

Abstract
Standard RNA-seq has a well know tendency to generate “ghost” antisense reads due to formation of spurious second strand cDNA in the sequencing process. We recently reported on a novel variant of RNA-seq coined “tagRNA-seq” introduced for the purpose of distinguishing primary from processed transcripts in bacteria. Incidentally, the additional information provided by the tags is also very suitable for detection of true anti-sense RNA transcripts and quantification of spurious antisense signals in a sample. We briefly explain how to perform such a detection and illustrate on previously published datasets.
Keywords: antisense RNA, complementary, DNA, spurious second strand cDNA, tagRNA-seq, transcriptome, transcript discovery
In the last decade, the so-called “Next Generation Sequencing” platforms have proven to be a valuable and very flexible tool for research in many areas of Life Sciences.1 One of their most prominent applications is the study of genetic expression using RNA sequencing (RNA-seq),2 which provides a far greater level of information than previous methods such as micro-array. Beside RNA abundance and among others,a RNA-seq allows to probe for post-transcriptional modifications such as splicing and RNA processing sites and predict locations of transcription start sites within a few nucleotides.3,4
While there exist many variants of RNA-seq, all common methods involve reverse transcription of RNA into DNA,5 a process that is well known to generate spurious second strand cDNA using freshly synthesized cDNA as template.5,6
We recently reported on a novel variant of RNA-seq coined “tagRNA-seq” introduced for the purpose of distinguishing primary from processed transcripts in bacteria.7 Briefly, the method consists in ligating short artificial RNA sequences called “tags” to 5′ ends of transcripts, with different tags for primary (triphosphate 5′ ends) and processed (monophosphate 5′ ends) RNAs.8 Incidentally, the additional information provided by the tag is also very suitable for detection of true antisense RNA transcripts (asRNA) in sequencing data as well as via classical RT-PCR methods.8 We here detail how this can be achieved in tagRNA-seq and demonstrate how to quantitatively estimate the level of artifactual antisense RNA reads in previously published data sets.
In tagRNA-seq, tags with an arbitrarily chosen sequence are ligated to the 5′ end transcripts (Fig. 1A). The ligation uses the T4 RNA ligase, an enzyme that ligates hydroxyl 3′ ends to monophosphate 5′ ends. Triphosphate 5′ ends are converted to monophosphate ends using the tobacco alkaline phosphatase (TAP) before a second iteration of the ligation. The method as presented here is limited to bacterial RNA and cannot be applied to eukaryotes without a redesign of the protocol, mainly due to the presence of the m7G cap on 5′ends of their mRNAs. The ligation of tags is in principle similar to the RNA hybridization performed in many modern single strand RNA-seq preparation protocols, like the Illumina single stranded RNA-seq,9 with 2 major differences. Firstly, the tag is ligated before RNA fragmentation and thus present only on the reads that correspond to an RNA extremity, i.e. a true transcription start or a processing site (Fig. 1B). Secondly, unlike the sequencing adapters which are used as an anchor for a primer initiating the sequencing process (by synthesis in Illumina or by ligation in SOLiD), the tag is sequenced and present in the final data delivered to the user. In the alignment process, reads are sorted based on the tags presence at the beginning of the read or their absence. The tag sequence is removed in silico and the remaining part of the read is aligned using standard methods.7
While a normal (untagged) read cannot be distinguished from its ghost antisense copy resulting from accidental second strand cDNA formation, such accidents on tagged RNAs will lead to reads carrying the reversed-complemented sequence of the tag toward the end of the read, instead of the tag sequence at the beginning (Fig. 1C, D). Therefore, an antisense RNA the 5′ end of which has been mapped by tagRNA-seq provides a much higher level of certainty on the existence of that transcript. It is worth noting that the T4 RNA ligase is known to act with a different efficiency on different RNA molecules due to differences in structure and accessibility of the 5′ end.7,10,11 As a result, some transcripts will be detected with proportionally more tags than others, but these shortcomings do not affect the detection scheme as the increased confidence on ‘true’ or ‘ghost’ antisense reads is derived from the presence of a tag or its reverse-complement, and does not draw any conclusions from their absence. In the extreme case where a given transcript receives no tags at all, the situation is identical to plain RNA-seq where ‘ghost’ and ‘true’ antisense reads cannot be distinguished.
We performed a search for reads containing reverse complemented tags in the 3 tagRNA-seq experiments performed on the SOLiD platform (5500 XL and Wildfire) described in Innocenti et al., 2014.7 While we previously searched for and sorted the tag sequence at the beginnings of reads using flexbar v2.5,12 and command line parameters –barcode-trim-end LEFT –barcode-threshold 1.6 –barcode-unassigned –barcode-min-overlap 9, reverse completed tags at the end of reads can be found in a similar way by simply changing the command line parameters to –barcode-trim-end RIGHT and using the reverse complemented tag sequences as input.
Overall, we observed that the fraction of reverse complemented tags found lies between 1.8 and 2.6% (Table 1). Those numbers are 3 to 4 time higher than levels of artifactual antisense reported in earlier studies for similar sequencing protocols on the Illumina platform.9 This confirms previous reports that the current single stranded RNA-seq protocols based on RNA hybridization generate small amounts of artifactual antisense RNA5 that limit the sensitivity and reliability of standard RNA-seq for antisense RNA discovery. We stress that none of these reads would have been called as from true antisense transcripts in our procedure precisely because they have the reverse complemented tag at the end. On the contrary, the presence of reads with the tags toward the 5′end confirms that the potential antisense transcripts considered are true ones.
Table 1.
Number of reads found with the tag sequence at the beginning (column “Tags”) and the reverse-complemented tag at the end of the read (“cTags”). The ratio of the latter number to the first is given in parenthesis in the “cTags” column
| Experiment | Tags | cTags |
|---|---|---|
| Rt | 5 557 183 | 98 598 (1.8%) |
| St | 3 597 350 | 87 941 (2.4%) |
| Coli | 4 359 587 | 114 918 (2.6%) |
While designed primarily for identifying 5′ ends and sorting them based on their nature (i.e., primary or processed RNAs), tagRNA-seq appears to be suitable for detection of antisense RNA transcripts. It provides increased confidence on the true existence of detected antisense signals, since their true transcription start and processing sites can be retrieved in a strand specific manner and distinguished from artifactual antisense signal originating from spurious second-strand cDNA. It also allows to perform a quick and simple global qualitative check on the amount of ghost antisense signal in a data set without any additional steps in the experimental protocol. Beyond prokaryotes, many antisense RNAs have also been reported in eukaryotes, solely based on RNA-seq data. An appropriate variant of the present method, enabling to selectively tag eukaryotic 5′RNA ends, would help to distinguish spurious from real antisense transcripts.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Figure 1.
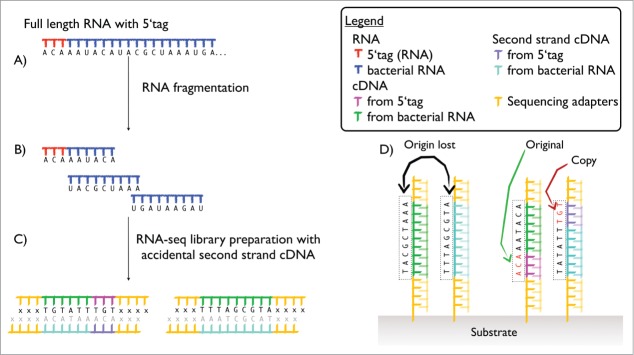
Schematic representation of how a 5′ tagged transcript, its cDNA copy and the corresponding hypothetical ghost resulting from second strand cDNA synthesis appear at different steps of the sequencing protocol. (A) Show the full length RNA after extraction and ligation of the tag to the 5′ end. (B) RNA after fragmentation: the fragment corresponding to the first part of the molecule carries the tag. (C) Schematic representation of the RNA library for tagged and untagged fragment with spurious second-strand cDNA copies. (D) Final DNA fragment on the substrate (beads for SOLiD 5500 and older platform, solid surface of the flow cell for Illumina and SOLiD Wildfire) ready for sequencing. The sequencing adapters hybridize with complementary sequence present on the substrate, the cDNA is used as template for elongation and discarded (semi-transparent in the figure). The figures illustrate how the original cDNA fragment of a tagged read can be distinguished from the second strand copy
Note
a. Annotated bibliography of *Seq assays, Lior Pachter: https://liorpachter.wordpress.com/seq/
References
- 1.Soon WW, Hariharan M, Snyder MP. High-throughput sequencing for biology and medicine. Mol Syst Biol 2013; 9(1):640; PMID:23340846; http://dx.doi.org/ 10.1038/msb.2012.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat Meth 2008; 5(7):621-8, 07; PMID:18516045; http://dx.doi.org/ 10.1038/nmeth.1226 [DOI] [PubMed] [Google Scholar]
- 3.Morton T, Petricka J, Corcoran DL, Li S, Winter CM, Carda A, Benfey PN, Ohler U, Megraw M. Paired-end analysis of transcription start sites in arabidopsis reveals plant-specific promoter signatures. Plant Cell 2014; 26(7):2746-60; PMID:25035402; http://dx.doi.org/ 10.1105/tpc.114.125617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wery M, Descrimes M, Thermes C, Gautheret D, Morillon A. Zinc-mediated RNA fragmentation allows robust transcript reassembly upon whole transcriptome RNA-seq. Methods 2013; 63(1):25-31, Diversity of the non-coding transcriptomes revealed by RNA-seq technologies.; PMID:23523657; http://dx.doi.org/ 10.1016/j.ymeth.2013.03.009 [DOI] [PubMed] [Google Scholar]
- 5.van Dijk EL, Jaszczyszyn Y, Thermes C. Library preparation methods for next-generation sequencing: Tone down the bias. Exp Cell Res 2014; 322(1):12-20; PMID:24440557; http://dx.doi.org/ 10.1016/j.yexcr.2014.01.008 [DOI] [PubMed] [Google Scholar]
- 6.Spiegelman S, Burny A, Das MR, Keydar J, Schlom J, Travnicek M, Watson K. DNA-directed DNA polymerase activity in oncogenic RNA viruses. Nature 1970; 227(5262):1029-31, 09; PMID:4317810; http://dx.doi.org/ 10.1038/2271029a0 [DOI] [PubMed] [Google Scholar]
- 7.Innocenti N, Golumbeanu M, Fouquier d'Hérouël A, Lacoux C, Bonnin RA, Kennedy SP, Wessner F, Serror P, Bouloc P, Repoila F, et al Whole-genome mapping of 5′ RNA ends in bacteria by tagged sequencing: a comprehensive view in Enterococcus faecalis. RNA 2015; 21(5):1018-30; PMID:25737579; http://dx.doi.org/ 10.1261/rna.048470.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fouquier d'Hérouel A, Wessner F, Halpern D, Ly-Vu J, Kennedy SP, Serror P, Aurell E, Repoila F. A simple and efficient method to search for selected primary transcripts: non-coding and antisense RNAs in the human pathogen Enterococcus faecalis. Nucleic Acids Res 2011; 39:e46; PMID:21266481; http://dx.doi.org/ 10.1093/nar/gkr012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA, Friedman N, Gnirke A, Regev A. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Meth 2010; 7(9):709-15, 09; PMID:20711195; http://dx.doi.org/ 10.1038/nmeth.1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhuang F, Fuchs RT, Sun Z, Zheng Y, Robb GB. Structural bias in T4 RNA ligase-mediated 3′-adapter ligation. Nucleic Acids Res 2012; 40:e54; PMID:22241775; http://dx.doi.org/ 10.1093/nar/gkr1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raabe CA, Tang TH, Brosius J, Rozhdestvensky TS. Biases in small RNA deep sequencing data. Nucleic Acids Res 2014; 42:1414-26; PMID:24198247; http://dx.doi.org/ 10.1093/nar/gkt1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dodt M, Roehr JT, Ahmed R, Dieterich C. FLEXBAR— Flexible Barcode and Adapter Processing for Next-Generation Sequencing Platforms. Biology 2012; 1:895-905; PMID:24832523; http://dx.doi.org/ 10.3390/biology1030895 [DOI] [PMC free article] [PubMed] [Google Scholar]