that potentially alter binding affinities of different transcription factors and thus affect transcription of the CRP gene (3). In addition to baseline CRP levels, CRP single nucleotide polymorphisms (SNPs) also contribute to variation of inflammatory responses to certain stimuli. The T-allele of SNP1444C>T (rs 1130864) has been reported to affect both baseline CRP and inflammatory responses to experimental lipopolysaccharide-induced endotoxemia in healthy adults (6). This allele is also associated with differential CRP responses in patients undergoing periodontal treatment and coronary artery bypass graft surgery (5,7). These findings prompted us to examine the effects of genetic variants of CRP gene on the response of CRP to fenofibrate that, in addition to their lipid-lowering action, have anti-inflammatory activity (8).
Fenofibrate is a drug of the fibrate class commonly used for management of dyslipidemia. A growing body of evidence has suggested that fibrates target both the atherogenic “lipid triad” (high triglycerides and low HDL with small dense LDL particles) and inflammation (8). Because both phenotypes are important components of diabetes and the metabolic syndrome and potentially link these two metabolic disorders to CVD (9), fibrates appear to be well suited for treating dyslipidemia associated with diabetes and the metabolic syndrome and more effective in reducing CVD in those high-risk populations (10). However, individual response to fenofibrate is highly variable. Prior studies have reported that genetic variants at genes involved in lipid metabolism modulate lipid response to fenofibrate (11). However, the effect of CRP variants on the anti-inflammatory effect of fenofibrate may be a significant determinant of its therapeutic success and remains to be investigated.
Therefore, the goals of our study were 1) to examine the effect of common CRP polymorphisms on baseline plasma CRP levels among white U.S. participants in the Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) Study and 2) to investigate the effect of these variants on the response of CRP to a 3-week fenofibrate treatment in GOLDN participants with the metabolic syndrome who, within the population, are the obvious targets of this family of drugs.
Research Design and Methods
The study population consisted of 539 men and 584 women who participated in the GOLDN study. The GOLDN study was a single-arm, uncontrolled, nonrandomized intervention funded by the National Heart, Lung, and Blood Institute with the purpose of identifying genetic variants associated with interindividual variability of triglyceride responses to a high-fat meal and fenofibrate. The design and methodology of the study have previously been described (12). Detailed materials and methods are provided in online supplement 1 (available at http://dx.doi.org/10.2337/dc07-1687). Briefly, participants were given open-labeled fenofibrate 160 mg tablets (TriCor; Abbott Laboratories, Chicago, IL) and instructed to take one tablet with a breakfast meal once daily for 3 weeks. For the study of cross-sectional association, a total of 1,123 subjects with complete genotype data, baseline CRP measurements, and relevant covariates were included in the analysis. For the study of the genetic effect on fenofibrate response, analysis was performed among 290 subjects classified as having the metabolic syndrome according to the 2005 National Cholesterol Education Program Adult Treatment Panel III guidelines with full data records for baseline and the post-treatment values. The protocol for this study was approved by the human studies committees of the institutional review boards at University of Minnesota, University of Utah, and Tufts University/New England Medical Center. Written informed consent was obtained from all participants.
Biochemical measurements
Plasma high-sensitivity CRP was measured using the K-Assay CRP(2), a latex particle– enhanced immunoturbidimetric assay on the Hitachi 911 (Kamiya Biomedical Company, Seattle, WA).
DNA isolation and genotyping
Genomic DNA was extracted from blood samples and purified using commercial Puregene reagents (Gentra System) following the manufacturer's instructions. Genotyping was performed using the 5′nuclease allelic discrimination Taqman assay with ABI 7900HT system (Applied Biosystems, Foster City, CA). The description of SNPs, primer and probe sequences, and ABI assay-on-demand IDs are presented in supplemental Table 1.
Statistical analyses
Statistical analyses were performed using SAS, version 9.0 (SAS Institute, Cary, NC). A χ2 test was used to determine whether genotype distribution followed Hardy-Weinberg equilibrium. A logarithmic transformation was applied to plasma CRP levels to normalize the distribution of data. For the additive model, the coding of the genotypes was based on the number of variant alleles at the polymorphic site. The genotypes were coded as the dummy variables in the codominant model. We used the generalized estimating equation approach with exchangeable correlation structure as implemented in the GENMOD procedure in SAS to adjust for correlated observations due to familial relationships. Potential confounding factors included age, sex, BMI, smoking status, alcohol consumption, physical activity, hormones, and use of aspirin and nonsteroidal anti-inflammatory drugs and drugs for lowering cholesterol, diabetes, and hypertension. Additional adjustments for baseline CRP levels and change of triglyceride and interleukin-6 levels were made for comparison of the mean change of CRP across genotypes. Subjects with plasma CRP levels > 10 mg/l at either baseline or posttreatment were excluded from the data analysis because this may indicate an ongoing acute inflammatory condition rather than low levels of chronic inflammation (13). Pairwise linkage disequilibrium (LD) between SNPs was estimated as a correlation coefficient (r2) in unrelated subjects using the Helixtree software (Golden Helix, Bozeman, MT). Haplotypes were estimated using the program MERLIN (14), which reconstructs haplotypes for a set of tightly linked markers from extended pedigrees. We used the generalized estimating equation model to examine the haplotype association, in which inferred haplotypes were considered a predictor and the aforementioned confounding factors were considered covariates. A two-tailed P value of <0.05 was considered statistically significant.
Results
Baseline characteristics of the GOLDN Study population
The demographic and biochemical characteristics of subjects at baseline (mean age ± SD, 49 ± 16 years; 52% female) are presented in Table 1. Among 1,123 subjects, 34% were defined as having the metabolic syndrome.
Table 1. Baseline characteristics of study participants.
| Total | Men | Women | |
|---|---|---|---|
| n | 1,123 | 539 | 584 |
| Age (years) | 48.6 ± 16.4 | 48.9 ± 16.4 | 48.2 ± 16.4 |
| BMI (kg/m2) | 28.3 ± 5.6 | 28.5 ± 4.9 | 28.0 ± 6.2 |
| Waist (cm) | 96.4 ± 16.9 | 100.6 ± 15.0 | 92.4 ± 17.7 |
| Triglycerides (mg/dl) | 139 ± 116 | 153 ± 142 | 125 ± 82 |
| Total cholesterol (mg/dl) | 191 ± 39 | 190 ± 38 | 191 ±40 |
| HDL cholesterol (mg/dl) | 47.1 ± 13 | 41.5 ± 9.8 | 52.2 ± 13.7 |
| LDL cholesterol (mg/dl) | 121 ± 31 | 123 ± 30 | 120 ± 32 |
| Fasting glucose (mg/dl) | 101 ± 19 | 106 ± 21 | 98 ± 16 |
| Fasting insulin (μU/ml) | 13.7 ± 8.2 | 14.2 ± 8.4 | 13.3 ± 7.9 |
| Systolic blood pressure (mmHg) | 115 ± 17 | 118 ± 15 | 112 ± 17 |
| Diastolic blood pressure (mmHg) | 68 ± 9 | 71 ± 9 | 66 ± 9 |
| C-reactive protein (mg/l) | 2.48 ± 5.0 | 1.88 ± 3.75 | 3.03 ± 5.78 |
| Interleukin-6 (ng/l) | 1.98 ±3.11 | 2.07 ± 4.15 | 1.89 ± 1.63 |
| Current smoker | 86 (7.7) | 42 (7.8) | 44 (7.6) |
| Current drinker | 562 (50) | 266 (49) | 296 (51) |
| Hormone treatment | 122 (11) | — | 122 (21) |
| Metabolic syndrome | 381 (34) | 205 (38) | 181 (31) |
Data are means ± SD or n (%).
LD patterns among SNPs at the CRP locus
Minor allele frequencies and SNP pairwise LD values were evaluated in a subset of 148 unrelated subjects (supplemental Table 2). Genotype distributions followed Hardy-Weinberg equilibrium (P > 0.05). SNPs m301G>A, i178T>A, 3u1273C>T, and 3u2131C>T were in strong LD (r2 = 0.4–0.9; P< 0.001). Them772A>G was in significant LD with the m301G>T (r2 = 0.935; P< 0.001) and in weak LD with the other three SNPs (r2 = 0.17; P < 0.05). Because the i178T>A and the 3u1273C>T were in complete LD (r2 = 1; P < 0.001) and, consequently, displayed the same pattern of phenotypic associations, results for only the i178T>A SNP are presented.
Association of CRP SNPs and haplotypes with baseline CRP levels
We examined the association of four SNPs and baseline CRP levels (Fig. 1A). Because no significant gene-sex interactions were observed, we analyzed men and women together. All SNPs except m772A>G showed significant associations with plasma CRP levels in multivariate adjusted models. For m301G>A>T, major allele homozygotes exhibited the lowest CRP level and heterozygotes had an intermediate level of CRP, whereas subjects with only minor alleles (T or A) had the highest CRP level (P = 0.003). The minor allele of i178T>A was associated with higher CRP compared with the common allele (P = 0.001). In contrast, the minor allele of 3u2131C>T was associated with lower CRP compared with the common allele (P < 0.001).
Figure 1.
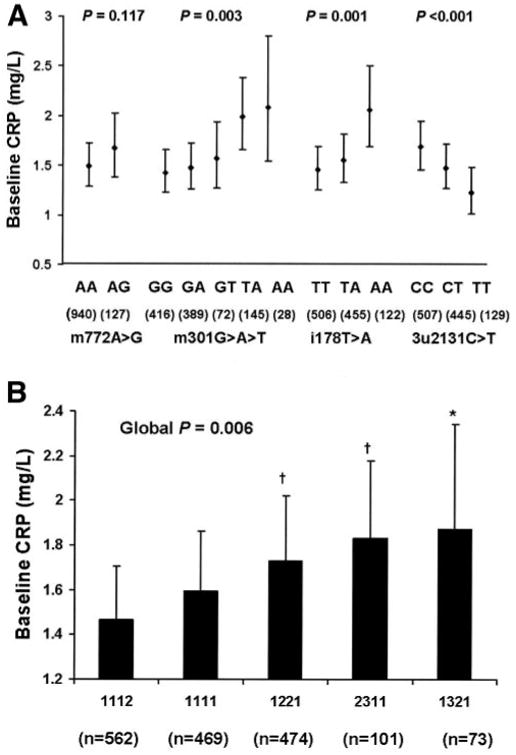
Associations of CRP polymorphisms (A) and haplotypes (B) with baseline plasma CRP levels (geometric means ± 95% CI). P values were obtained from the generalized estimating equation model adjusting for age, sex, BMI, plasma triglyceride and interleukin-6, smoking status, alcohol consumption, physical activity, and hormones. Further adjustment for use of aspirin and nonsteroidal anti-inflammatory drugs and drugs for lowering cholesterol, diabetes, and hypertension did not modify the statistical significance. *P < 0.05 and †P < 0.001 compared with haplotype 1112.
The haplotype consisted of four SNPs at the CRP locus ordered from 5′ to 3′ along the chromosome in the following order: m772A>G, m301G>A>T, i178T>A, and 3u2131C>T (with 1 representing the major allele, 2 the minor allele, and 3 the minor allele with the lowest frequency). A total of five major haplotypes with frequency ≥5% was identified, representing 97% of the observed haplotypes. Baseline CRP levels differed significantly across these common haplotypes (global P = 0.006) (Fig. 1B). Haplotypes 1221, 2311, and 1321 had significantly higher CRP compared with haplotype 1112 (P < 0.001, P < 0.001, and P = 0.040, respectively) in the multivariate-adjusted model. Compared with all other haplotypes combined, haplotype 1112 was associated with low CRP levels (P = 0.001), whereas haplotypes 1221 and 2311 were associated with high CRP levels (P = 0.035 and P = 0.032, respectively). Haplotype 1321 was associated with high CRP levels when compared with all other haplotypes combined; however, the association did not reach statistical significance (P = 0.248).
Effect of CRP SNPs on CRP response to fenofibrate among subjects with the metabolic syndrome
We further examined the effects of CRP SNPs on the CRP response after a 3-week fenofibrate treatment among subjects with the metabolic syndrome (Table 2). Because sex did not modify genotype effects, we analyzed men and women together. Common allele homozygotes for the i178T>A displayed the highest reduction of CRP, whereas heterozygotes and minor allele homozygotes displayed an intermediate and the smallest reduction of CRP, respectively (−30 for TT, − 19 for TA, and −11% for AA; P for the additive model = 0.004). For m301G>A>T, genotypes were categorized into three groups based on the number of the common G-alleles: homozygous GG group, heterozygous GA or GT group, and noncarriers group (TA or AA). The major allele carriers displayed higher reductions of CRP than noncarriers (−20 for GG, −15 for GA and GT, and −0.3% for TA and AA; P for the additive model = 0.02). There were no significant differences in CRP response across genotype groups for 3u2131C>T and m772A>G. Further haplotype analyses showed no significant effect on plasma CRP responses after treatment (supplemental Table 3). In addition, there was no significant effect of CRP genotypes on lipids or glucose and insulin responses (supplemental Tables 4–6).
Table 2. Plasma CRP response to fenofibrate among subjects with the metabolic syndrome according to CRP genotypes.
| SNPs | Genotype | n | Before drug | After drug | Responses | |||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
||||||
| Geometric mean (95% CI) | P† | Geometric mean (95% CI) | P† | % change (95% CI) | P† | |||
| i178T>A | TT | 139 | 2.76 (2.15–3.55) | 0.168 | 1.77 (1.30–2.41) | 0.008 | −30 (−40 to −18) | 0.004 |
| TA | 118 | 2.6 (2.11–3.21) | 1.93 (1.50–2.48) | −19 (−29 to − 6) | ||||
| AA | 32 | 3.91 (2.83–5.42) | 3.07 (2.11–4.48) | −11 (−30 to 13) | ||||
| 3u2131C>T | CC | 141 | 2.85 (2.23–3.65) | 0.034 | 2.00 (1.46–2.74) | 0.025 | −23 (−33 to −10) | 0.147 |
| CT | 116 | 2.68 (2.19–3.28) | 1.83 (1.43–2.36) | −22 (−33 to −10) | ||||
| TT | 32 | 1.92 (1.35–2.74) | 1.30 (0.86–1.97) | −34 (−47 to −17) | ||||
| m772A>G | AA | 246 | 2.67 (2.15–3.31) | 0.299 | 1.90 (1.44–2.49) | 0.861 | −21 (−32 to −9) | 0.249 |
| AG | 39 | 3.16 (2.20–4.54) | 1.96 (1.25–3.08) | −31 (−45 to −14) | ||||
| m301G>A>T | GG | 110 | 2.77 (2.15–3.58) | 0.066 | 1.84 (1.34–2.53) | 0.008 | −20 (−31 to −7) | 0.02 |
| GA + GT | 125 | 2.54 (2.06–3.14) | 1.76 (1.36–2.28) | −15 (−25 to −4) | ||||
| TA + AA | 49 | 3.86 (2.90–5.14) | 3.08 (2.18–4.35) | −0.3 (−19 to 22) | ||||
Data are adjusted for baseline CRP levels, change of triglyceride, change of interleukin-6, age, sex, BMI, smoking status, alcohol intake, physical activity, hormone treatment in women, and use of aspirin and nonsteroidal anti-inflammatory drugs and drugs for lowering cholesterol, diabetes, and hypertension. Subjects with CRP > 10 mgl at baseline or after the treatment were excluded from the analysis.
Additive model.
Conclusions
In the current study, we demonstrate that common CRP polymorphisms were associated with baseline CRP levels in white U.S. participants in the GOLDN Study. Specifically, carriers for either the A- or T-allele of m301G>A>T exhibited higher CRP levels than carriers of the G-allele. The minor allele of i178 T>A was associated with high CRP levels, whereas the minor allele of 3u2131C>T was associated with low CRP levels. The results were consistent with previous reports (3–5). Furthermore, the CRP variants modulated plasma CRP response to a 3-week fenofibrate intervention among subjects with the metabolic syndrome. Thus, G-allele carriers for the m301G>A>T SNP displayed greater reduction of CRP levels than non-carriers. Similarly, TT subjects for the i178T>A SNP had greater reduction in CRP levels than TA and AA subjects.
The observed association of m301G> A>T with baseline CRP levels could be due to its suggested functionality. Studies have shown that m301G>A>T resides within the hexameric core of transcription factor–binding elements, and the mutation alters a transcription factor–binding motif for upstream stimulating factor 1 (USF1), thus affecting the transcriptional activity of the CRP gene (3). Because the i178 T>A is in nearly complete LD with m301G>T>A, the observed association for i178T>A could simply be attributable to this functional triallelic SNP. Interestingly, our analysis of the CRP region for transcription factor motifs using the program MAPPER (15) indicates that i178T>A contains a binding motif for the transcription repressor growth factor independent 1, which has been shown in animal models to limit inflammatory responses (16). Presence of the major T-allele maintains the integrity of the binding site for growth factor independent 1, whereas the minor A-allele abolishes this binding site. This prediction is in concordance with our observed genotypic association; the minor allele was associated with high CRP in a genotype dose-dependent manner. However, until further functional work substantiates these potential mechanisms, caution should be taken to interpret such a polymorphism as a functional variant. In contrast to the above two SNPs, the common allele for 3u2131C>T was associated with high CRP levels. The mechanism underlying this association could be through its LD with functional alleles, such as the triallelic SNP. As a result of the moderate correlation between these two SNPs (r2 = 0.437), it is also likely that the SNP is functional because it lies in the 3′ untranslated region and potentially could affect mRNA structure and stability.
We have also observed strong associations between common haplotypes and baseline CRP levels. Haplotype 1112 (m772A-m301G-i178T-3u2131T) was associated with low CRP level, whereas haplotypes 1221 (m772A-m301A-i178A-3u2131C) and 2311 (m772G-m301T-i178T-3u2131C) were associated with high CRP levels. These three haplotypes were comparable with haplotypes H3, H2, and H5, as reported by Miller et al. (4), who demonstrated that H3 associated with lower CRP, and H2 and H5 with high CRP, in three cohorts including the Women's Health Study, the Physician's Health Study, and the study of Pravastatin Inflammation/CRP Evaluation. Notably, the minor allele for m301G>A>T and the common allele for 3u2131C>T always occur on haplotypes associated with high CRP levels. This finding further supports the functional importance of these two SNPs. However, analysis of two haplotypes that only differ for 3u2131C>T showed marginally significant difference of plasma CRP levels (P = 0.09), suggesting that the effect of this SNP may be subject to the haplotypic context.
Studies have shown that fenofibrate along with other fibrates lowers inflammatory markers such as CRP in patients with dyslipidemia associated with insulin resistance, obesity, and the metabolic syndrome (17,18). The anti-inflammatory action is mediated by the activation of peroxisome proliferator–activated receptor α (PPAR-α), which functions as a negative regulator of genes involved in the inflammatory response by antagonizing the activity of transcription factors such as nuclear factor-κB and activator protein-1 (19). However, individual response, in particular plasma CRP response to fenofibrate, is highly variable and potentially subjected to genetic regulation.
In our study among subjects with the metabolic syndrome, major allele carriers for i178T>A (in complete LD with 3u1273C>T) had a favorable response to treatment compared with minor allele carriers. We also observed similar results for m301G>A>T, in which major allele carriers had greater CRP reduction than nonmajor carriers. It is of interest that the alleles associated with high baseline inflammatory status appear to be more resistant to the anti-inflammatory effect of fenofibrate. Notably, under the opposite condition, such as in the presence of an inflammatory stimuli, these alleles, such as the minor allele of 3u1273C>T (previously reported as 1444>T, rs1130864), have been shown to have a greater CRP rise (5,7). The mechanism underlying the modulation of genetic variants on CRP response to fenofibrate is undefined. However, as both SNPs appear to be involved in transcription factor–binding motifs, it is possible that the interaction of these transcription factors, in particular USF1 with PPAR-α, might underlie some of the genetic effect. We postulate that the allele-specific binding of USF1 may directly interfere with PPAR-α–mediated transrepression of inflammatory gene expression and, thus, potentially counteract PPAR-α ligand activity. It is also possible that USF1 may enhance the availability and/or DNA binding capacity of nuclear factor-κB to the CRP promoter and result in the decreased response to anti-inflammatory effect of PPAR-α. In this regard, future in vitro studies focusing on the protein-protein interaction and how this interaction determines the anti-inflammatory effect of fenofibrate are warranted.
We believe our findings are of practical importance for several reasons. First, although a series of large-scale intervention trials using fibrates has established the role of fibrates in normalizing lipid profiles, the results regarding their efficacy on the reduction of CVD events is inconsistent (10). Further subgroup analyses revealed that features of the metabolic syndrome modify the effect of fibrates on CVD such that cardiovascular benefits are largely confined to subjects with features of the metabolic syndrome (10). Our data provide additional insight into the heterogeneity of the treatment response, suggesting that genetic difference could further differentiate individual response to fenofibrate and, thus, may affect the disease outcome among subjects at high risk. Second, if the reduction of CRP could directly lead to the decreased rate of recurrent events of CVD and the progression of coronary atherosclerosis, as reported from multiple randomized clinical trials using statins (20), individuals carrying certain genotypes that potentially impact on CRP response to treatment may have a different trajectory of cardiovascular events from non-carriers and, therefore, may require different lifestyle modifications or therapeutic regimes.
In summary, we demonstrated that common genetic polymorphisms at the CRP locus have a significant influence on baseline CRP levels and further modulate CRP response to fenofibrate among subjects with the metabolic syndrome. These findings have some implications for refining the assessment of susceptibility to CVD. More importantly, this information may assist in future development of more personalized therapeutic strategies to CVD on a genetic basis.
Supplementary Material
Acknowledgments
This study was supported by contract 53-K06-5-10 from the National Institutes of Health (NIH) and 58-1950-9-001 from the U.S. Department of Agriculture (Agriculture Research Service) and by NIH Heart, Lung and Blood Institute Grant U 01 HL72524, Genetic and Environmental Determinants of Triglycerides.
We acknowledge Abbott Laboratories (Abbott Park, IL) for their supply of study medication for this project.
Abbreviations
- CVD
cardiovascular disease
- CRP
C-reactive protein
- GOLDN
Genetics of Lipid Lowering Drugs and Diet Network
- LD
linkage disequilibrium
- PPAR-α
peroxisome proliferator–activated receptor α
- SNP
single nucleotide polymorphism
- USF1
upstream stimulating factor 1
Footnotes
Additional information for this article can be found in an online appendix at http://dx.doi.org/10.2337/dc07-1687.
References
- 1.Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002;347:1557–1565. doi: 10.1056/NEJMoa021993. [DOI] [PubMed] [Google Scholar]
- 2.Haffner SM. The metabolic syndrome: inflammation, diabetes mellitus, and cardiovascular disease. Am J Cardiol. 2006;97:3A–11A. doi: 10.1016/j.amjcard.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 3.Carlson CS, Aldred SF, Lee PK, Tracy RP, Schwartz SM, Rieder M, Liu K, Williams OD, Iribarren C, Lewis EC, Fornage M, Boerwinkle E, Gross M, Jaquish C, Nickerson DA, Myers RM, Siscovick DS, Reiner AP. Polymorphisms within the C-reactive protein (CRP) promoter region are associated with plasma CRP levels. Am J Hum Genet. 2005;77:64–77. doi: 10.1086/431366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller DT, Zee RY, Suk Danik J, Kozlowski P, Chasman DI, Lazarus R, Cook NR, Ridker PM, Kwiatkowski DJ. Association of common CRP gene variants with CRP levels and cardiovascular events. Ann Intern Med. 2005;69:623–638. doi: 10.1111/j.1529-8817.2005.00210.x. [DOI] [PubMed] [Google Scholar]
- 5.Brull DJ, Serrano N, Zito F, Jones L, Montgomery HE, Rumley A, Sharma P, Lowe GD, World MJ, Humphries SE, Hingorani AD. Human CRP gene polymorphism influences CRP levels: implications for the prediction and pathogenesis of coronary heart disease. Arterioscler Thromb Vasc Biol. 2003;23:2063–2069. doi: 10.1161/01.ATV.0000084640.21712.9C. [DOI] [PubMed] [Google Scholar]
- 6.Marsik C, Sunder-Plassmann R, Jilma B, Kovar FM, Mannhalter C, Wagner O, Rumpold H, Endler G. The C-reactive protein (+)1444C/T alteration modulates the inflammation and coagulation response in human endotoxemia. Clin Chem. 2006;52:1952–1957. doi: 10.1373/clinchem.2006.069823. [DOI] [PubMed] [Google Scholar]
- 7.D'Aiuto F, Casas JP, Shah T, Humphries SE, Hingorani AD, Tonetti MS. C-reactive protein (+1444>T) polymorphism influences CRP response following a moderate inflammatory stimulus. Atherosclerosis. 2005;179:413–417. doi: 10.1016/j.atherosclerosis.2004.10.036. [DOI] [PubMed] [Google Scholar]
- 8.Libby P, Plutzky J. Inflammation in diabetes mellitus: role of peroxisome proliferator-activated receptor-alpha and peroxisome proliferator-activated receptor-gamma agonists. Am J Cardiol. 2007;99:27B–40B. doi: 10.1016/j.amjcard.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 9.Roberts AW, Evans M. The metabolic syndrome, inflammation and cardiovascular disease in type 2 diabetes. Curr Opin Lipidol. 2004;15:89–91. doi: 10.1097/00041433-200402000-00016. [DOI] [PubMed] [Google Scholar]
- 10.Barter PJ, Rye KA. Is there a role for fibrates in the management of dyslipidemia in the metabolic syndrome? Arterioscler Thromb Vasc Biol. 2008;28:39–46. doi: 10.1161/ATVBAHA.107.148817. [DOI] [PubMed] [Google Scholar]
- 11.Brisson D, Ledoux K, Bosse Y, St-Pierre J, Julien P, Perron P, Hudson TJ, Vohl MC, Gaudet D. Effect of apolipoprotein E, peroxisome proliferator-activated receptor alpha and lipoprotein lipase gene mutations on the ability of fenofibrate to improve lipid profiles and reach clinical guideline targets among hypertriglyceridemic patients. Pharmacogenetics. 2002;12:313–320. doi: 10.1097/00008571-200206000-00007. [DOI] [PubMed] [Google Scholar]
- 12.Corella D, Arnett DK, Tsai MY, Kabagambe EK, Peacock JM, Hixson JE, Straka RJ, Province M, Lai CQ, Parnell LD, Borecki I, Ordovas JM. The −256T>C polymorphism in the apolipoprotein A-II gene promoter is associated with body mass index and food intake in the genetics of lipid lowering drugs and diet network study. Clin Chem. 2007;53:1144–1152. doi: 10.1373/clinchem.2006.084863. [DOI] [PubMed] [Google Scholar]
- 13.Pearson TA, Mensah GA, Alexander RW, Anderson JL, Cannon RO, 3rd, Criqui M, Fadl YY, Fortmann SP, Hong Y, Myers GL, Rifai N, Smith SC, Jr, Taubert K, Tracy RP, Vinicor F. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107:499–511. doi: 10.1161/01.cir.0000052939.59093.45. [DOI] [PubMed] [Google Scholar]
- 14.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 15.Marinescu VD, Kohane IS, Riva A. MAPPER: a search engine for the computational identification of putative transcription factor binding sites in multiple genomes. BMC Bioinformatics. 2005;6:79. doi: 10.1186/1471-2105-6-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karsunky H, Zeng H, Schmidt T, Zevnik B, Kluge R, Schmid KW, Duhrsen U, Moroy T. Inflammatory reactions and severe neutropenia in mice lacking the transcriptional repressor Gfi1. Nat Genet. 2002;30:295–300. doi: 10.1038/ng831. [DOI] [PubMed] [Google Scholar]
- 17.Despres JP, Lemieux I, Pascot A, Almeras N, Dumont M, Nadeau A, Bergeron J, Prud'homme D. Gemfibrozil reduces plasma C-reactive protein levels in abdominally obese men with the atherogenic dyslipidemia of the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2003;23:702–703. doi: 10.1161/01.ATV.0000062990.62034.64. [DOI] [PubMed] [Google Scholar]
- 18.Rosenson RS, Wolff DA, Huskin AL, Helenowski IB, Rademaker AW. Fenofibrate therapy ameliorates fasting and postprandial lipoproteinemia, oxidative stress, and the inflammatory response in subjects with hypertriglyceridemia and the metabolic syndrome. Diabetes Care. 2007;39:1945–1951. doi: 10.2337/dc07-0015. [DOI] [PubMed] [Google Scholar]
- 19.Kleemann R, Gervois PP, Verschuren L, Staels B, Princen HM, Kooistra T. Fibrates down-regulate IL-1-stimulated C-reactive protein gene expression in hepatocytes by reducing nuclear p50-NFkappa B-C/EBP-beta complex formation. Blood. 2003;101:545–551. doi: 10.1182/blood-2002-06-1762. [DOI] [PubMed] [Google Scholar]
- 20.Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, McCabe CH, Pfeffer MA, Braunwald E. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20–28. doi: 10.1056/NEJMoa042378. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.