

Abstract
Background: The intestinal microbiota is associated with human health and diseases. The luminal microbiota (LM) and the mucosal-associated microbiota (MAM) are 2 distinct ecosystems with different metabolic and immunological functions. Aim: To characterize the intestinal LM and MAM in humans using high throughput sequencing of the 16S rRNA gene. Methods: Fresh fecal samples and distal colonic mucosal biopsies collected from 24 healthy subjects before (fecal) and during (mucosa) a flexible sigmoidoscopy of an un-prepared bowel. High throughput sequencing of the 16S rRNA gene was used to characterize bacterial communities. Sequences were processed using the QIIME pipeline. Results: LM and MAM populations were significantly different (ANOSIM: R = 0.49, P = 0.001). The LM displayed tighter clustering compared to the MAM (average weighted UniFrac distances 0.27 ± 0.05 vs. 0.43 ± 0.09, P < 0.001, respectively), and showed higher diversity (Shannon diversity index: 4.96 ± 0.37 vs 4.14 ± 0.56, respectively, P < 0.001). The dominant phyla in the LM and MAM were significantly different: Firmicutes (41.4% vs. 29.1%, FDR < 0.0001, respectively), Bacteroidetes (20.2% vs. 26.3%, FDR < 0.05, respectively), Actinobacteria (22% vs. 12.6%, FDR < 0.0001, respectively) and Proteobacteria (9.3% vs. 19.3%, FDR < 0.0001, respectively). The abundance of 56 genera differed significantly (FDR < 0.1) between the 2 niches. All of the genera in the fecal microbiota were present in the MAM while 10 genera were found to be unique to the MAM. Conclusion: The LM and MAM are distinct microbial ecosystems that differ significantly from each other in microbial diversity and composition. These two microbial niches should be investigated independently to better understand the role of the intestinal microbiota in health and disease.
Keywords: human microbiota, high throughput sequencing, intestinal microbiota, mucosal microbiota, 16S rRNA gene
Abbreviations
- IBD
Inflammatory bowel diseases
- IBS
Irritable bowel syndrome
- IRB
Internal Review Board
- LM
Luminal microbiota
- MAM
Mucosal-associated microbiota
- UNC
University of North Carolina
Introduction
Emerging data implicate the importance of the intestinal microbiota with human health and suggest that alterations in the intestinal microbiota have a role in the pathogenesis of gastrointestinal (GI) and non-GI diseases.1
The intestinal microbiota is a complex community of bacteria, archaea, viruses and eukarya. The wide variety of the bacterial species in the GI tract exert numerous effects on the host and influence a variety of GI functions including metabolic activity, immune responses and motor-sensory functions.2,3 The intestinal microbiota consists of 2 separate microbial populations, the luminal microbiota (LM) and the mucosal-associated microbiota (MAM).4-6 Studies investigating the intestinal microbiota in humans have often used fecal samples because they are easily collected and only a few studies have investigated the MAM.7-12 Recent studies comparing the two microbial ecosystems demonstrated compositional and diversity differences between fecal microbiota (representing the luminal niche) and MAM in humans4,10,12 suggesting different roles for these two distinct microbial populations within the intestinal microbiota ecosystem. However, the majority of the studies that investigated the MAM in humans were performed on mucosal samples collected from the large bowel following bowel preparation for endoscopic procedures7-15 which has been shown to significantly affect the composition of the mucosal microbiota.15 Furthermore, the majority of studies that specifically investigated the MAM in humans have used non-sequenced based microbiology techniques such as microbial culture, qPCR12,16 and terminal restriction fragment length polymorphism (T-RFLP).17,18 To our knowledge there has been no study that investigated and directly compared the two microbial populations of the intestinal microbiota from unprepped bowels in humans using deep sequencing technology. Thus the degree to which the composition of the LM differs from the MAM in humans remains unclear.
The goal of the present study was to investigate and compare the intestinal luminal- and mucosal- associated microbiota in humans. We used high throughput pyrosequencing of the bacterial 16S rRNA gene to characterize the fecal- and mucosal-associated microbial communities and examine how these microbial communities differ in regard to their overall microbial richness, diversity and relative abundances of taxa.
Results
Study population
We investigated a total of 24 subjects. All subjects provided a fecal sample and 20 subjects also provided a colonic mucosal sample. The study population consisted of 79.2% females with a mean age of 34 (21–58) years and a body mass index (BMI) of 26.3 (18.1-53) kg/m2.
Analysis of the microbial richness and diversities
A total of 413,669 16S rRNA sequences were obtained from the V1–3 16S rRNA gene regions, with an average of 9,401 reads per sample in the whole cohort (range: 871–47,053). The average number of reads per sample in the stool and mucosal samples was 9,185 and 9,661 respectively.
Rarefaction curves showed a significant higher bacterial community richness in the fecal compared to the mucosal samples (Fig. 1A). Similarly, the Shannon diversity index showed that the diversity of the fecal microbiota was significantly higher than that of the MAM (4.96 ± 0.37 vs 4.14 ± 0.56, respectively, P < 0.001) (Fig. 1B). Additionally, the fecal microbiota displayed significantly tighter clustering compared to the MAM with average weighted UniFrac distances of 0.27 ± 0.05 vs. 0.43 ± 0.09, P < 0.001, respectively (Fig. 2B). However, a significantly less tighter clustering in fecal microbiota compared to the MAM was found using unweighted UniFrac analysis with average distances of 0.64 ± 0.02 vs. 0.62 ± 0.02, P < 0.01, respectively (Fig. 2D).
Figure 1.
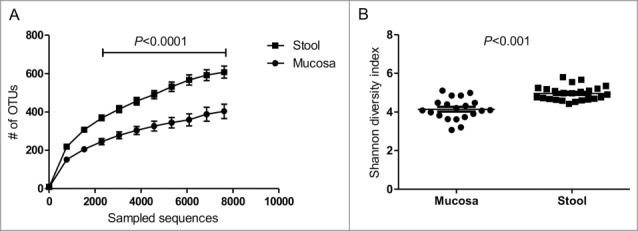
Microbial richness and diversities of fecal and mucosal associated microbiota. (A) Rarefaction curves demonstrating an increased diversity for fecal bacteria. Sequences were rarefied at 10 sequencing depths. The average number of the observed OTUs for each sequencing depth is presented. (B) Shannon diversity index for fecal and mucosal samples from healthy individuals: the diversity was significantly higher in fecal samples compared to mucosal samples. Data are presented as mean ± Standard deviation of the mean for each of the niches; student's t test was calculated using GraphPad prism for unpaired normally distributed samples.
Figure 2.
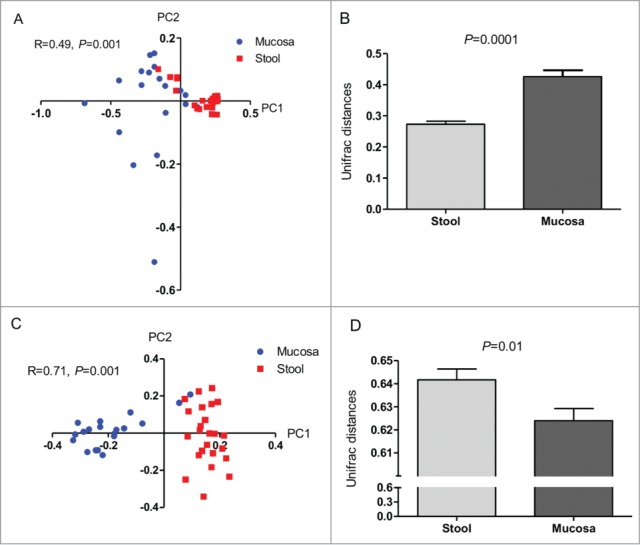
Variation in bacterial composition between fecal and mucosal microbiota. PCoA plots demonstrating that the fecal microbiota and MAM are significantly different in weighted (A) and unweighted (C) analysis. Fecal microbiota displayed significantly tighter clustering compared to the MAM (B). A significantly less tighter clustering in fecal microbiota compared to the MAM was found using unweighted UniFrac analysis with average distances (D). Data are presented as mean ± Standard deviation of the mean for each of the niches, Mann-Whitney test was used to calculate P values.
Taxa analysis
The fecal microbiota and MAM were found to be significantly different (ANOSIM, weighted: R = 0.49, P = 0.001; unweighted: R = 0.71, P = 0.001) (Fig. 2A and C). Further comparisons of the proportions of dominant bacterial taxa (taxa that appear in at least 25% of samples) between the fecal and mucosal microbiota revealed significant differences in the abundance of dominant phyla: Firmicutes 41.4% vs. 29.1%, FDR < 0.0001, respectively), Bacteroidetes (20.2% vs. 26.3%, FDR < 0.05, respectively), Actinobacteria (22% vs. 12.6%, FDR < 0.0001, respectively) and Proteobacteria (9.3% vs. 19.3%, FDR < 0.0001, respectively), as depicted in Figure 3.
Figure 3.
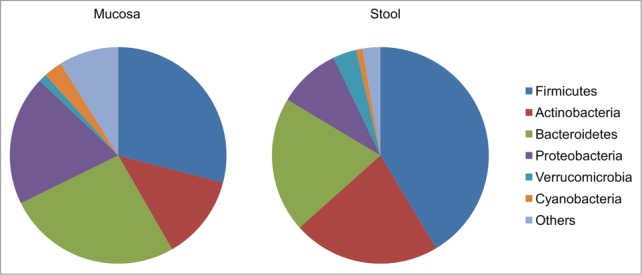
Proportions of core bacterial taxa in fecal and colonic mucosal samples from healthy individuals. Compared to mucosal samples, fecal samples had a significantly higher abundance of Firmicutes (41.4% vs. 29.1%, FDR < 0.0001, respectively) and of Actinobacteria (22% vs. 12.6%, FDR < 0.0001, respectively) compared to mucosal samples, and a significantly lower abundance of Bacteroidetes (20.2% vs. 26.3%, FDR < 0.05, respectively), and Proteobacteria (9.3% vs. 19.3%, FDR < 0.0001, respectively).
The dominant taxa were comprised of 68 genera. Only 11 genera (Streptococcus, Blautia, Coprococcus, Dorea, Ruminococcus, Eubacterium and Faecalibacterium) were found in all 44 samples. The abundance of 56 genera differed significantly between the 2 niches. All genera encompassed within the Proteobacteria (n = 9) and Bacteroidetes (n = 6) phyla and most genera within the Firmicutes phyla (23 out of 34) were more abundant in the MAM niche compared to the LM while all genera within the Actinobacteria (n = 6) phyla were more abundant in the LM Figure 4. A list of taxa abundances at the genus level is detailed in Table 1. In addition, 10 genera were found to be unique to the MAM; however, all of the genera (of the dominant taxa) found in the fecal microbiota were also present in the MAM. The relative proportion of some genera was more than 10 times higher in the MAM compared to the fecal microbiota, these included- Finegoldia, Anaerococcus and Peptoniphilus.
Figure 4.
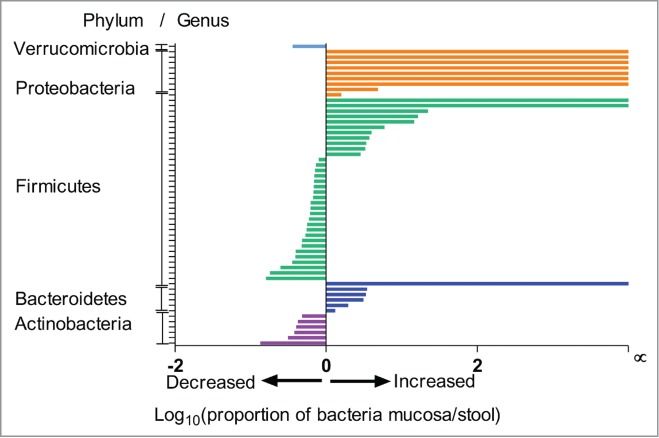
Ratio of bacterial abundance in mucosa compared to stool. Ratio of bacterial abundance in mucosa compared to stool displaying major differences in dominant taxa at the Phylum/Genus level.
Table 1.
Ratio of proportions of genera between mucosa to stool*
| Phylum | Genus | Mucosal | Fecal | P value | FDR* | Ratio mucosa/stool |
|---|---|---|---|---|---|---|
| Actinobacteria | Bifidobacterium | 8.03E-04 | 5.98E-03 | 5.70E-03 | 1.02E-02 | 1.34E-01 |
| Actinobacteria | 4.89E-03 | 1.56E-02 | 4.54E-04 | 1.10E-03 | 3.13E-01 | |
| Actinobacteria | Adlercreutzia | 6.04E-03 | 1.59E-02 | 6.90E-04 | 1.62E-03 | 3.79E-01 |
| Actinobacteria | Collinsella | 8.55E-03 | 2.13E-02 | 3.56E-03 | 6.92E-03 | 4.02E-01 |
| Actinobacteria | Eggerthella | 5.42E-03 | 1.29E-02 | 1.17E-02 | 1.95E-02 | 4.21E-01 |
| Actinobacteria | Actinomyces | 2.69E-03 | 5.59E-03 | 3.09E-02 | 4.20E-02 | 4.81E-01 |
| Bacteroidetes | Bacteroides | 3.94E-02 | 3.00E-02 | 2.33E-02 | 3.45E-02 | 1.31E+00 |
| Bacteroidetes | 5.81E-03 | 2.96E-03 | 8.08E-02 | 9.81E-02 | 1.96E+00 | |
| Bacteroidetes | Odoribacter | 5.95E-03 | 1.92E-03 | 2.86E-02 | 4.13E-02 | 3.09E+00 |
| Bacteroidetes | Prevotella | 2.51E-02 | 7.49E-03 | 1.47E-05 | 6.26E-05 | 3.35E+00 |
| Bacteroidetes | Butyricimonas | 5.96E-03 | 1.71E-03 | 1.57E-02 | 2.49E-02 | 3.48E+00 |
| Bacteroidetes | Porphyromonas | 7.10E-03 | 0.00E+00 | 1.06E-04 | 3.13E-04 | |
| Firmicutes | Lachnobacterium | 1.23E-03 | 7.73E-03 | 4.02E-03 | 7.59E-03 | 1.59E-01 |
| Firmicutes | cc_115 | 9.86E-04 | 5.43E-03 | 7.77E-03 | 1.32E-02 | 1.81E-01 |
| Firmicutes | SMB53 | 4.03E-03 | 1.62E-02 | 2.29E-04 | 5.98E-04 | 2.49E-01 |
| Firmicutes | Turicibacter | 3.30E-03 | 9.31E-03 | 2.27E-02 | 3.43E-02 | 3.55E-01 |
| Firmicutes | Anaerostipes | 6.12E-03 | 1.57E-02 | 1.29E-03 | 2.83E-03 | 3.91E-01 |
| Firmicutes | 7.60E-03 | 1.92E-02 | 2.84E-05 | 1.07E-04 | 3.96E-01 | |
| Firmicutes | 1.16E-02 | 2.45E-02 | 5.77E-06 | 2.80E-05 | 4.73E-01 | |
| Firmicutes | 5.92E-03 | 1.23E-02 | 2.97E-02 | 4.12E-02 | 4.80E-01 | |
| Firmicutes | 1.74E-02 | 3.26E-02 | 1.63E-04 | 4.43E-04 | 5.35E-01 | |
| Firmicutes | Clostridium | 1.57E-02 | 2.85E-02 | 3.28E-05 | 1.12E-04 | 5.52E-01 |
| Firmicutes | 1.19E-02 | 2.12E-02 | 3.50E-03 | 7.00E-03 | 5.60E-01 | |
| Firmicutes | Coprococcus | 2.65E-02 | 4.48E-02 | 1.95E-08 | 2.66E-07 | 5.91E-01 |
| Firmicutes | Blautia | 2.88E-02 | 4.73E-02 | 1.33E-08 | 2.27E-07 | 6.10E-01 |
| Firmicutes | Lachnospira | 1.54E-02 | 2.48E-02 | 5.32E-03 | 9.78E-03 | 6.20E-01 |
| Firmicutes | [Ruminococcus] | 2.11E-02 | 3.37E-02 | 2.16E-06 | 1.22E-05 | 6.26E-01 |
| Firmicutes | Roseburia | 1.95E-02 | 2.90E-02 | 2.58E-03 | 5.49E-03 | 6.74E-01 |
| Firmicutes | 3.25E-02 | 4.78E-02 | 2.42E-08 | 2.74E-07 | 6.79E-01 | |
| Firmicutes | Faecalibacterium | 2.87E-02 | 4.20E-02 | 4.47E-05 | 1.45E-04 | 6.83E-01 |
| Firmicutes | 3.70E-02 | 5.35E-02 | 1.00E-07 | 8.54E-07 | 6.92E-01 | |
| Firmicutes | Dorea | 2.25E-02 | 3.22E-02 | 9.52E-04 | 2.16E-03 | 6.98E-01 |
| Firmicutes | 3.55E-02 | 4.98E-02 | 1.21E-06 | 7.49E-06 | 7.12E-01 | |
| Firmicutes | Ruminococcus | 2.56E-02 | 3.50E-02 | 3.48E-03 | 7.17E-03 | 7.33E-01 |
| Firmicutes | Streptococcus | 2.01E-02 | 2.51E-02 | 5.71E-02 | 7.19E-02 | 7.99E-01 |
| Firmicutes | Staphylococcus | 3.84E-03 | 1.35E-03 | 2.89E-02 | 4.10E-02 | 2.85E+00 |
| Firmicutes | Clostridium | 6.67E-03 | 2.02E-03 | 5.10E-02 | 6.67E-02 | 3.30E+00 |
| Firmicutes | Lactobacillus | 8.40E-03 | 2.48E-03 | 5.97E-03 | 1.04E-02 | 3.39E+00 |
| Firmicutes | Mogibacterium | 3.11E-03 | 8.29E-04 | 5.29E-02 | 6.78E-02 | 3.75E+00 |
| Firmicutes | Parvimonas | 4.34E-03 | 1.10E-03 | 4.59E-02 | 6.12E-02 | 3.96E+00 |
| Firmicutes | Anaerotruncus | 5.12E-03 | 8.60E-04 | 1.69E-02 | 2.62E-02 | 5.96E+00 |
| Firmicutes | Finegoldia | 1.82E-02 | 1.24E-03 | 2.19E-10 | 1.49E-08 | 1.46E+01 |
| Firmicutes | Anaerococcus | 1.07E-02 | 6.49E-04 | 1.08E-06 | 7.37E-06 | 1.64E+01 |
| Firmicutes | Peptoniphilus | 1.71E-02 | 7.70E-04 | 3.47E-10 | 1.18E-08 | 2.22E+01 |
| Firmicutes | 1–68 | 8.24E-03 | 0.00E+00 | 1.02E-04 | 3.16E-04 | 1.00E+04 |
| Firmicutes | WAL_1855D | 1.06E-02 | 0.00E+00 | 2.22E-05 | 8.86E-05 | |
| Proteobacteria | Sutterella | 2.05E-02 | 1.29E-02 | 1.34E-02 | 2.17E-02 | 1.59E+00 |
| Proteobacteria | 1.62E-02 | 3.35E-03 | 2.73E-04 | 6.88E-04 | 4.84E+00 | |
| Proteobacteria | Comamonas | 4.59E-03 | 0.00E+00 | 1.10E-04 | 3.12E-04 | |
| Proteobacteria | Ralstonia | 7.13E-03 | 0.00E+00 | 3.08E-05 | 1.10E-04 | |
| Proteobacteria | Campylobacter | 7.96E-03 | 0.00E+00 | 6.08E-06 | 2.75E-05 | |
| Proteobacteria | Alishewanella | 9.79E-03 | 0.00E+00 | 7.95E-08 | 7.73E-07 | |
| Proteobacteria | Acinetobacter | 6.37E-03 | 0.00E+00 | 2.69E-06 | 1.41E-05 | |
| Proteobacteria | 7.36E-03 | 0.00E+00 | 2.87E-07 | 2.17E-06 | ||
| Proteobacteria | Pseudomonas | 1.57E-02 | 0.00E+00 | 4.72E-10 | 1.07E-08 | |
| Verrucomicrobia | Akkermansia | 2.05E-03 | 5.73E-03 | 7.78E-02 | 9.62E-02 | 3.58E-01 |
Only statistically significant (false discovery rate (FDR) < 0.1) groups are included in the table.
The five genera within the MAM that exhibited the highest average proportion were Bacteroides (3.9%), an un-annotated genus from the Ruminococcaceae family (3.7%), an un-identified genus of the Lachnospiraceae family (3.54%), an un-annotated genus from the Clostridiales order (3.25%) and Blautia (2.88%). In contrast, the dominant genera from the fecal microbiota included: an un-annotated genus from the Ruminococcaceae family (5.34%), an un-identified genus of the Lachnospiraceae family (5%), an un-annotated genus from the Clostridiales order (4.78%), Blautia (4.72%), and Coprococcus (4.4%).
Correlation analysis
Correlation analysis on the abundance of genera in the fecal microbiota and MAM revealed that 17 genera significantly correlated between the two niches (Table 2). Among these genera, the two most prominent correlations belonged to the Actinobacteria phyla, and to the Coriobacteriaceae family: these included- Collinsella (r = 0.92, FDR < 0.0001), Adlercreutzia (r = 0.85, FDR < 0.0001). However, some genera from all major phyla (Firmicutes, Bacteriodetes, Proteobacteria, and Verrucomicrobia) were found to be correlated between the two niches.
Table 2.
Bacterial taxa that significantly correlate between the enteric mucosal and luminal niches
| Phylum | Family | Genus | R* | P value | FDR* |
|---|---|---|---|---|---|
| Actinobacteria | Coriobacteriaceae | Adlercreutzia | 0.85 | 0.0000 | 0.0000 |
| Collinsella | 0.92 | 0.0000 | 0.0000 | ||
| Bacteroidetes | Porphyromonadaceae | Parabacteroides | 0.57 | 0.0037 | 0.0171 |
| Firmicutes | Gemellaceae | unannotated | 0.51 | 0.0110 | 0.0338 |
| Christensenellaceae | unannotated | 0.60 | 0.0021 | 0.0119 | |
| Clostridiaceae | unannotated | 0.73 | 0.0001 | 0.0006 | |
| Clostridium | 0.48 | 0.0182 | 0.0524 | ||
| Lachnospiraceae | Dorea | 0.56 | 0.0047 | 0.0196 | |
| Roseburia | 0.54 | 0.0062 | 0.0221 | ||
| Peptostreptococcaceae | unannotated | 0.45 | 0.0262 | 0.0710 | |
| Ruminococcaceae | Ruminococcus | 0.67 | 0.0003 | 0.0023 | |
| Veillonellaceae | Dialister | 0.59 | 0.0025 | 0.0127 | |
| Phascolarctobacterium | 0.61 | 0.0015 | 0.0101 | ||
| Erysipelotrichaceae | cc_115 | 0.73 | 0.0001 | 0.0008 | |
| Coprobacillus | 0.52 | 0.0097 | 0.0317 | ||
| Proteobacteria | Alcaligenaceae | Sutterella | 0.55 | 0.0058 | 0.0223 |
| Verrucomicrobia | Verrucomicrobiaceae | Akkermansia | 0.70 | 0.0001 | 0.0013 |
r-correlation coefficient; FDR-false discovery rate.
Functional genes analysis
PICRUSt predicted that 34 functional gene categories significantly differed between the 2 niches (FDR < 10%). Within the mucosal adherent bacteria niche there was an increased proportion of genes responsible for bacterial invasion into epithelial cells, protein digestion, and fatty acids metabolism. A complete list of genes categories that differed between the niches is presented in supplemental Table 2.
Discussion
Previous studies comparing human fecal and mucosal microbial ecosystems suggest diversity and compositional differences between these 2 distinct niches.10,12,16-18 However, these previous studies were performed on mucosal samples collected following bowel preparation for endoscopic procedures10,12,16-18 or used non-sequenced based microbiology techniques.16-18 Thus, we sought to add to the current knowledge by conducting a deep sequencing analysis on mucosal samples from unprepped colons and fecal samples collected from all participants at the same day of the study visit. Our study included a cohort of healthy individuals that were carefully screened to exclude GI diseases, chronic symptoms, and recent use of interventions (e.g., antibiotics, probiotics, antidiarrheal, pro-motility and anti-inflammatory agents) that can alter their intestinal microbiota.
In a previous study we used T-RFLP to compare the fecal and mucosal microbiota from unprepped colons of 16 patients with D-IBS and 21 healthy controls.17 We demonstrated a significantly higher level of microbial biodiversity in the fecal microbiota compared to MAM within both, samples collected from the D-IBS patients and samples collected from healthy controls. The increase in biodiversity in fecal compared to mucosal samples was greater in the healthy controls than in D-IBS patients. Another study using T-RFLP compared bacterial diversity and composition from rectal swabs and rectal mucosal biopsies from unprepped colons of 11 healthy volunteers.18 This study also found that the overall microbial diversity was higher in rectal swabs compared with rectal biopsies. Consistent with these studies our current study, using high throughput sequencing of the 16S rRNA gene, demonstrates higher levels of microbial richness (based on the number of observed species) and diversity (based on the Shannon index of diversity) in fecal samples compared to mucosal samples from healthy individuals (Fig. 1). Additionally, weighted Unifrac distances exhibited tighter clustering within fecal samples compared to the mucosal samples (Fig. 2). UniFrac distances represent the fraction of branch length shared by any 2 samples' communities in a 16S rRNA-based phylogenetic tree generated by sequence data obtained from all samples. Weighted UniFrac analyses indicate microbiota community differences between sample groups due to differences in relative taxon abundance, whereas unweighted UniFrac analyses indicate community differences between sample groups based on the presence or absence of taxa.19 This suggests that despite high diversity levels, there is also higher similarity of dominant OTUs among fecal samples compared to mucosal samples. Our study also supports the notion that the LM and MAM are distinct niches in terms of bacterial composition (Fig. 3). The dominant genera were different between the fecal microbiota and MAM. Out of 68 dominant genera, the proportion of 56 genera differed significantly between the 2 niches. Interestingly, of the genera that were more abundant in the MAM (n = 26) compared to the fecal microbiota, some were more than 10 fold higher, suggesting that these genera may be of greater importance at the mucosal level where their microbiologic, metabolic and/or immunologic effects may be more pronounced. In addition, it is important to note that while all of the dominant genera that were found in the fecal microbiota were also present in the MAM, certain dominant genera were represented solely at the MAM niche. This suggests that while studying the intestinal microbiota with the current technologies (down to the genus level) sampling and analyzing the fecal microbiota may not be sufficient as additional information can be generated when the MAM niche is studied. This principle was illustrated in determining dysbiosis in newly diagnosed IBD patients.10 Furthermore, our study also suggests that when investigating specific genera within the intestinal microbiota, one of the niches (LM or MAM) may be more informative than the other. For instance all genera belonging to the Proteobacteria phylum are more abundant at the MAM niche. Within this phylum most of the genera (7 out of 9, such as Acinetobacter, Ralstonia and Pseudomonas) were not represented in the LM niche. In contrast, all the genera of the Actinobacteria phylum were more abundant in the LM niche. For instance the relative proportion of Bifidobacterium was more than 5 fold higher in the LM compared to the MAM niche.
The observed differences in microbial compositions between the fecal and mucosal niches may be a consequence of the different environments surrounding the 2 niches and are likely to have different physiological implications. For example, the differences in bacterial composition may relate to differences in oxygen tension between the fecal and mucosal niches.20 Consistent with the recent report by Albenberg et at.21 the higher oxygen tension in the mucosal niche may be responsible for our finding of increase in proportion of organisms from the Proteobacteria phyla that are rich in aerotolerant bacteria (such as Campylobacter genus- Table 1). Other relevant factors including differences in nutrient availability, antibacterial components (e.g., defensins)22 and the effects of the intestinal mucous factors (e.g., MUC2 mucins) may influence the composition of the MAM.23
Comparison of bacterial groups in our study with those found by Albenberg et at.,21 further support the finding that specific bacterial groups are unique to the mucosal niche. For example, in our study Proprionibacterium (Actinobacteria) was found in 8 mucosal samples and no fecal sample, and Corynbacterium (Actinobacteria) was found in 7 mucosal samples and only 1 fecal sample. Both aerotolerant bacterial groups are not mentioned in Table 1, as we could not report a statistical difference between LM and MAM niches. A potential mechanism for an increase of specific bacteria in the mucosal niche (Albenberg et al.)21 includes expansion of asaccharolytic bacteria (bacteria that require proteinaceous substrates, that are thought to be more common in the mucosal niche due to shedding of intestinal epithelial cells) as energy sources. Indeed, the genera Finegoldia, Anaerococcus and Peptoniphilus (all belonging to the Firmicutes phylum- Table 1) and the genus Porphyoromonas (Bacteroidetes phylum) are all significantly more abundant in the mucosal niche compared to the fecal niche and are all asaccharolytic, which is consistent with Albenberg et al.'s study.21 Some of these findings were supported by gene functional analysis using the PICRUSt software that predicts gene function according to the 16S rRNA sequences. However, it should be noted that the PICRUSt software is limited in its accuracy (about 80%) for prediction of bacterial metagenomes and their function. Despite the differences between the two niches, 11 genera were common to all 44 samples. However, correlation analysis revealed that out of the 68 dominant genera only 4 genera demonstrate a strong (r=0.70) and significant correlation between the abundance of genera within the fecal and the MAM. This may suggest that these genera may have important dominant functional role at both the luminal and mucosal microbial niches and perhaps fecal studies of these genera may be extrapolated to the mucosal niche.
Our study has several strengths including the carefully selected population to exclude chronic or recurring GI symptoms, GI diseases, conditions (e.g., celiac disease, lactose malabsorption) or recent interventions (e.g., antibiotic treatment or intentional consumption of probiotics) that could affect the intestinal microbiota; collection of fecal and mucosal samples on site at the same day of the analysis; collection of colonic biopsies from unprepped colon; biopsies taken at a single, consistent location in the colon, consistent DNA isolation and sequencing techniques for both niches; and deep sequencing analysis by 454 sequencing. Our study limitations include the LM was investigated from fecal samples representing the overall end product of the entire GI tract luminal microbiota. Furthermore, the biostructure of a fecal sample has been previously documented24 and had shown different bacterial composition in the inside and outside layers of the sample. Thus, a homogenized fecal sample is a mix of different microbial niches within the fecal sample while the mucosal biopsy represents a single microbial niche from a single site at the distal part of the colon.
In conclusion, taken together our findings demonstrate that the intestinal luminal microbiota (represented by fecal samples) and MAM (represented by the colonic biopsy samples) are distinct microbial ecosystems that differ significantly from each other in diversity and microbial composition.
Our findings highlight the importance of sampling and investigating both the LM and the MAM in intestinal microbiota-based studies. These two microbial ecosystems should be investigated independently to better understand the role of the intestinal microbiota in health and disease conditions.
Methods
Study population
We studied 24 healthy individual volunteers. Inclusion criteria comprised of subjects age 18 or older, any gender, race, or ethnicity. All subjects were evaluated by a physician to exclude GI disease conditions. Healthy individuals had no significant chronic or recurring GI symptoms. Subjects with a history of GI tract surgery other than appendectomy or cholecystectomy, a history of inflammatory bowel diseases (IBD), celiac disease, lactose malabsorption, or other diagnoses that could affect intestinal microbiome were excluded from the study. In addition, participants were excluded if they had a history of antibiotic treatment or intentional consumption of probiotics 2 months prior to the enrolment in the study. Subjects were recruited from the Chapel Hill general population by advertising and from the University of North Carolina (UNC) at Chapel Hill outpatient clinics. The study was approved by the UNC Internal Review Board and all subjects signed a consent form prior to participation in the study.
Sample collection and preparation
Colonic mucosal samples were collected from all subjects during an un-sedated flexible sigmoidoscopy. To avoid the possible effects of a colonic cleansing preparation on the mucosal associated microbiota, all endoscopic procedures were performed on un-prepped colons. Colonic mucosal biopsies were taken from the distal colon just above the rectosigmoid junction using cold biopsy forceps. Once removed from the colon, each biopsy was washed and swirled in 1 ml of sterile PBS to remove non-adherent fecal material. The biopsy samples were then weighed, flash-frozen in liquid nitrogen and stored at −80°C for further DNA extraction and molecular microbiological analysis.
Fresh stool samples were collected from the same participants on site immediately prior to the flexible sigmoidoscopy or at home at the same morning of the procedure. Following collection each fecal sample was immediately transferred on ice to the laboratory where it was homogenized, divided into aliquots and stored at −80°C for DNA extraction and sequencing analysis. Fecal samples were collected and handled following a recently validated protocol.25
Extraction of DNA
Bacterial DNA was isolated from a total of 20 paired mucosal and fecal and 4 additional fecal samples, for which mucosal samples were not available due to technical reasons. DNA extraction was carried out using a phenol/chloroform extraction method combined with physical disruption of bacterial cells and a DNA clean-up kit (Qiagen DNeasy® Blood and Tissue extraction kit [Qiagen, 69504]) as previously described.26 In brief, 100 mg of frozen feces or a mucosal biopsy was suspended in 750 μl of sterile bacterial lysis buffer (200 mM NaCl, 100 mM Tris [pH 8.0], 20 mM EDTA, 20 mg mL−1 lysozyme) and incubated at 37°C for 30 min. Next, 40 μl of proteinase K (20 mg mL−1) and 85 μl of 10% SDS was added to the mixture and incubated at 65°C for 30 min. 300 mg of 0.1 mm zirconium beads (BioSpec Products, 11079101z) was then added and the mixture and homogenized in a bead beater (BioSpec Products, 112011) for 2 min. The homogenized mixture was cooled on ice and then centrifuged at 14,000 rpm for 5 min. The supernatant was transferred to a new 1.5 ml microfuge tube and fecal DNA was further extracted by phenol/chloroform/iso-amyl alcohol (25:24:1) and then chloroform/iso-amyl alcohol (24:1). Following extraction the supernatant was precipitated by absolute ethanol at −20°C for 1 hour. The precipitated DNA was suspended in DNase free H2O and then cleaned using the DNeasy® Blood and Tissue extraction kit (Qiagen, 69504) from step 3 as per the manufacturer's instructions.
454 pyro-sequencing of 16S rRNA genes
Bacterial community composition in isolated DNA samples was characterized by amplification of the V1-3 (forward, 8f:5′-AGAGTTTGATCMTGGCTCAG-3′; reverse 518r: 5′-ATTACCGCGGCTGCTGG-3′) variable regions of the 16S rRNA gene by polymerase chain reaction (PCR). Forward primers were tagged with 10 bp unique barcode labels at the 5′ end along with the adaptor sequence (5′-CCATCTCATCCCTGCGTGTCTCCGACTCAG-3′) to allow multiple samples to be included in a single 454 GS FLX Titanium sequencing plate as previously described.27,28 16S rRNA PCR products were quantified, pooled, and purified for the sequencing reaction. 454 GS FLX Titanium sequencing was performed on a 454 Life Sciences Genome Sequencer FLX machine (Roche, Florence, SC) at the microbiome core at UNC-Chapel Hill (http://www.med.unc.edu/microbiome).
Analysis of 16S rRNA sequences using the QIIME pipeline
16S rRNA sequence data generated by the 454 GS FLX Titanium sequencer was processed by the quantitative insights into microbial ecology (QIIME) pipeline.29 Briefly, sequences that were less than 200 bp or greater than 1,000 bp in length, contained incorrect primer sequences, or contained more than 1 ambiguous base were discarded. Operational taxonomic units (OTUs) were picked using BLAST and the Greengenes reference database at a level of 97% similarity.30 For β diversity (UniFrac distances) we analyzed 1,500 sequences from each sample (losing only one sample with less than 1,000 sequences that was not included in the analysis). This depth was chosen to encompass the majority of samples (n = 43) at an acceptable sequencing depth. UniFrac analysis was used to calculate the distance between OTUs/bacterial communities on a phylogenetic tree and principal coordinates were generated using un-weighted and weighted UniFrac distances for all samples.19,31 Weighted and un-weighted UniFrac distances represent compositional dissimilarity/heterogeneity (often refer to as β diversity). However, un-weighted analysis considers only the presence or absence of bacterial groups thus, all bacterial groups have similar impact on the UniFrac distances. In weighed analysis the relative abundance of the bacterial groups within the communities is being accounted for and thus, the more abundant bacterial groups have a greater impact on the UniFrac distances. Principal coordinates Analysis (PCoA) plots were used to visualize the similarities or dissimilarities between the fecal and MAM that best represent the pair-wise distances between sample groups. Statistical differences in weighted and un-weight UniFrac distances between groups (β diversity) were tested using analysis of similarity (ANOSIM – available through QIIME) by permutation of group membership with 999 replicates. The test statistic R, which measures the strength of the results, ranges from −1 to 1: R = 1 signifies differences between groups, while R = 0 signifies that the groups are identical.
Rarefaction curves were generated by the QIIME software. All reads were pooled for each of the two niches. Sequences were rarefied at 10 sequencing depths in order to best visualize the change in diversity with respect to sampling depth. Samples that had less reads than the sequencing depth of a group sampled- were not included in that group. For example, if a sample has 1,500 reads, it was not included in the statistical analysis of the rarefied data of samples with 2,293, 4,576 or 7,620 reads. The upper bound of rarefaction is the median number of sequences for all samples (7,620). Ten sampling intervals were used. The number of observed bacterial species and the Shannon index of diversity (a measure of diversity taking into account richness and evenness of OTUs)27 were compared following rarefaction of OTUs using a Student's t test. The proportions of dominant bacterial taxa (taxa that appear in at least 25% of samples of within either fecal or mucosal niche) were normalized as previously described28 and compared using a student's t test. Briefly, to account for variability in depth of sequencing the abundance of each taxon (OTU/Phylum/etc.) in a given sample was normalized by calculating the logged sequence abundance using the following formula: Log10[(Frequency / # sequences in sample)*Average # of sequences per sample +1]. The relative proportions of the abundances were calculated and used for all statistics presented. Normalized taxa abundances from both niches were correlated with each other and using Pearson correlation. All results were corrected for multiple comparisons and were considered significant if false discovery rate (FDR) was <0.1.
Gene function prediction
We used PICRUSt32 to predict and categorize gene function of mucosal and fecal bacteria. We compared categories of bacterial gene function between the mucosa and stool setting a threshold of >2 fold differences between niches with a P < 0.5. Significant differences were reported if an FDR <10% was achieved.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank Raad Gharaibeh PhD for his help with the microbiota analysis. The authors would also like to thank the UNC Microbiome Core for their help with this study.
Funding
This study was partially funded by DK084294 and DK075621 grants from the National Institutes of Health awarded to YR. This study was also partially supported by Award Number UL1RR025747 from the National Center for Research Resources.
References
- 1.Bäckhed F, Fraser-Liggett C, Ringel Y, Sanders ME, Sartor RB, Sherman PM, Versalovic J, Young V, Finlay BB. Defining a healthy human gut microbiome: current concepts, future directions, and clinical applications. Cell Host Microbe 2012; 15(5):611-22; PMID:23159051; http://dx.doi.org/ 10.1016/j.chom.2012.10.012 [DOI] [PubMed] [Google Scholar]
- 2.Kamada N, Seo SU, Chen GY, Núñez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol 2013; 13(5):321-35; PMID:23618829; http://dx.doi.org/ 10.1038/nri3430 [DOI] [PubMed] [Google Scholar]
- 3.Ringel Y, Maharshak N. Intestinal microbiota and immune function in the pathogenesis of irritable bowel syndrome. Am J Physiol Gastrointest Liver Physiol 2013; 305(8):G529-41; PMID:23886861; http://dx.doi.org/ 10.1152/ajpgi.00207.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science 2005; 309(5728):1635-8; PMID:15831718; http://dx.doi.org/ 10.1126/science.1110591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van den Abbeele P, Van de Wiele T, Verstraete W, Possemiers S. The host selects mucosal and luminal associations of coevolved gut microorganisms: A novel concept. FEMS Microbiol Rev 2011; 35(4):681-704; PMID:21361997; http://dx.doi.org/ 10.1111/j.1574-6976.2011.00270.x [DOI] [PubMed] [Google Scholar]
- 6.Walker AW, Sanderson JD, Churcher C, Parkes GC, Hudspith BN, Rayment N, Brostoff J, Parkhill J, Dougan G, Petrovska L. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and noninflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol 2011; 11:7; PMID:21219646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sokol H, Lepage P, Doré J, Marteau P. Molecular comparison of dominant microbiota associated with injured versus healthy mucosa in ulcerative colitis. Gut 2007; 56(1):152-4; PMID:17172591; http://dx.doi.org/ 10.1136/gut.2006.109686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hong PY, Croix JA, Greenburg E, Gaskins HR, Mackie RI. Pyrosequencing-based analysis of the mucosal microbiota in healthy individuals reveals ubiquitous bacterial groups and micro-heterogeneity. PLoS One 2011; 6(9):e25042; PMID:21966408; http://dx.doi.org/ 10.1371/journal.pone.0025042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parkes GC, Rayment NB, Hudspith BN, Petrovska L, Lomer MC, Brostoff J, Whelan K, Sanderson JD. Distinct microbial populations exist in the mucosa-associated microbiota of sub-groups of irritable bowel syndrome. Neurogastroenterol Motil 2012; 24(1):31-9; PMID:22070725; http://dx.doi.org/ 10.1111/j.1365-2982.2011.01803.x [DOI] [PubMed] [Google Scholar]
- 10.Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, et al.. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host & Microbe 2014; 15(3):382-92; PMID:24629344; http://dx.doi.org/ 10.1016/j.chom.2014.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 2007; 104(34):13780-5; PMID:17699621; http://dx.doi.org/ 10.1073/pnas.0706625104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyra A, Forssten S, Rolny P, Wettergren Y, Lahtinen SJ, Salli K, Cedgård L, Odin E, Gustavsson B, Ouwehand AC. Comparison of bacterial quantities in left and right colon biopsies and faeces. World J Gastroenterol 2012; 18(32):4404-11; PMID:22969206; http://dx.doi.org/ 10.3748/wjg.v18.i32.4404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Momozawa Y, Deffontaine V, Louis E, Medrano JF. Characterization of bacteria in biopsies of colon and stools by high throughput sequencing of the V2 region of bacterial 16S rRNA gene in human. PLoS One 2011; 6(2):e16952; PMID:21347324; http://dx.doi.org/ 10.1371/journal.pone.0016952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Brien CL, Allison GE, Grimpen F, Pavli P. Impact of colonoscopy bowel preparation on intestinal microbiota. PLoS One 2013; 8(5):e62815; PMID:23650530; http://dx.doi.org/ 10.1371/journal.pone.0062815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrell L, Wang Y, Antonopoulos D, Young V, Lichtenstein L, Huang Y, Hanauer S, Chang E. Standard colonic lavage alters the natural state of mucosal-associated microbiota in the human colon. PLoS One 2012; 7(2):e32545; PMID:22389708; http://dx.doi.org/ 10.1371/journal.pone.0032545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carroll IM, Chang YH, Park J, Sartor RB, Ringel Y. Luminal and mucosal-associated intestinal microbiota in patients with diarrhea-predominant irritable bowel syndrome. Gut Pathog 2010; 2(1):19; PMID:21143915; http://dx.doi.org/ 10.1186/1757-4749-2-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carroll IM, Ringel-Kulka T, Keku TO, Chang YH, Packey CD, Sartor RB, Ringel Y. Molecular analysis of the luminal- and mucosal-associated intestinal microbiota in diarrhea-predominant irritable bowel syndrome. Am J Physiol Gastrointest Liver Physiol 2011; 301(5):G799-807; PMID:21737778; http://dx.doi.org/ 10.1152/ajpgi.00154.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Araújo-Pérez F, McCoy AN, Okechukwu C, Carroll IM, Smith KM, Jeremiah K, Sandler RS, Asher GN, Keku TO. Differences in microbial signatures between rectal mucosal biopsies and rectal swabs. Gut Microbes 2012; 3(6):530-5; PMID:23060016; http://dx.doi.org/ 10.4161/gmic.22157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carroll IM, Ringel-Kulka T, Siddle JP, Klaenhammer TR, Ringel Y. Characterization of the fecal microbiota using high-throughput sequencing reveals a stable microbial community during storage. PLoS ONE 2012; 7(10):e46953; PMID:23071673; http://dx.doi.org/ 10.1371/journal.pone.0046953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marteyn B, West NP, Browning DF, Cole JA, Shaw JG, Palm F, Mounier J, Prévost MC, Sansonetti P, Tang CM. Modulation of Shigella virulence in response to available oxygen in vivo. Nature 2010; 465(7296):355-8; PMID:20436458; http://dx.doi.org/ 10.1038/nature08970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Albenberg L, Esipova TV, Judge CP, Bittinger K, Chen J, Laughlin A, Grunberg S, Baldassano RN, Lewis JD, Li H, et al.. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology 2014; 147 (5):1055-63; PMID:25046162; http://dx.doi.org/ 10.1053/j.gastro.2014.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramasundara M, Leach ST, Lemberg DA, Day AS. Defensins and inflammation: the role of defensins in inflammatory bowel disease. J Gastroenterol Hepatol 2009; 24(2):202-8; PMID:19215333; http://dx.doi.org/ 10.1111/j.1440-1746.2008.05772.x [DOI] [PubMed] [Google Scholar]
- 23.Kim YS, Ho SB. Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr Gastroenterol Rep 2010. Oct; 12(5):319-30; PMID:20703838; http://dx.doi.org/ 10.1007/s11894-010-0131-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swidsinski A, Loening-Baucke V, Verstraelen H, Osowska S, Doerffel Y. Biostructure of fecal microbiota in healthy subjects and patients with chronic idiopathic diarrhea. Gastroenterology 2008; 135(2):568-79; PMID:18570896; http://dx.doi.org/ 10.1053/j.gastro.2008.04.017 [DOI] [PubMed] [Google Scholar]
- 25.Carroll IM, Ringel-Kulka T, Siddle JP, Ringel Y. Alterations in composition and diversity of the intestinal microbiota in patients with diarrhea-predominant irritable bowel syndrome. Neurogastroenterol Motil 2012; 24(6):521-30; PMID:22339879; http://dx.doi.org/ 10.1111/j.1365-2982.2012.01891.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Preheim SP, Perrotta AR, Friedman J, Smilie C, Brito I, Smith MB, Alm E. Computational methods for high-throughput comparative analyses of natural microbial communities. Methods Enzymol 2013; 531:353-70; PMID:24060130; http://dx.doi.org/ 10.1016/B978-0-12-407863-5.00018-6 [DOI] [PubMed] [Google Scholar]
- 27.Arthur JC, Gharaibeh RZ, Uronis JM, Perez-Chanona E, Sha W, Tomkovich S, Mühlbauer M, Fodor AA, Jobin C. VSL#3 probiotic modifies mucosal microbial composition but does not reduce colitis-associated colorectal cancer. Sci Rep 2013. Oct 8; 3:2868; PMID:24100376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caporaso JG, Kuczynski J, Strombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al.. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010; 7:335-6; PMID:20383131; http://dx.doi.org/ 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. An improved greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. Isme J 2012; 6:610-8; PMID:22134646; http://dx.doi.org/ 10.1038/ismej.2011.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lozupone CA, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 2005; 71(12):8228-35; PMID:16332807; http://dx.doi.org/ 10.1128/AEM.71.12.8228-8235.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lozupone CA, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. Isme J 2011; 5:169-72; PMID:20827291; http://dx.doi.org/ 10.1038/ismej.2010.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D. Reyes J, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, et al.. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013. 8:1-10; PMID:23975157 [DOI] [PMC free article] [PubMed] [Google Scholar]