

Abstract
The human sphingomyelin synthase 1 gene (SGMS1) encodes an essential enzyme that is involved in the synthesis of sphingomyelin and diacylglycerol from phosphatidylcholine and ceramide. Among the products of SGMS1, we found new transcripts, circular RNAs (circRNAs), that contain sequences of the gene's 5′ untranslated region (5′UTR). Some of them include the gene's coding region and fragments of introns. An analysis of the abundance of circRNAs in human tissues showed that the largest transcripts were predominantly found in different parts of the brain. circRNAs of rat and mouse sphingomyelin synthase 1 orthologous genes were detected and are highly similar to the human SGMS1 gene transcripts. A quantitative analysis of the abundance of such transcripts also revealed their elevated amount in the brain. A computational analysis of sequences of human circRNAs showed their high potential of binding microRNAs (miRNAs), including the miRNAs that form complexes with Ago proteins and the mRNA of SGMS1. We assume that the circRNAs identified here participate in the regulation of the function of the SGMS1 gene in the brain.
Keywords: brain-specific expression, circular RNAs, microRNAs, sphingomyelin synthase 1, 5′UTR
Introduction
The sphingomyelin synthase 1 gene (SGMS1) encodes a transmembrane protein that is conserved among mammalians and participates in the synthesis of sphingomyelin, cholesterol metabolism, the regulation of intracellular molecular transport, cell proliferation, apoptosis and other essential processes.1-4 Previously, we studied the structure of the human SGMS1 gene and characterized transcripts of this gene that are potentially able to direct protein synthesis. We established that the transcriptional diversity of the SGMS1 gene is provided by the presence of alternative promoters, alternative polyadenylation and exon splicing.5-9 In human tissues, transcripts with a 5′ untranslated region (5′UTR) that includes exons 1–6 and exon 7, with a coding region that is localized in exons 7–10 and in a portion of exon 11, and with a 3′UTR that stems entirely to the remaining portion of exon 11 are mostly abundant.8 The multiplicity and complexity of the biological processes that involve the sphingomyelin synthase 1 (SMS1) protein suggest the high specificity of the control of the function of its encoding gene in the individual cells and tissues of organisms. However, almost no information is available regarding the specific regulatory mechanisms of this gene. In this study, among the transcripts of the SGMS1 gene, we found circRNAs that were nascent from the 5′ untranslated region and that were highly abundant in the brain.
In eukaryotic organisms, long non-coding RNAs that form circular structures were isolated as a fraction that is resistant to RNase R treatment, which degrades linear forms of RNA.10 Whole-transcriptome sequencing of eukaryotic cells revealed the existence of thousands of circular RNAs (circRNAs).11-16 As an example, it was shown that circRNAs in humans, mouse and drosophila account for 10% of the total number of the linear transcripts.13 circRNA expression proceeds in a complex tissue-, cell-type- or developmental-stage-specific manner and has been observed for orthologous genes of humans and mice.12,13,15-17 Repeated sequences have been shown to participate in pre-mRNA structure formation, which contributes to splicing.18 For instance, inverted repeats can contribute to circRNA formation.12,15,19 Currently, the biological role of circRNAs has not been established and is being actively studied. These species are thought to play a regulatory role and have been ascribed the function of “molecular sponges.”15,16 It was shown that the formation of “circRNA–microRNA” complexes suppresses the function of microRNAs (miRNAs), which allows circRNA to act as a mediator of miRNA activity in cells.20,21 circRNAs containing sequences of introns exercise transcriptional control because of the sorption of RNA-binding proteins15 and snRNA.22
In this study, we established the structure of the circRNA of the human SGMS1 gene and analyzed its homologous circRNAs in rat and mouse tissues. We studied the abundance of some circRNAs in animal tissues and found that their amount in the brain was elevated. A computational analysis was used to detect Alu repeats in human SGMS1 introns that participate in the formation of circRNAs, and a large number of miRNAs that interact with circRNAs were found. We discuss the structure, expression features and possible functional role of circRNAs of the sphingomyelin synthase 1 gene. The circRNAs identified are likely to be involved in the regulation of the expression mechanism of this gene.
Results
Detection of circRNAs among the transcripts of the human SGMS1 gene
We found several sequences in the SRA (http://blast.ncbi.nlm.nih.gov/Blast.cgi/) and circBase (http://www.circbase.org/) databases that coincided with individual exons of the SGMS1 gene (Table S1). These exons may have resulted from back-splicing and have a circular topology. As the greatest part of the circRNAs stored in these databases included those stemming from the RNA-Seq analysis of RNase R-treated RNA from human cellular lines and were obtained by computerized treatment of short RNA-Seq reads, experimental confirmation of their presence in human tissues was required.
To confirm the circular topology of the new transcript variants of the SGMS1 gene, the Northern blot hybridization method was applied (Fig. 1). Hybridization of human frontal cortex RNA preparations was performed using radioactive probes that were complementary to the gene's coding region (exons 7–8), and 3′UTR (exons 7–11) and 5′UTR (exons 2–6). Protein-coding transcripts hybridized with all probes and were detected after fractioning in agarose gel as a band in the region of 3300–4000 nt (Fig. 1A). An additional radioactive signal corresponding to transcripts with lengths of 200–500 nt was only detected in the case of hybridization with a probe that was complementary to the sequence of exons 2–6. The high intensity of the signal implied a high content of short transcripts bearing 5′UTR exons of the gene in the RNA sample studied. Treatment of a frontal cortex RNA preparation with RNase R, which depletes linear transcripts, resulted in the disappearance of the upper band in a similar experiment; however, the bottom band, which corresponded to the short products, was retained (Fig. 1A). This observation indicated the circular topology of the transcripts detected in this region. The fractioning of a frontal cortex RNA preparation in PAAG and subsequent hybridization with a probe that was complementary to the exon 2–6 sequence (5′UTR of the gene) led to the detection of individual circular transcripts (Fig. 1B). Thus, we showed that the frontal cortex RNA preparation contained circRNAs with a size that varied considerably and with a sequence that coincided with the 5′UTR of the SGMS1 gene.
Figure 1.
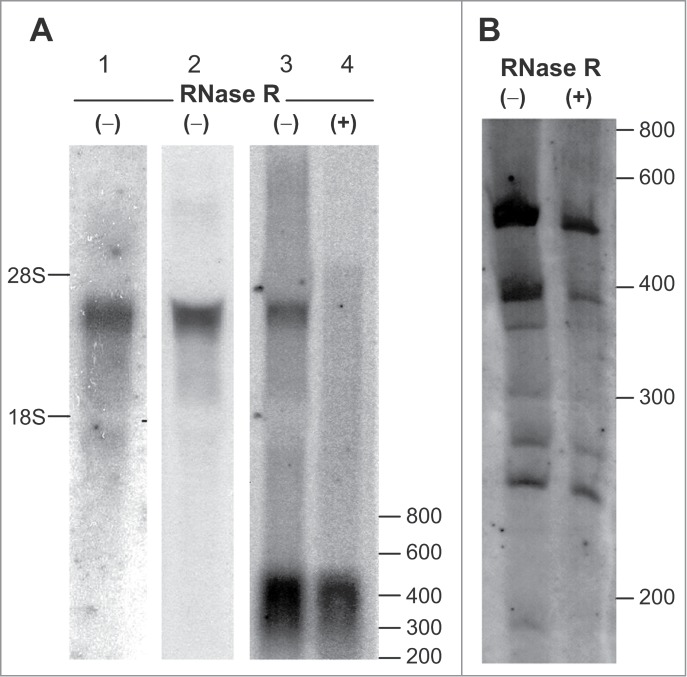
Northern blot analysis of transcripts of the SGMS1 gene in the human frontal cortex. Total RNA (−) and RNase R-treated RNA (+) were hybridized with (α-32P)dATP-labeled probes. (A) RNA fractionation in 1.2% agarose gel; hybridization with probes complementary to the regions of 1—the coding region (exons 7–8); 2—the coding region and 3′UTR (exons 7–11); 3 and 4—5′UTR (exons 2–6). (B) Fractionation of the RNA preparation in 3.5% PAAG; hybridization with a probe complementary to the 5′UTR region of the gene (exons 2–6). The positions of the marker bands of 28S and 18S rRNA, and also of the RiboRuler Low-Range RNA Ladder are shown (nt).
A subsequent search of circRNAs carrying individual exons of the SGMS1 gene was performed using PCR. Amplification of a frontal cortex cDNA synthesized from RNase R-treated RNA was carried out in the presence of outward-facing primers corresponding to the sequence of each of the 11 exons. Supplemental Table 2 shows the sequences of the primers used, and Figure 2A illustrates their localization relative to the exons. When the outward-facing primers corresponding to exons 1 and 8–11 were used, no PCR products were detected; in contrast, PCR products were detected in the presence of primers for exons 2–7, which confirmed the circular topology of the matrix (Fig. 2B). We must note that the transcripts obtained using primers to exon 7 were detected at a lower level. During the subsequent amplification of the frontal cortex cDNA synthesized from RNA without preliminary RNase R treatment in the presence of inward-facing primers, we observed the synthesis of individual PCR products corresponding to linear protein-encoding mRNAs (Fig. 2C). Concomitantly, PCR products were detected in the presence of outward-facing primers, which confirmed the presence of new variants of the alternative splicing with a circular topology in RNA preparations. Thus, circRNAs carrying sequences of exons 2–7 of the gene were present in frontal cortex RNA preparations.
Figure 2.
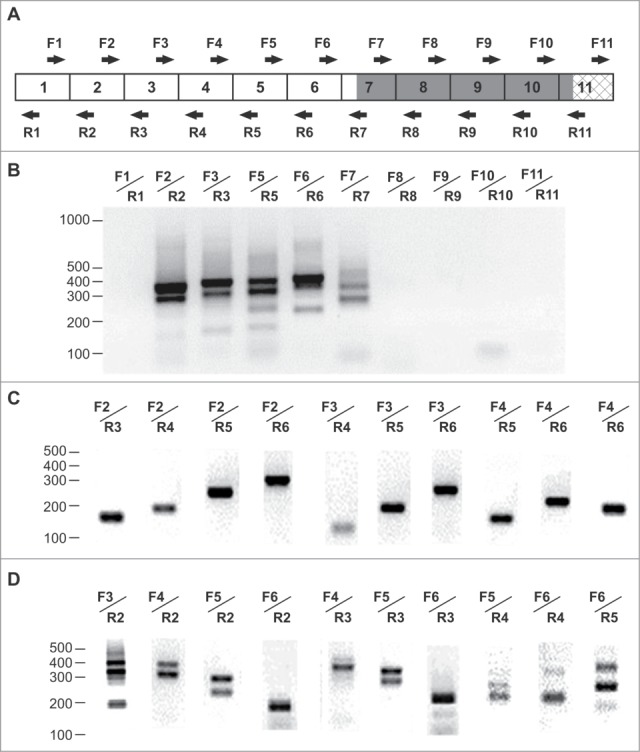
RT–PCR analysis of transcripts of the SGMS1 gene in the human frontal cortex. (A) The structure of the major coding transcripts; 5′UTR exons are marked by open rectangular boxes, coding exons are marked by dark rectangular boxes and 3′UTR exons are marked by cross-hatched rectangular boxes; the direction of the arrows indicates the 5′→3′ orientation of the primers relative to the exons. (B) Electrophoretic separation of the amplification products of cDNAs synthesized from an RNase R-treated RNA. Outward-facing primers corresponding to the sequence of exons 1–11 were used. (C, D) Electrophoretic separation of the amplification products of cDNAs synthesized from an RNase R-untreated RNA in the presence of inward-facing primers and outward-facing primers (D) corresponding to the exon sequence of the SGMS1 gene. The position of the bands of the Log Scale DNA Ladder marker is shown (nt).
Sequencing of individual PCR products in the presence of outward-facing primers allowed the clarification of the structural organization of several circRNAs of the human SGMS1 gene, as well as the identification of the regions of introns that are part of the circRNAs (Fig. 3A). We determined the nucleotide sequence of 11 circular transcripts of the SGMS1 gene (Fig. 3B). Six circRNAs (circSGMS1_2–6, circSGMS1_2–5, circSGMS1_4–6, circSGMS1_2–3, circSGMS1_4–5 and circSGMS1_3–6) included different combinations of exons 2–6 (5′UTR) of the SGMS1 gene. Transcript circSGMS1_7 represented the sequence of exon 7 shaped as a closed circle. Three RNAs (circSGMS1_1c-2–3, circSGMS1_2–2c-3 and circSGMS1_2–3–3c) included sequences of introns (1c, 2c and 3c, respectively), which have been spliced with the known exons 2 and 3. The sequence of circSGMS1_7–7b included exons 7 and 7b. Exon 7b was previously identified in an alternative transcript of the SGMS1 gene.6 According to the data of the Human Splicing Finder Program, exons 1c, 2c, 3c and 7b contain canonic splicing sites at their ends. Table 1 shows the characterization of the new exons 1c, 2c, 3c and 7b. The GenBank database records the circRNAs detected by us as KP710931–KP710938 and KR020014–KR020016 (Table S1).
Figure 3.

Structural organization of the new transcripts of the human SGMS1 gene. (A) Exon–intron structure of the human SGMS1 gene. Exons are marked by rectangular boxes, introns by dashed lines. (B) Structure of the circRNAs of the human SGMS1 gene. The numbers in the circles indicate the length of the circRNAs (nt). (C) 1—electrophoretic separation of the amplification products of a cDNA synthesized from an RNA from the human frontal cortex using the F1a/R8 primers. The structure of the PCR products and their sizes are shown on the right (nt). M—λ phage DNA treated with the PstI restriction nuclease was used as a marker.
Table 1.
Characterization of the new exons
| Name | Length (nt) | Position in genomea |
|---|---|---|
| 1c | 43 | 50598823–50598781 |
| 2c | 80 | 50527947–50527868 |
| 3c | 112 | 50516434–50516323 |
| 7b | 78 | 50340464–50340387 |
GenBank accession number NC_000010.11
A search for linear transcripts lacking sequences of exons 2–7 was carried out using RT–PCR in frontal cortex RNA preparations. Similar RNAs are formed according to the exon-skipping principle and may be accompanied by the formation of circRNAs. Several such variants were detected among transcripts starting from exon 1a. For instance, shorter PCR products were detected in parallel to the products (1538 nt) corresponding to the full-size transcripts during the amplification of frontal cortex cDNAs in the presence of F1a/R8 primers. The sequencing of these products detected transcripts lacking exon 7 (684 nt), as well as transcripts lacking exons 2–7 (232 nt). The latter were detected at much lower levels (Fig. 3C). In similar studies, linear transcripts were found that included exon 1a but had no sequences of exons 2–5 and 2–6. In the GenBank database, the transcripts that were detected here have accession numbers KR020022–KR020025.
RT–PCR analysis of the abundance of circRNAs in human tissues
An analysis of the abundance of the circRNAs detected in 9 human brain tissues and 9 human non-brain tissues was carried out via RT–PCR using primers F2/R2, F5/R5 and F7/R7. Figure 4 shows the electrophoretic separation of the amplification products, and it is obvious that the amounts of some circRNAs in human tissues varied markedly. The long circSGMS1_2–6 (452 nt) and circSGMS1_2–5 (371 nt) species were mostly represented in brain tissues (Fig. 4A and B). New exon-containing circSGMS1_2–3–3c was also represented more often in the brain tissues compared with other tissues (Fig. 4A). In the majority of tissues, circSGMS1_7–7b was detected at a low level (Fig. 4C). Primers F1/R3, which were specific to the coding transcript containing exon 1, were employed for the endogenous control of the expression level of the SGMS1 gene (Fig. 4D).
Figure 4.
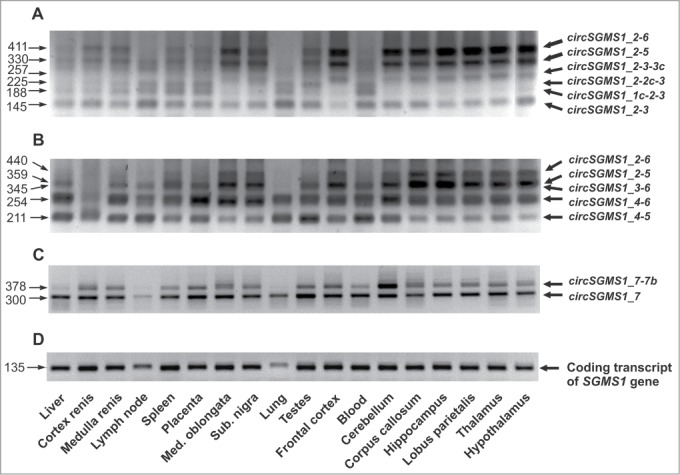
Electrophoretic separation of the PCR products corresponding to the circRNAs of the SGMS1 gene. Electrophoretic separation of the PCR products corresponding to circRNAs was carried out after 40 amplification cycles. For the amplification, outward-facing primers corresponding to the sequences of exons 2 (A), 5 (B) and 7 (C) were used. (D) For the control of the amount of RNA in a sample, the level of the coding transcript of the SGMS1 gene was assessed using the inward-facing F1/F3 primers (28 amplification cycles). circRNAs are shown to the right of the electrophoretogram, and the lengths of PCR products are shown to its left (nt); tissues from which RNA was isolated are shown at the bottom.
Analysis of homologous circRNAs of the sphingomyelin synthase 1 gene in rats and mice
We discovered several new circRNAs in rats and mice using PCR. Outward-facing primers corresponding to exons 2, 5 and 6 (F2r/R2r, F2m/R2m; F5r/R5r, F5m/R5m; and F6r/R6r, F6m/R6m) were used to amplify cDNA fragments of the total brains of rats (Fig. 5) and mice (Fig. S1). These primers' sequences are shown in Supplemental Table 2, and their localization relative to exons in rats and mice is shown in Figure 5A and Supplemental Figure 1A, respectively. Sequencing of individual PCR products showed the existence of several circRNAs in the rat (Fig. 5B and C) and in the mouse (Fig. S1B and C). These circRNAs included sequences of the 5′UTR (exons 2–5) and of exon 6 containing a fragment of the Sgms1 coding region of the rat and mouse. In the GenBank database, the accession numbers of the rat and mouse circRNAs identified here are KP710939–KP720941, KR020017 and KR020018 (rat); and KP710942 and KR020019–KR020021 (mouse) (Table S1).
Figure 5.
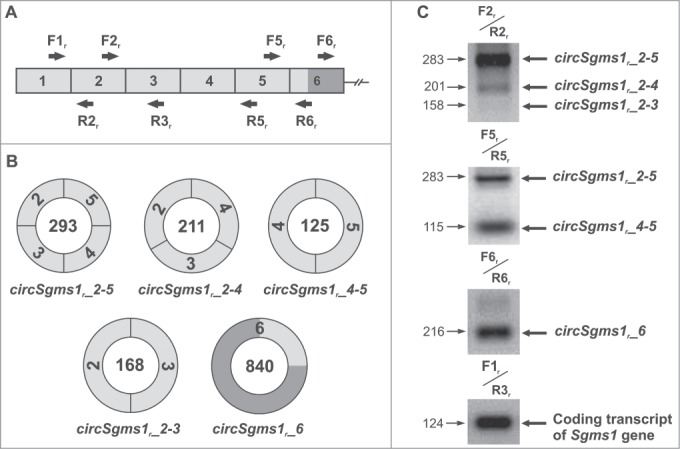
Analysis of circRNAs of the rat Sgms1 gene. (A) A fragment of the structure of the coding transcript of the rat Sgms1 gene. (B) Structure of the detected circRNAs of the rat Sgms1 gene. The numbers in the circles designate the length of circRNAs (nt). 5′UTR exons are shown in white, and coding exons are shown in black. (C) Electrophoretic separation of the amplification products of a cDNA synthesized from RNA from total rat brain. For amplification, outward-facing primers corresponding to the sequences of exons 2, 5 and 6 were used. For the control of RNA content in samples, the level of the Sgms1 coding transcript was assessed using the inward-facing F1r/R3r primers. circRNAs are shown to the right of the electrophoretogram, and the lengths of the PCR products are shown to its left (nt).
The use of bioinformatics to compare the exon 2–7 nucleotide sequences belonging to human circRNAs and the exon 2–6 nucleotide sequences of the rat and mouse Sgms1 gene revealed a high similarity of individual sequences, both in length and nucleotide content. A high homology (73%–94%) between the exons of rats and mice was observed. In rodents, homologues of human exons 2, 3, 4, 6 and 7 (2, 3, 4, 5 and 6, respectively) were found, with a degree of homology of 51%–81%; however, exons homologous to human exon 5 were absent (Fig. S2).
circRNAs of the rat and mouse were similar, as they included homologous exons. The single-exon RNAs circSgms1r_6 and circSgms1m_6 of rodents were highly homologous (88%–89%) to human circSGMS1_7 RNA (Fig. S3). The multi-exon circRNAs of rodents were less homologous (38%–58%) to human circRNAs, mainly because of the absence of an exon homologous to human exon 5 in these rodent circRNAs. Nonetheless, the observation that formation of some of them was associated with the back-splicing of their homologous exons was important. For instance, circSgms1r_2–5 and circSgms1m_2–5 of rats were similar to human circSGMS1_2–6, and circSgms1r_4–5 and circSgms1m_4–5 were similar to human circSGMS1_4–6 (Fig. S3).
Analysis of the quantitative abundance of circRNAs in tissues of humans, rats and mice
The quantitative abundance of major coding transcripts and circRNAs of the sphingomyelin synthase 1 gene in different tissues of humans (rats and mice) was assessed using real–time RT–PCR technique relative to the average mRNA level of 3 housekeeping genes LDHA, GAPDH, and RPL3 (Ldha, Gapdh, and Rpl3), which was set as 100% (Fig. 6). Primers used for major coding transcripts and circRNAs are listed at Supplemental Table 3. Figure 6A shows that transcript encoding the human SMS1 varied from 5.8% (frontal cortex) to 23.1% (corpus callosum) in brain tissues, and was comparable to non-brain tissues, where it varied from 5.2% (liver) to 16.6% (spleen). Figure 6D shows that the amount of human circRNAs varied from 1.0% (cerebellum) to 4.9% (corpus callosum), whereas it did not exceed 0.4% in non-brain tissues (lymph node). Similar results for major coding transcripts and circRNAs of Sgms1 are shown for the rat (Fig. 6B and E) and mouse (Fig. 6C and F).
Figure 6.

Analysis of the quantitative abundance of coding transcript and homologous circRNAs in the sphingomyelin synthase 1 gene of humans, mice and rats. The content of major coding transcript of gene encoding SMS1 for human (A), rat (B), mouse (C), and circSGMS1_2–6 (D), circSgms1r_2–5 (E), circSgms1m_2–5 (F) referred to GAPDH, RPL3 and LDHA (Gapdh, Rpl3 and Ldha) genes in tissues. (G) The content of circSGMS1_2–6 referred to the major coding transcript SGMS1 in human tissues; circSgms1r_2–5 (H) and circSgms1m_2–5 (I) referred to the major coding transcript Sgms1 in rat and mouse tissues, respectively. The level of reference genes set to be equal to 100%. The data are presented as the mean ± standard error of the mean. Results for brain tissues are enclosed in the gray background.
We also assessed the quantitative abundance of circSGMS1_2–6, circSgms1r_2–5 and circSgms1m_2–5 in human, rat and mouse tissues relative to the basic protein-encoding transcript, which level was taken as 100%. As shown in Figure 6G, the amount of circSGMS1_2–6 in the brain tissues varied from 7.8% (cerebellum) to 37.3% (thalamus). In non-brain tissues, its amount was considerably lower and varied from 0.7% (placenta) to 3.3% (lymph node). In tissues of rats and mice we found, a considerably larger amount of circRNAs in brain tissues compared with non-brain tissues. The amount of rat brain circSgms1r_2–5 varied from 2.8% (cerebellum) to 9.2% (subcortex), whereas it did not exceed 1% in non-brain tissues (lung) (Fig. 6H). In the total brain sample of mice, the level of the homologous circular circSgms1m_2–5 was 2.1%, whereas it did not exceed 0.6% in non-brain tissues (Fig. 6I).
Thus, the circRNAs of all studied organisms were more abundant in brain tissues compared with non-brain tissues. In humans and rats, it was shown that the amount of circRNAs in subcortical structures of the brain (thalamus, hypothalamus and hippocampus) was, on average, greater than that detected in the frontal cortex and in the cerebellum.
Analysis of repeats in the structure of the sphingomyelin synthase 1 gene
Several tens of Alu repeats were detected in human SGMS1 introns with the help of the UCSC browser. Most of them were about 300 nucleotides long. Analysis of the 5′- and 3′-end sequences of these repeats allowed us to divide them into 2 groups. One consisted of Alu repeats with predominant amounts of T nucleotides at the 5′ end, whereas the other consisted of Alu repeats with predominant amounts of A nucleotides at the 3′ end. The Global Sequence Alignment Tool (NCBI) program was used to show the high similarity of the nucleotide sequence of Alu repeats in each group, whereas Alu repeats from different groups exhibited high complementarity. We showed that some Alu repeats from different groups were located near exons involved in cyclization (Fig. S4A). For instance, AluSx was located before exon 2 and AluY was located after exon 6, with a complementarity of 80%. The interaction between these Alu repeats might contribute to the approximation of the 5′ end of exon 2 to the 3′ end of exon 6, to their splicing and to the formation of circSGMS1_2–6 (Fig. S4B and C). Exons 1c, 2c, 3c and 7b detected in circRNAs were also located near Alu repeats. AluSp was located 1349 nucleotides away from the 3′ end of the exon 7b. Both exons 1c and 2c were surrounded by homologous Alu repeats. Regarding exon 1c, AluSg and AluSp were located 778 and 745 nucleotides away from the 5′ and 3′ ends of the sequence, respectively. In the case of exon 2c, AluJr and AluSc were located 677 and 799 nucleotides away from the 5′ and 3′ ends of the sequence, respectively. Homologous Alu repeats may participate in the regulation of the incorporation of an additional exon into circRNAs, because of their competition for the interaction with the oppositely oriented Alu.
Dozens of repeats (Alu, B1, B2, B4, ID families) were detected in rat and mouse Sgms1 introns using the UCSC browser. The Table S4 shows the characteristics of repeats, which are located near the exons, involved in circularization, and may allow the formation of the secondary structure of pre-mRNAs, which contributes to back-splicing and to the emergence of circRNAs. Thus, the complexes of repeats that flank the sequences of these circRNAs and underlie the preferential yield of specific splicing variants are likely to be involved in the formation of the circular transcripts detected here (Fig. S4, Table S4).
Analysis of circRNA sequences of the human SGMS1 gene for the presence of miRNAs and protein-binding sites
Using a bioinformatics approach, we estimated the potential of circRNAs of the human SGMS1 gene to bind miRNAs. According to mirBase, their sequences can bind dozens of miRNAs. The STarMirDB database was used to search for miRNA-binding sites in the sequence of exons 1–7 of the human SGMS1 gene (GenBank accession number NM_147156). Figure 7A shows the total number of miRNA sites for which the start site is found in the specified interval of 25 nt. It is obvious that the largest number of miRNA-binding sites was observed in the area of exons 5, 6 and 7, whereas their smallest number was detected in that of exon 1. In the 5′UTR of exon 7, their amount exceeded the number of binding sites found in the coding region. We searched for miRNA-binding sites that interacted with Ago proteins in the mRNA sequence of the SGMS1 gene. Unique miRNAs were found that were able to cooperate with Ago and interact with the mRNA of the SGMS1 gene in the protein-coding region and in the 3′UTR. Subsequently, we determined the amount of individual miRNAs that were able to interact with both Ago proteins and the exon 1–7 sequence. We only studied those miRNAs with binding sites that were located entirely within the zone of a specified exon. The number of miRNA-binding sites of the circRNA sequences was determined by adding the number of miRNA-binding sites to the number of exons that were part of the cycle. Figure 7B shows the number of entry sites in individual miRNAs on some of the circRNAs detected here (circSGMS1_2–6, circSGMS1_2–5, circSGMS1_2–3 and circSMGS1_7) and in exon 1, which was not included in circRNAs. Exon 1 obviously had a minimal number of similar sites, and the circRNAs containing exons 5, 6 and 7 had a larger amount of miRNA-binding sites that were able to interact with Ago proteins.
Figure 7.

Search of miRNA-binding sites in the sequences of exons 1–7 of the human SGMS1 gene. (A) The total number of sites of the miRNA in which the start site falls within the specified 25-nt-long interval of the mRNA (GenBank accession number NM_147156). (B) Curves of the potential binding of individual miRNAs involved in the formation of complexes with Ago proteins, circRNAs and the sequence of exon 1.
Using RBPDB, we estimated the potential of the binding of exons 1c, 2c, 3c and 7b of circRNAs to RNA-binding proteins (RBPs). The largest number of RBP binding sites was found in the sequence of exon 3c, in particular in the MBNL1, EIF4B, YTHDC1, ELAVL1, KHDRBS3 and SFRS1 proteins, which are involved in the control of transcription, splicing and translation.
Discussion
Evidence of a newly discovered class of RNA in eukaryotes was reported recently. These transcripts are formed during the ligation of linear RNA exons, so that the 5′ end of the pre-mRNA exon binds the 3′ end of the upstream exon.16,24 They are circular and non-coding RNAs. 16 The abundance of circRNAs has been shown using RNA-Seq technology.12,14,23 In this study, we confirmed 7 circRNAs and discovered new 4 circRNAs for the SGMS1 gene in RNA preparations of human tissues. It should be noted that, in human tissues studied, we failed to detect the circRNAs from the circBase database that included the sequences of exons 1, 8, 9, 10 and 11 of the SGMS1 gene. Simultaneously, circRNAs, which contain exons 1c, 2c, 3c and 7b have been found by us for the first time. Many circRNAs from circBase database are probably either specific for definite human cellular lines or do not exist in vivo, such as those resulting from computational data processing of short RNA-Seq reads (Table S1). Over half the circRNAs that are known currently contain exons that only code for proteins.17 In the case of the SGMS1 gene, its circRNAs contained mostly exon sequences from the 5′UTR. The fact that the 5′UTR of SGMS1 consists of several exons separated from each other by large introns led us to assume that this type of 5′UTR organization of genes in eukaryotic cells might be used, via back-splicing, for the synthesis of non-coding circRNAs that are able to participate in the regulation of the function of these genes.
The splicing of exons separated by large introns can involve complementary interspersed repetitive elements. These allow intron splicing sites to be found more quickly by folding the intronic RNA upon itself at smaller intervals, thus reducing the distance between donor and acceptor sites.18 The formation of circRNAs sometimes also includes the connection of splicing sites that are located far away from each other and can be controlled at the level of pre-mRNA splicing.25,26 In this process, Alu repeats can play the role of repeated sequences.12,24,27,28 Using ZKSCAN1, H1PK3 and EPHB4 as examples, it was discovered that the removal of Alu elements in flanking introns interrupted exon circularization.19 The exon-flanking introns of the human SGMS1 gene are tens of times longer than its exons. They include a large number of Alu repeats placed in an orientation that allows the formation of the secondary structure of pre-mRNAs, which contributes to back-splicing and to the emergence of the transcripts discovered by the authors. The incorporation of additional exons (1c, 2c, 3c and 7b) surrounded by Alu repeats in circRNAs was another indication of the involvement of Alu repeats in the formation of circRNAs.
Alternative promoters located inside introns also take part in the synthesis of the transcripts of the SGMS1 gene.8 Compared with the distal promoter located at the 5′ end of the gene's locus, the contribution of the alternative promoters to the synthesis of the SGMS1 mRNA is not large. One can suggest that they participate in the synthesis of circRNAs. For instance, the detection of linear transcripts in the human frontal cortex, starting from exon 1a but missing exon 7 or exons 2–7, suggests the involvement of an exon 1a-containing promoter in the synthesis of circRNAs (Fig. 3C). Such transcripts can result from an exon-skipping process accompanied by the circulation of lost exons. Whether alternative promoters participate in the synthesis of the mRNA and circRNAs of SGMS1 is a problem that remains open for discussion and requires further investigation.
We detected similar circRNAs in rats and mice. Detection of highly complementary repeats in the orientation, which may allow the convergence of back-splicing sites, points to a similar principles of circRNAs formation in human and rodents. At the same time some homologous circRNAs nascent from the 5′UTR of the SGMS1 gene were more abundant in the brain compared with other tissues. This phenomenon is in good agreement with the evidence that circRNAs can be expressed from orthologous genes and that their expression can proceed in a tissue-specific manner.13,17 circRNAs may be localized in neuronal cell bodies and synaptic processes (axons and dendrites), and change at developmental stages that correspond to synapse formation and follow homeostatic plasticity.29 A high content of circRNAs has been shown in the human and murine brain.20,21,23 There is evidence that circRNAs may exist at different levels in various brain parts.13 For instance, a large amount of the antisense ciRS-7 RNA of the CDR1 gene has been reported in the developing mouse midbrain and in several other animals.13 In the case of humans and rats, we observed that the amount of the circRNAs studied can also be different in different brain parts.
The mechanisms of formation of circRNAs are probably similar in the human, rat and mouse brain, which raises the problem of the biogenesis of the elevated amounts of circRNAs in the brain. The regulation of these processes can occur at multiple levels and involve the synthesis, transport and degradation of circRNAs. Pre-mRNA splicing provides one of the most important factors of biogenesis. Apparently, back-splicing of pre-mRNAs in the brain has more competitive advantages than it does in non-brain tissues. The elevated quantity of circRNAs from the 5′UTR of the sphingomyelin synthase 1 gene observed here might point to their functional significance in the brain.
The biological significance of circRNAs is being studied actively. Regarding exonic circRNAs, the fact itself of their existence may mean a reduction of the amount of full-size mRNAs, and, consequently, can affect protein levels. Concomitantly, circRNAs are resistant to the action of nucleases, and some of them can act as molecular sponges for miRNAs.15,16,30 In particular, the function of circRNAs as miRNA activity mediators was studied in the instance of the circular antisense ciRS-7 RNA, which is highly expressed in the human and mouse brain.20,21 It has been shown that ciRS-7 binds to miR-7 and is subsequently destroyed in an Ago-dependent manner. There is a correlation between the level of miRNAs and that of the circRNA ciRS-7. For instance, the amount of the circRNA ciRS-7 is decreased in the hippocampus of patients with Alzheimer's disease, whereas the level of miR-7 increases at the same time, which probably affects the expression of several Alzheimer's disease-specific factors.31-33 We presumed that the exonic circRNAs of the human SGMS1 gene can also bind miRNAs. We discovered that circular transcripts that include exons 5, 6 and 7 exhibited the highest capacity for sorption of miRNAs. In addition, these circRNAs can absorb miRNAs that interact with Ago proteins and with the mRNA of the SGMS1 gene. Proteins of the Ago family are well known to act as RISC complex factors, which cut the RNA target.34 Sorption of miRNAs that interact with Ago proteins can reduce the quantity of active miRNA–Ago complexes and contribute to the protection of the mRNA target. circSGMS1_2–6 and circSGMS1_2–5 displayed a high level of representation in the brain, and their contribution to the reduction of the pool of miRNAs might be considerable.
The discovery of the incorporation of intronic fragments in circular transcripts seems of interest as far as the functions of these RNAs are concerned. Their structure suggests a putative combination of functions that are typical of exonic and intronic circRNAs. Exons can act as sponges for miRNAs, whereas an intronic fragment, like intronic circRNAs, can take part in the sorption of RNA-binding proteins.15 According to the RBPDB, the sequence of exon 3c, for instance, as a part of the circSGMS1_2–3–3c that prevails in the brain, can adsorb proteins that are involved in the control of transcription, splicing and translation. The results obtained here allowed us to assume that the circRNAs of the sphingomyelin synthase 1 gene can act as cis-acting factors for the regulation of gene expression.
In conclusion, we note that the significance of the majority of circular transcripts remains obscure, and that, sometimes, they are interpreted as inconsequential side products of pre-mRNA splicing.17 The high amount of circRNAs from the 5′UTR of the sphingomyelin synthase 1 gene observed in the brain, which was typical in the mouse, rat and in humans, testifies in favor of the functional significance of this class of RNA. The discovery of circular non-coding RNAs in the sphingomyelin synthase 1 gene not only expands our knowledge about its organization, but also helps answer questions about the functional significance of multi-exonic 5′UTRs.
Materials and Methods
Animals
Experiments on animals were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publ. no 80–23, revised 1996). White male rat of the Wistar line (weight, 200–250 g) and white outbred mice (weight, 20 g) were maintained on a 12 h light/dark cycle at a temperature of 22–24°C, with free access to food and water.
RNA isolation
The study was performed after approval from the local ethics committee, was obtained. Total RNA was isolated from human placenta, blood and post-mortem tissues (the time from accidental death to autopsy was 4 h), as well as from rat and mouse tissues using TRIzol reagent (Invitrogen, USA) and an acid guanidine thiocyanate–phenol–chloroform extraction.35 The isolated RNA was treated with DNase I (ThermoFisher scientific, USA) in the presence of RiboLock (ThermoFisher scientific), according to the protocol recommended by the manufacturer. Deproteinization was performed using a 1:1 phenol:chloroform mixture. The isolated RNA was precipitated with sodium acetate (3.0 M, pH 5.2) and ethanol. RNA integrity was checked by analysis of ratio between the bands of 28S and 18S rRNAs after denaturing agarose gel electrophoresis.36
Total RNA RNase R treatment
To carry out the synthesis of cDNA, 2 μg of total RNA was treated with 10 μl of the reaction mixture and 10 U of RNase R (Epicentre) in 1× RNase R-buffer. The mixture was incubated at 37°C for 1 h. Subsequently, 1 μl of 1 mM EDTA was added and used for the synthesis of cDNA. For Northern blot analysis, RNase R-treated RNA was subjected to deproteinization with a 1:1 phenol:chloroform mixture and re-precipitated from isopropanol.
cDNA synthesis
The synthesis of cDNA was carried out in 20 μl of reaction mixture containing 2 μg of RNA using the reagents of a RevertAid First Strand cDNA Synthesis Kit and short random oligodeoxyribonucleotide primers (ThermoFisher scientific), in accordance with the manufacturer's instructions.
RT–PCR and sequencing of PCR products
To obtain samples for sequencing, 20 μl of reaction mixture was prepared for PCR, containing 2 μl of 0.2× reverse transcriptase reaction sample, forward and reverse primers (5 μmol each) (Table S2), 0.2 mM of dNTP, 1× High-Fidelity PCR Buffer with MgCl2, High-Fidelity PCR Enzyme Mix (5 U/μL) (ThermoFisher scientific) and nuclease-free water (up to 20 μL). Specific primers were selected using the OLIGO Primer Analysis Software 6.31 and synthesized by the Evrogen Joint Stock Company, Russia (Table S2). Amplification of cDNA was performed on a multichannel amplifier (DNA technology, Russia) in the following mode: stage 1 (denaturation): 95°C, 5 min; and stage 2 (amplification): 95°C, 30 s; 65°C, 1 min; 72°C, 40 s (35 cycles). The electrophoretic separation of PCR products in an agarose gel was performed in 1× TAE buffer.37 Sequencing of PCR products using the Sanger method was performed by the Evrogen Joint Stock Company.
Northern blot analysis
For Northern blot hybridization, frontal cortex RNA samples that were fractionated in agarose or polyacrylamide gel (PAAG) were used. Total RNA (12 μg) and 12 μg of RNase R-treated RNA were separated on a denaturing 1.2% agarose gel containing 40 mM MOPS (pH 7.0), 10 mM sodium acetate, 1 mM EDTA and 2.2 M formaldehyde, as described in,37 and were transferred to a Hybond N+ membrane (GE Healthcare, USA) via the downward alkaline capillary method described previously.38 Total RNA (30 μg) and 30 μg of RNase R-treated RNA were separated in a denaturing 3% PAAG containing 48% carbamide, 12.1 g/L of tris(hydroxymethyl)aminomethane, 5.1 g/L of boric acid and 0.4 g/L of EDTA according to the procedure described in,37 and transferred to a Hybond N+ membrane (GE, Healthcare, USA) via electrotransfer.
To produce a radioactively labeled probe for exons 2–6 of the SGMS1 gene, cDNA synthesized from a total RNA sample from the frontal cortex was amplified in the presence of forward (5′–GGAGAAAGAAGACAGCCATCA–3′) and reverse (5′–GCTTCTCCCATTGTTCCAGTA–3′) primers. The 338-nt-long fragment of cDNA amplicon comprised 29 nt of the 3′-end region of exon 2, the full length of exons 3, 4 and 5 and 33 nt of the 5′-end region of exon 6. To obtain a radioactively labeled probe for exons 7–8, cDNA synthesized from a total RNA sample from the frontal cortex was amplified in the presence of forward (5′–GGCGTAGACATCCCCACC–3′) and reverse (5′–TACAGCGTGCCAACTATGC–3′) primers. The 407-nt-long cDNA fragment contained 362 nt of the 3′-end region of exon 7 and 45 nt of the 5′-end region of exon 8. To generate a radioactively labeled probe for exons 7–11, exons of cDNA synthesized from a total RNA sample from the frontal cortex were amplified in the presence of forward (5′–TGGTTAATTCAGTGGCTGCTC–3′) and reverse (5′–ATGGTGGTTGCGGGTTAT–3′) primers. The 918-nt-long cDNA fragment contained 32 nt of the 3′-end region of exon 7, the full length of exons 8, 9 and 10 and 447 nt of the 5′-end region of exon 11. Amplification of cDNA was performed in the following mode: stage 1 (denaturation): 95°C, 5 min; and stage 2 (amplification): 95°C, 30 s; 65°C, 1 min; 72°C, 40 s (35 cycles). The fragments obtained were isolated from the gel, purified, sequenced by Sanger method and used for the production of probes via asymmetric amplification using the corresponding reverse primers. Reaction mixtures of 20 μl contained 20 ng of matrix, 150 nM of primers, 0.1 mM of each of the 3 nucleotides dGTP, dCTP and dTTP, 2 μM dATP, 2 μM (α-32P)dATP (5 MBq), 1.5 mM MgCl2 and 1 U of a mixture of DNA polymerases (High Fidelity PCR Enzyme Mix, Thermo Scientific) in the buffer supplied with the mixture of polymerases. Reaction conditions were as follows: initial denaturation of the matrix, 94°C, 4 min; followed by 94°C, 30 s; 58°C, 1 min; 72°C, 1 min (25 cycles). The product of the 3 reactions was extracted with a 24:1 chloroform:isoamyl alcohol mixture, and ammonium acetate was added to 2 M; subsequently, it was precipitated with isopropanol and dissolved in water, denatured by heating at 95°C for 5 min and added to 6 mL of hybridization buffer (0.5 M phosphate buffer (pH 7.2), 7% SDS, 1% BSA and 1 mM EDTA). Blots were hybridized at 65°C for 12 h. The signal was recorded using a Typhoon FLA 9500 apparatus (GE Healthcare).
Real-time RT–PCR
The 25 μl PCR mixture contained 2 μl of 0.2× revertase reaction sample, forward and reverse primers (5 μmol each), 5 μl of 5× reaction mixture (Evrogen, Russia) including PCR buffer, Taq DNA polymerase, dNTPs and the intercalating dye SYBR Green I. Primers specific to the genes studied were selected using OLIGO Primer Analysis Software 6.31 and were synthesized by the Evrogen Joint Stock Company (Table S3). The amplification of cDNAs was performed on a StepOnePlus™ Real-Time PCR System (Applied Biosystems, USA) in the following mode: stage 1 (denaturation), 95°C, 10 min; and stage 2 (amplification with fluorescence measured), 95°C, 1 min; 65°C, 1 min; 72°C, 1 min (40 cycles). Each cDNA sample was analyzed 3 times.
Data analysis of real-time RT–PCR and statistics
The average cycle threshold (Ct) was calculated for each triplicate. Three reference genes GAPDH, RPL3, LDHA (human) and Gapdh, Rpl3, Ldha (rat and mouse) were used to normalize the cDNA samples in the quantification of the transcripts encoding the SMS1 protein and circRNAs.39 Calculations were performed using BestKeeper v140 and Relative Expression Software Tool (REST) 2009 software. The manual at the site “REST.-gene-quantification.info” (http://www.gene-quantification.de/rest-2009.html) was used to evaluate expression target genes relative to the expression levels of the reference genes. The values were calculated as
where E is the PCR efficiency, Ct(tar) is the average Ct of the target gene, and ECt(ref) is the geometric average ECt of the reference genes. The PCR efficiencies for all primer pairs were estimated using a cDNA dilution series amplification in which 3 replicate reactions were performed for each dilution. PCR efficiencies were assessed using the amplification of a series of standard dilutions of cDNAs and computed using the REST software.41 The efficiency values for all PCR reactions were in the range of 1.88–1.99. Additional calculations were performed using Microsoft Excel.
Computational methods of analysis
circRNAs were analyzed with the help of SRA-Blast from NCBI (search database SRA using Megablast, available at http://blast.ncbi.nlm.nih.gov/Blast.cgi) and circBase, available at http://www.circbase.org.42 Analysis of the sequencing results was performed using Chromas Lite software. The comparison between the sequencing results and the sequence of the genome was performed using Blast from NCBI. The search of the splicing sites was carried out using Human Splicing Finder, v-2.4.1, available at www.umd.be/HSF. The search of repeats was accomplished using the UCSC browser, available at http://genome.ucsc.edu. Alignments of repeat sequences and the determination of the homology between the sequences of different animals were carried out using the Global Sequence Alignment Tool (NCBI). The search of potential miRNA-binding sites was performed using mirBase, available at http://www.mirbase.org and STarMirDB,43 available at http://sfold.wadsworth.org/cgi-bin/starmir.pl. The STarMirDB database carries information on the V-CLIP project, which pertains to miRNA site-containing Ago crosslink-centered regions (CCRs).44 Possible interactions with RNA-binding proteins were studied using RBPDB, available at http://rbpdb.ccbr.utoronto.ca.45 Additional calculations were performed using Microsoft Excel.
Data access
The annotated sequences were deposited into the GenBank with the accession numbers KP710931–KP710943, KR020014–KR020025.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank members of the Human Molecular Genetics Department for useful discussions.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Ethical Statement
The experiments comply with the current laws of the country in which they were performed.
Authors' Contributions
IBF, OYuS, SAL, and LVD conceived and designed the experiments; IBF and OYuS performed the experiments; IBF and OYuS analyzed the data; IBF and LVD wrote the paper. SAL coordinated the study, participated in study design and edited the manuscript.
Funding
This work was supported by a grant from the Molecular and Cellular Biology Program of the Russian Academy of Sciences and by grant from the Russian Foundation for Basic Research (#14–04–00487), and by the Federal Program for Support of Scientific Schools of the Russian Ministry of Science and Education.
References
- 1.Guillen N, Navarro MA, Surra JC, Arnal C, Fernandez-Juan M, Cebrian-Perez JA, Osada J. Cloning, characterization, expression and comparative analysis of pig Golgi membrane sphingomyelin synthase 1. Gene 2007; 388(1–2):117-24; PMID:17156943; http://dx.doi.org/ 10.1016/j.gene.2006.10.013 [DOI] [PubMed] [Google Scholar]
- 2.Shakor AB, Taniguchi M, Kitatani K, Hashimoto M, Asano S, Hayashi A, Nomura K, Bielawski J, Bielawska A, Watanabe K et al.. Sphingomyelin synthase 1-generated sphingomyelin plays an important role in transferrin trafficking and cell proliferation. J Biol Chem 2011; 286(41):36053-62; PMID:21856749; http://dx.doi.org/ 10.1074/jbc.M111.228593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Subathra M., Qureshi A, Luberto C. Sphingomyelin synthases regulate protein trafficking and secretion. PLoS One 2011; 6(9):e23644; PMID:21980337; http://dx.doi.org/ 10.1371/journal.pone.0023644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yan N, Ding T, Dong J, Li Y, Wu M. Sphingomyelin synthase overexpression increases cholesterol accumulation and decreases cholesterol secretion in liver cells. Lipids Health Dis 2011; 10:46; PMID:21418611; http://dx.doi.org/ 10.1186/1476-511X-10-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vladychenskaya IP, Dergunova LV, Dmitrieva VG, Limborska SA. Human gene MOB: structure specification and aspects of transcriptional activity. Gene 2004; 338(2):257-65; PMID:15315829; http://dx.doi.org/ 10.1016/j.gene.2004.06.003 [DOI] [PubMed] [Google Scholar]
- 6.Rozhkova AV, Dmitrieva VG, Zhapparova ON, Sudarkina OY, Nadezhdina ES, Limborska SA, Dergunova LV. Human sphingomyelin synthase 1 gene (SMS1): organization, multiple mRNA splice variants and expression in adult tissues. Gene 2011; 481(2):65-75; PMID:21549185; http://dx.doi.org/ 10.1016/j.gene.2011.04.010 [DOI] [PubMed] [Google Scholar]
- 7.Dergunova LV, Rozhkova AV, Sudarkina OYu, Limborska SA. The use of alternative polyadenylation in the tissue-specific regulation of human SMS1 gene expression. Mol Biol Rep 2013; 40(12):6685-90; PMID:24062078; http://dx.doi.org/ 10.1007/s11033-013-2783-0 [DOI] [PubMed] [Google Scholar]
- 8.Rozhkova AV, Filippenkov IB, Sudarkina OYu, Limborska SA, Dergunova LV. Alternative promoters located in SGMS1 gene introns participate in regulation of its expression in human tissues. Mol Biol (Mosk) 2015; 49(2):287-94; http://dx.doi.org/ 10.1134/S002689331501015X [DOI] [PubMed] [Google Scholar]
- 9.Sudarkina OY, Filippenkov IB, Brodsky IB, Limborska SA, Dergunova LV. Comparative analysis of sphingomyelin synthase 1 gene expression in human tissues at transcriptional and translational levels. Mol Cell Biochem 2015; PMID:25912551; http://dx.doi.org/ 10.1007/s11010-015-2427-x [DOI] [PubMed] [Google Scholar]
- 10.Suzuki H, Zuo Y, Wang J. Zhang MQ, Malhotra A, Mayeda A. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res 2006; 34(8):e63; PMID:16682442; http://dx.doi.org/ 10.1093/nar/gkl151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 2012; 7(2):e30733; PMID:22319583; http://dx.doi.org/ 10.1371/journal.pone.0030733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013; 19(2):141-57; PMID:23249747; http://dx.doi.org/ 10.1261/rna.035667.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al.. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013; 495(7441):333-8; PMID:23446348; http://dx.doi.org/ 10.1038/nature11928 [DOI] [PubMed] [Google Scholar]
- 14.Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. Cell-type specific features of circular RNA expression. PLoS Genet 2013; 9(9):e1003777; PMID:24039610; http://dx.doi.org/ 10.1371/journal.pgen.1003777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH, Zhu S, Yang L, Chen LL. Circular intronic long noncoding RNAs. Mol Cell 2013; 51(6):792-806; PMID:24035497; http://dx.doi.org/ 10.1016/j.molcel.2013.08.017 [DOI] [PubMed] [Google Scholar]
- 16.Lasda E, Parker R. Circular RNAs: diversity of form and function. RNA 2014; 20(12):1829-42; PMID:25404635; http://dx.doi.org/ 10.1261/rna.047126.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo JU, Agarwal V, Guo H, Bartel DP. Expanded identification and characterization of mammalian circular RNAs. Genome Biol 2014; 15(7):409; PMID:25070500; http://dx.doi.org/ 10.1186/s13059-014-0409-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shepard S, McCreary M, Fedorov A. The peculiarities of large intron splicing in animals. PLoS One 2009; 4(11):e7853; PMID:19924226; http://dx.doi.org/ 10.1371/journal.pone.0007853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang D, Wilusz JE. Short intronic repeat sequences facilitate circular RNA production. Genes Dev 2014; 28(20):2233-47; PMID:25281217; http://dx.doi.org/ 10.1101/gad.251926.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hansen TB, Wiklund ED, Bramsen JB, Villadsen SB, Statham AL, Clark SJ, Kjems J. miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J 2011; 30(21):4414-22; PMID:21964070; http://dx.doi.org/ 10.1038/emboj.2011.359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature 2013; 495(7441):384-8; PMID:23446346; http://dx.doi.org/ 10.1038/nature11993 [DOI] [PubMed] [Google Scholar]
- 22.Li Z, Huang C, Bao C, Chen L, Lin M, Wang X, Zhong G, Yu B, Hu W, Dai L, et al.. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol 2015; 22(3):256-64; PMID:25664725; http://dx.doi.org/ 10.1038/nsmb.2959 [DOI] [PubMed] [Google Scholar]
- 23.Rybak-Wolf A, Stottmeister C, Glažar P, Jens M, Pino N, Giusti S, Hanan M, Behm M, Bartok O, Ashwal-Fluss R, et al.. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol Cell 2015; PMID:25921068; http://dx.doi.org/ 10.1016/j.molcel.2015.03.027 [DOI] [PubMed] [Google Scholar]
- 24.Wilusz JE, Sharp PA. Circuitous route to noncoding RNA. Science 2013; 340(6131):440-1; PMID:23620042; http://dx.doi.org/ 10.1126/science.1238522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N, Kadener S. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 2014; 56(1):55-66; PMID:25242144; http://dx.doi.org/ 10.1016/j.molcel.2014.08.019 [DOI] [PubMed] [Google Scholar]
- 26.Starke S, Jost I, Rossbach O, Schneider T, Schreiner S, Hung LH, Bindereif A. Exon circularization requires canonical splice signals. Cell Rep 2015; 10(1):103-111; PMID:25543144; http://dx.doi.org/ 10.1016/j.celrep.2014.12.002 [DOI] [PubMed] [Google Scholar]
- 27.Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol 2014; 32(5):453-61; PMID:24811520; http://dx.doi.org/ 10.1038/nbt.2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. Complementary sequence-mediated exon circularization. Cell 2014; 159(1):134-47; PMID:25242744; http://dx.doi.org/ 10.1016/j.cell.2014.09.001 [DOI] [PubMed] [Google Scholar]
- 29.You X, Vlatkovic I, Babic A, Will T, Epstein I, Tushev G, Akbalik G, Wang M, Glock C, Quedenau C, et al.. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci 2015; 18(4):603-10; PMID:25714049; http://dx.doi.org/ 10.1038/nn.3975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomas LF, Saetrom P. Circular RNAs are depleted of polymorphisms at microRNA binding sites. Bioinformatics 2014; 30(16): 2243-6; PMID:24764460; http://dx.doi.org/ 10.1093/bioinformatics/btu257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bingol B, Sheng M. Deconstruction for reconstruction: the role of proteolysis in neural plasticity and disease. Neuron 2011; 69(1):22-32; PMID:21220096; http://dx.doi.org/ 10.1016/j.neuron.2010.11.006 [DOI] [PubMed] [Google Scholar]
- 32.Lonskaya I, Shekoyan AR, Hebron ML, Desforges N, Algarzae NK, Moussa CE. Diminished parkin solubility and co-localization with intraneuronal amyloid-β are associated with autophagic defects in Alzheimer disease. J Alzheimers Dis 2013; 33(1):231-47; PMID:22954671 [DOI] [PubMed] [Google Scholar]
- 33.Lukiw WJ. Circular RNA (circRNA) in Alzheimer disease (AD). Front Genet 2013; 4:307; PMID:24427167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Valencia-Sanchez MA, Liu J, Hannon GJ, Parker R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev 2006; 20(5):515-24; PMID:16510870; http://dx.doi.org/ 10.1101/gad.1399806 [DOI] [PubMed] [Google Scholar]
- 35.Chomczynski P, Sacchi N. The single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction: twenty-something years on. Nat Protoc 2006; 1(2):581-5; PMID:17406285; http://dx.doi.org/ 10.1038/nprot.2006.83 [DOI] [PubMed] [Google Scholar]
- 36.Masek T, Vopalensky V, Suchomelova P, Pospisek M. Denaturing RNA electrophoresis in TAE agarose gels. Anal Biochem 2005; 336(1):46-50; PMID:15582557; http://dx.doi.org/ 10.1016/j.ab.2004.09.010 [DOI] [PubMed] [Google Scholar]
- 37.Sambrook J, Russell DW Molecular Cloning: A Laboratory Manual, 3rd Ed. Cold Spring Harbor, NY:Cold Spring Harbor Laboratory Press, 2001; pp. 7.42–7.45. [Google Scholar]
- 38.Chomczynski P. One-hour downward alkaline capillary transfer for blotting of DNA and RNA. Anal Biochem 1992; 201(1):134-9; PMID:1621951; http://dx.doi.org/ 10.1016/0003-2697(92)90185-A [DOI] [PubMed] [Google Scholar]
- 39.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, et al.. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 2009; 55(4):611-22; PMID:19246619; http://dx.doi.org/ 10.1373/clinchem.2008.112797 [DOI] [PubMed] [Google Scholar]
- 40.Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper – Excel-based tool using pair-wise correlations. Biotechnol Lett 2004; 26(6):509-15; PMID:15127793; http://dx.doi.org/ 10.1023/B:BILE.0000019559.84305.47 [DOI] [PubMed] [Google Scholar]
- 41.Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 2002; 30(9):e36; PMID:11972351; http://dx.doi.org/ 10.1093/nar/30.9.e36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glazar P, Papavasileiou P, Rajewsky N. circBase: a database for circular RNAs. RNA 2014; 20(11): 1666-70; PMID:25234927; http://dx.doi.org/ 10.1261/rna.043687.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rennie W, Liu C, Carmack C, Wolenc A, Kanoria S, Lu J, Long D, Ding Y. STarMir: a web server for prediction of microRNA binding sites. Nucleic Acids Res 2014; 42:W114-8; PMID:24803672; http://dx.doi.org/ 10.1093/nar/gku376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu C, Mallick B, Long D, Rennie W, Wolenc A, Carmack C, Ding Y. CLIP-based prediction of mammalian microRNA binding sites. Nucleic Acids Res 2013; 41(14):e138; PMID:23703212; http://dx.doi.org/ 10.1093/nar/gkt435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cook KB, Kazan H, Zuberi K, Morris Q, Hughes TR. RBPDB: a database of RNA-binding specificities. Nucleic Acids Res 2011; 39:D301-8; PMID:21036867; http://dx.doi.org/ 10.1093/nar/gkq1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.