

Abstract
Current RNA expression profiling methods rely on enrichment steps for specific RNA classes, thereby not detecting all RNA species in an unperturbed manner. We report strand-specific RNAome sequencing that determines expression of small and large RNAs from rRNA-depleted total RNA in a single sequence run. Since current analysis pipelines cannot reliably analyze small and large RNAs simultaneously, we developed TRAP, Total Rna Analysis Pipeline, a robust interface that is also compatible with existing RNA sequencing protocols. RNAome sequencing quantitatively preserved all RNA classes, allowing cross-class comparisons that facilitates the identification of relationships between different RNA classes. We demonstrate the strength of RNAome sequencing in mouse embryonic stem cells treated with cisplatin. MicroRNA and mRNA expression in RNAome sequencing significantly correlated between replicates and was in concordance with both existing RNA sequencing methods and gene expression arrays generated from the same samples. Moreover, RNAome sequencing also detected additional RNA classes such as enhancer RNAs, anti-sense RNAs, novel RNA species and numerous differentially expressed RNAs undetectable by other methods. At the level of complete RNA classes, RNAome sequencing also identified a specific global repression of the microRNA and microRNA isoform classes after cisplatin treatment whereas all other classes such as mRNAs were unchanged. These characteristics of RNAome sequencing will significantly improve expression analysis as well as studies on RNA biology not covered by existing methods.
Keywords: strand-specific RNA-sequencing, RNAome, non-coding RNA, whole transcriptome, RNA expression, RNA abundance
Abbreviations
- lncRNAs
long non-coding RNA
- eRNA
enhancer RNA
- NGS
next generation sequencing
- poly(A)
poly-adenylation
- mRNASeq
mRNA sequencing
- smallRNASeq
small non-coding RNA sequencing
- DEGs
differentially expressed genes
- rRNA
ribosomal RNA
- snoRNAs
small nucleolar RNAs
- isomiRs
microRNA isoforms.
Introduction
The discovery of thousands of non-coding RNAs, both small and large, has reshaped RNA biology. These non-coding RNAs have been implicated in numerous biological processes and diseases.1-7 A significant part of non-coding RNA function is controlling gene expression, e.g. microRNAs have been established as such regulators,3,8-11 but it is becoming clear that long non-coding RNAs (lncRNAs), including non-polyadenylated transcripts ranging from several hundred to thousands of nucleotides in length also regulate gene expression.12-14 An example is the recently identified enhancer RNA (eRNA) class, which are mostly non-polyadenylated lncRNAs transcripts ∼50 to 2000 nucleotides in length generated at enhancer sites of active promoters.5,15-17 Thus, systematic quantitative expression analysis of non-coding RNA classes in combination with mRNA expression will therefore assist in unraveling RNA networks in much greater detail and boost our understanding of cellular processes and diseases.
Gene expression profiling by microarray technology has substantially transformed biology by systematically monitoring the global gene expression, but also has some limitations such as the quality of the capture probes and novel RNA discovery. The emergence of next generation sequencing (NGS) technology has enormously improved these limitations of arrays and further revolutionized the deciphering of RNA networks by sequencing millions of RNA-derived cDNA (cDNA) molecules. Established NGS protocols monitoring RNA expression rely on enrichment of specific RNA classes, e.g., poly-adenylation (poly(A)) selection for mRNA sequencing (mRNASeq) or gel-size selection for small non-coding RNA sequencing (smallRNASeq).
Our objective was to set up RNAome sequencing (RNAomeSeq), which we defined as sequencing rRNA (rRNA)-depleted total RNA, both small and large RNAs, coding and non-coding in a single sequencing run. Sequencing of rRNA-depleted total RNA has been performed before to discover novel non-coding RNA species.18-20 In contrast to these methods, RNAomeSeq also includes small RNA analysis in the sequence run and does not fractionate rRNA-depleted RNA into a large and small RNA sample before sequencing, which could lose important information about the abundance of RNA classes. While there are several RNA sequencing analysis algorithms available, none of these can simultaneously analyze both small and large RNAs from a single sample. Therefore, we developed a robust and reliable RNA expression analysis tool named TRAP (Total Rna Analysis Pipeline), which is also compatible with existing RNA sequencing protocols. We show the improvements of RNAomeSeq over existing profiling protocols, i.e. mRNASeq, smallRNASeq and microarray, in mouse embryonic stem (mES) cells after cisplatin treatment.
Results
To obtain material for all omics protocols, mES cells were thawed, grown for 2 passages and subsequently either cisplatin- or mock-treated. Eight hours later total RNA was isolated. This complete procedure was repeated 4 times to obtain biological replicates for statistical analysis (Fig. 1A). Cisplatin treatment was chosen due to its well-documented transcriptional response in mES cells.21 Samples received rigorous DNase treatment during total RNA isolation to eliminate genomic DNA contamination. Then, total RNA from each sample was aliquoted for usage in all omics protocols, i.e., RNAomeSeq, mRNASeq, smallRNASeq and Affymetrix gene expression arrays (Table S1). The latter 3 were processed according to manufacturer's instruction (see Material and Methods).
Figure 1.
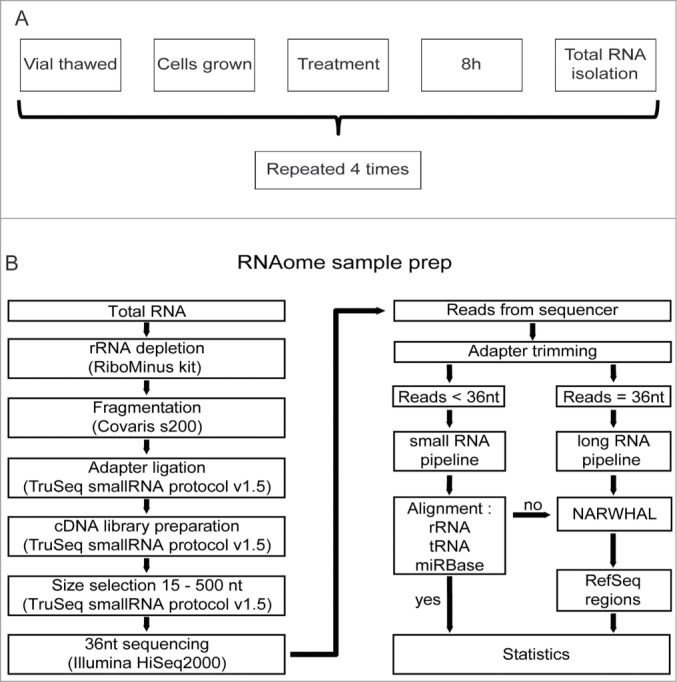
RNAomeSeq set up and analysis. (A). Diagram of biological replicate sample preparation from mES cells treated with 2.7 μM cisplatin or mock-treated (equal volume DMSO) for 8 hours. This procedure was repeated 4 times to obtain 4 independent biological replicates. All omics methods were performed on the exact same samples. (B). Schematic of the RNAomeSeq method. Total RNA was depleted of rRNA, fragmented and adapters were ligated to prepare a compatible cDNA library followed by fractionation on gel. Short sequencing reads (<36 nucleotides) were trimmed for adapter sequences and further processed by TRAP (Fig. 2). 36 nucleotide sequencing reads were processed as long RNAs.
Subsequently, total RNA aliquots for RNAomeSeq were depleted of highly abundant rRNA, using biotin-labeled LNA probes specific for rRNAs (i.e. 5 S, 5.8 S, 18 S and 28 S), and the remaining RNA was fragmented by sonication. All steps in this procedure were highly reproducible (Figure S1). Sequencing adapters were ligated to the fragmented RNA allowing the generation of a cDNA library. Finally, adapter dimers (fragments < 145 nt) were removed by gel size selection and the cDNA library was sequenced (36 nucleotides reads) (Fig. 1B, Table S2).
While there are several RNA sequencing analysis algorithms available, none of these can reliably and simultaneously analyze both small and large RNAs from a single sample. Therefore, we developed TRAP (Total Rna Analysis Pipeline), which extracts data from sequence files, categorizes RNAs in classes, identifies post-transcriptional sequence modifications of small RNAs and performs statistical analysis. Moreover, TRAP is also compatible with standard mRNASeq and smallRNASeq (Fig. 2). Briefly, prior to the analysis with TRAP, datasets containing small RNAs (i.e., the RNAomeSeq or smallRNASeq) were trimmed for adapter sequences. Then, sequence reads were divided into a small RNA category with RNA species length between 14 and 36 nucleotides after adapter trimming or into a group in which RNA species length is at least 36 nucleotides. The latter group was aligned to the reference genome with NARWHAL automation software.22 Expressed transcripts and regions were divided by RefSeq identifiers into 4 categories, i.e. coding transcripts, non-coding transcripts, intergenic or intronic transcripts (Fig. 2A). All reads in the small RNA category were first aligned to rRNA sequences (5 s and 5.8 s), tRNA sequences, miRBase database (v19)23 for microRNA identification and aligned to the genome using NARWHAL.22 Reads that aligned to the genome (small RNAs) were further processed as the longer RNA category in TRAP. The modular structure of TRAP also allows easy adjustments regarding transcript identifiers (e.g. GENCODE instead of RefSeq) or statistical algorithms (e.g., DESeq instead of edgeR). The detection of short transcripts, such as snoRNAs, resulted in an overestimation of these transcripts when normalizing on transcripts length (such as RPKM or FPKM). Therefore, we only used statistical analysis algorithms with raw reads as input. There are several statistical analysis algorithms available for RNA sequencing data sets.24-27 We tested the performance of 4 algorithms in the mRNASeq dataset and determined overlap in the microarray data set (Table S3). Three had similar performance, identifying 2055 to 2836 differentially expressed genes (DEGs), which were highly overlapping with the microarray results (74.2% – 76.8%). We used edgeR26 as the standard statistical analysis algorithm in TRAP for further analyses.
Figure 2.

Schematic of the Total RNA Analysis Pipeline, TRAP, for analysis of sequencing datasets. (A). Modules for long RNA analysis, script 1 for RefSeq annotated exonic transcripts and script 2 for RefSeq annotated non-exonic regions. (B). Modules for small RNA analysis, script 3 to align trimmed reads to first rRNA, then tRNA sequences and the microRNA database, miRBase version 19.
Subsequently, we analyzed the RNAomeSeq dataset. The reads obtained from RNAomeSeq allowed us to measure the abundance of all RNA classes found in mES cells (Fig. 3A). Only 7.8% of the reads mapped to rRNA sequences (7.7% 45 s in the large fraction, 0.1% 5/5.8 s rRNA in small fraction), showing efficient depletion of rRNA. The percentage of reads that aligned (Table S4) and did not align to the genome was similar to the mRNASeq and smallRNASeq data sets (Figure S2). These unaligned reads are likely to result from SNP-rich regions (TRAP's default settings allows 2 mismatches to the reference genome), small RNA fragments (TRAP's default settings only include RNA molecules >14 nucleotides), reference genome differences or sequencing errors (Figure S2). In the RNA fraction with a length of at least 36 nucleotides from RNAomeSeq we identified exonic reads, which refers to annotated, for function coding, transcripts (coding transcripts, mitochondrial transcripts, small nucleolar RNAs (snoRNAs) and annotated long non-coding RNAs (including e.g. pre-microRNAs)) and transcripts originating from intronic or intergenic regions (Fig. 3A), which is similar to previously published long RNA classes distribution.28 The small RNA fraction contained mature microRNAs, microRNA isoforms (isomiRs) and additional small RNA molecules. In these non-microRNA/isomiR classes of small RNAs we identified fragments of tRNAs and small RNAs from coding, non-coding, intergenic and intronic regions (Fig. 3A). The abundance of RNA classes found by mRNASeq (Fig. 3B) and smallRNASeq (Fig. 3C) showed the expected RNA classes enriched for poly(A)-coding transcripts and small RNAs, respectively.
Figure 3.

(See previous page). The proportion of RNA species found in mES cells. (A). The proportion of RNA classes detected by the RNAomeSeq protocol with a minimum of one read per million found across all biological replicates from at least one of the experimental groups. Detecting small RNA classes (right panel): tRNA fragments (0.2%), small coding (0.2%), small non-coding (0.3%), mature microRNA (miR) (0.7%), microRNA isoforms (isomiR) (0.9%), small intergenic (1.7%), small intronic (2.0%); and long RNA classes (left panel): non-coding transcripts also containing complete tRNAs (12.2%), coding transcripts (2.2%), snoRNA (19.4%), mitochondrial (1.9%), histones (0.2%), intronic region (37.4%), intergenic region (20.7%) classes. (B). The proportion of RNA species detected by the mRNASeq protocol with a minimum of 5 reads found across all biological replicates from at least one of the experimental groups. Detecting coding transcripts (71.0%), non-coding transcripts (1.2%) and reads from mitochondrial (2.3%), histones (0.1%), intronic regions (9.3%) and intergenic regions (16.2%). (C). The proportion of small RNA species detected by the smallRNASeq protocol with a minimum of 5 reads found across all biological replicates from at least one of the experimental groups. Detecting small RNA classes: tRNA fragments (4.0%), small coding (2.0%), small non-coding (17.6%), mature microRNA (miR) (27.9%), microRNA isoforms (isomiR) (25.7%), small intergenic (10.6%) and small intronic (12.1%). The indicated % represents the total aligned RNAs from that particular class compared to the total number of reads, excluding rRNA reads.
In addition, we noticed another important characteristic of RNAomeSeq, which is an almost perfect strand-specificity. We observed a 99.8% accurate orientation of sequence reads that align to the correct direction of gene transcription (Table S5). This feature will allow discrimination between overlapping reads from opposite strands. For example, reads aligning to the 3′UTR of Prpf39 and reads aligning to the overlapping last exon and 3′UTR of the Fkbp3 gene on the opposite strand could be separated from each other (Fig. S3).
To assess reliability, we determined correct RNA class representation in RNAomeSeq. Experimental verification of correct class representation is difficult to assess for most RNA classes. Poly(A) RNA however, can be quantitatively measured in a sample. Our results indicate that ∼90% of total RNA represents rRNA (Additional file 2), ∼2.2% of all reads referred to coding transcripts (Fig. 3A) and mRNASeq that is based on poly(A) selection, indicated that ∼71% of all reads map to coding regions (Fig. 3B). This suggests that approximately 0.3% of total RNA represents poly(A) RNA. We measured poly(A) RNA content of our samples directly by poly(dT) beads isolation followed by bioanalyzer analysis (Fig. S4). This analysis indicate that indeed ∼0.3% poly(A) RNA is present in total RNA, which is in line with our RNAomeSeq results. Additional cell lines from human and mouse origin had similar poly(A) RNA content, indicating that this observation is not specific for mES cells (Fig. S4).
Reliability is also determined by putative biases introduced by RNAomeSeq compared to standard mRNASeq or smallRNASeq. First, we analyzed the representation of transcripts in RNAomeSeq and mRNASeq by plotting the percentage of detected transcripts in transcript length bins (Fig. 4A). A >99% overlap of coding transcripts was observed between RNAomeSeq and mRNASeq without any differences in transcript length distribution. Secondly, we determined gene expression correlation between RNAomeSeq and mRNASeq by plotting read count per million (CPM) per coding transcript in a XY-scatterplot (Fig. 4B). Quantitative gene expression levels detected by RNAomeSeq were highly similar to mRNASeq (Pearson correlation coefficient R = 0.86; P < 2.2e–16). There was a noticeable difference: a class of coding transcripts was highly expressed in RNAomeSeq (Fig. 4B, red circle), but hardly expressed in mRNASeq. This group consisted of histones, which have very short or absent poly-A tails and are therefore hard to detect with standard mRNASeq. Thirdly, we determined the distribution of sequence reads mapping to coding transcripts across the gene body (Fig. 4C). In contrast to mRNASeq in which read density was equal across the gene body except for the 5′ and 3′ transcript ends, RNAomeSeq harbored several specific peaks. These peaks were produced by intronic snoRNAs, which transcripts overlap with exons from host genes. Therefore, these sequences were automatically included in this analysis. Removal of intronic snoRNAs from the analysis, which are also not detected by mRNASeq, abolished these peaks and produced a similar distribution as seen in mRNASeq. Finally, we determined any bias for small or large transcripts in the detected sequence reads. The percentage of detected sequence reads was plotted for transcript length bins (Fig. 4D). A slight deviation was observed compared to mRNASeq, which could be explained by intronic snoRNAs and histone sequences (Fig. 4D). In toto, RNAomeSeq performs equally compared to standard mRNASeq without any biases in detecting coding transcripts.
Figure 4.

Representation of coding transcripts. (A) Coding transcript length distribution of the whole genome or detected by mRNASeq and RNAomeSeq. (B) The Pearson-correlation between and X-Y scatter plot of coding transcript expression between RNAomeSeq and mRNASeq, histones encircled in red. (C) Distribution of reads along the body of all coding transcript for mRNASeq, RNAomeSeq and RNAomeSeq depl (depleted of histones and transcripts with intronic snoRNA). (D) Distribution of reads aligning to the detected coding transcripts by mRNASeq, RNAomeSeq and RNAomeSeq depl (depleted of histones and transcripts with intronic snoRNA) in regard to transcript length.
Subsequently, we determined putative biases in microRNA and isomiR detection by RNAomeSeq. By plotting the percentage of detected transcripts in transcript length bins, we observed that the representation of transcripts in RNAomeSeq and smallRNASeq was similar (Fig. 5A). There was however, a clear shift toward increased microRNA length in both smallRNASeq and RNAomeSeq compared to mirBase (v19), which could be explained by a lack of isomiRs in miRbase. Quantitative microRNA and isomiR expression correlation between RNAomeSeq and smallRNASeq was also very similar (Pearson correlation coefficient R = 0.76; P < 2.2e–16) between RNAomeSeq and smallRNASeq as seen in a XY-scatterplot in which CPM per microRNA/isomiR has been plotted (Fig. 5B). Finally, we determined any bias for microRNA/isomiR length in the detected sequence reads by plotting the percentage of detected microRNA/isomiR transcripts per length (Fig. 5C). A slight deviation was observed between the 2 methods, i.e., a decrease in microRNA/isomiRs with a length of 21 nucleotides and an increase in 24 nucleotide long microRNAs/isomiRs. Sample preparation differences such as gel excision (smallRNASeq) might explain the differences. RNA fractionation as performed in RNAomeSeq could result in fragments of long transcripts in the small RNA compartment that align to the genome and thereby generate observed differences between RNAomeSeq and smallRNASeq (Fig. 3A, C). We did not observe any obvious expression correlation in coding, non-coding, intergenic and intronic transcript levels between the small and large fractions in RNAomeSeq (Fig. S5). Taken together, this data indicate that RNAomeSeq correctly represents small RNA expression as well.
Figure 5.

Representation of microRNAs and isomiRs. (A) Length distribution of the microRNA/isomiRs transcripts in the miRbase database or detected by smallRNASeq and RNAomeSeq. (B) The Pearson-correlation between and X-Y scatter plot of microRNA/isomiRs expression between RNAomeSeq and smallRNASeq. (C) Distribution of microRNA/isomiRs reads detected by smallRNASeq and RNAomeSeq in regard to length.
We continued by analyzing expression level correlations between the biological replicates from coding transcripts in mRNASeq, microRNAs in smallRNASeq and both coding transcripts and microRNAs in RNAomeSeq. We observed very high and significant correlations for all replicates, which was on average a 0.99 and 0.95 correlation coefficient for the existing protocols and RNAomeSeq, respectively (Pearson rank correlation, all samples P-values < 2e–16) (Table 1), indicating that the RNAomeSeq procedure in itself is very reliable and can be used for expression profiling. We performed statistical analysis between cisplatin and mock treatment and compared the results from RNAomeSeq to mRNASeq and microarray (Fig. S6). First, we compared DEGs between microarray and mRNASeq, since both rely on poly(A) selection and are therefore expected to be most similar. For comparisons with the microarrays, probes were first filtered for correct annotation, i.e. probes annotated in the RefSeq database. RefSeq annotated probes specific for microarrays and not found in mRNASeq were mostly low intensity signals and therefore likely not expressed (Fig. S6A). 77% of the DEGs found by microarray (n = 4/group) were also significantly regulated in mRNASeq (n = 3/group). Moreover, DEG fold changes were highly correlated as well (Fig. S6B). We identified genes and enriched pathways as previously reported for cisplatin treatment in mES cells,21 indicating, together with the highly overlapping DEGs between microarray and mRNASeq, correct performance of the experiment and TRAP. High DEG fold change correlations were also observed between RNAomeSeq and microarray (Fig. S6C) and between RNAomeSeq and mRNASeq (Fig. S6D). Thus, we conclude that differential expression is also preserved in RNAomeSeq.
Table 1.
The Pearson-correlation between replicate samples in RNAomeSeq, mRNASeq and smallRNASeq, for the coding transcripts and/or microRNAs, all correlations had P-value < 2.2E–16.
| coding |
microRNA |
||||
|---|---|---|---|---|---|
| Pearson correlation | mRNASeq | RNAomeSeq | smallRNASeq | RNAomeSeq | |
| Replicate 1 vs 2 | 0.997 | 0.999 | 0.996 | 0.949 | Cisplatin |
| Replicate 1 vs 3 | 0.996 | 0.992 | 0.996 | 0.868 | |
| Replicate 2 vs 3 |
0.999 |
0.983 |
0.994 |
0.976 |
|
| Replicate 1 vs 2 | 0.999 | 0.997 | 0.999 | 0.973 | Control |
| Replicate 1 vs 3 | 0.996 | 0.831 | 0.998 | 0.983 | |
| Replicate 2 vs 3 | 0.996 | 0.969 | 0.997 | 0.970 | |
Since RNAomeSeq quantitatively preserves all RNA species in a single sequence run, we compared all RNA classes in mES cells with and without cisplatin treatment. We observed a specific global repression of the microRNA and isomiR classes after cisplatin treatment (Fig. 6). This observation is in agreement with observations that key components of the microRNA biogenesis pathway are targeted by caspases during apoptosis,29,30 which is consistent with the onset of apoptosis of cisplatin-treated mES cells. This demonstrates that RNAomeSeq can be used to study behavior of complete RNA classes.
Figure 6.
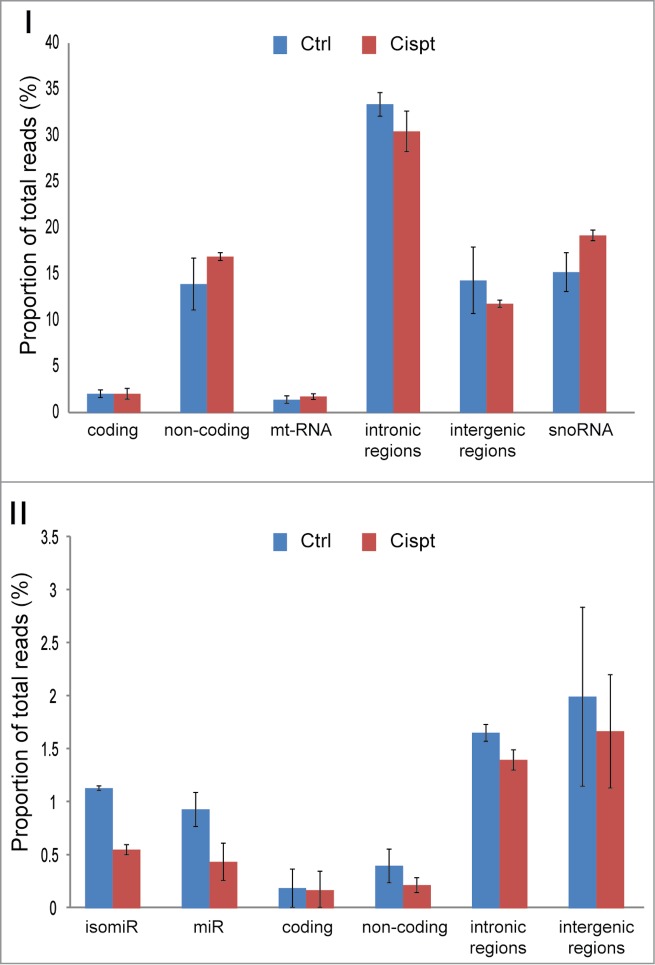
Quantitative preservation of all RNA species. Total proportion of RNA classes before and after cisplatin treatment. Panel I, long RNA classes, Panel II small RNA classes. Error bars represent standard deviations.
Discussion
Here we demonstrated that RNAomeSeq is a strand-specific (99.8% correct orientation, Table S5), robust and reliable method to sequence both small and large RNAs, coding and non-coding, in a single sequencing run. Expression correlations with standard smallRNASeq and mRNASeq were very high. In addition, we found that isomiRs are abundantly present in mES cells, which can be well documented by RNAomeSeq as well as standard smallRNASeq. Although the exact function of isomiRs is not known,31,32 TRAP can provide a thorough isomiR overview (Supplemental Dataset 1). Our approach allows simultaneous analysis of RNA expression, identification of novel RNAs and transcripts and a comparison between RNA classes.
As far as we can determine, the RNAomeSeq method does not introduce additional biases in quantitative transcript expression within a RNA class, such as microRNAs or coding transcripts, compared with standard smallRNASeq and mRNASeq. Next to a high transcript expression correlation between RNAomeSeq and mRNASeq / smallRNASeq, we did not observe a transcript length bias or differences in read distribution across transcripts. There were some noticeable differences between RNAomeSeq and mRNASeq, mostly in the detection of specific RNA classes (see Fig. 3). RNAomeSeq was able to identify non-polyadenylated RNAs, including histones and snoRNAs, and improved detection of annotated long non-coding RNAs. The completeness of RNAomeSeq also provides a disadvantage: sequencing depth should be sufficient, an estimated 180–200 million reads (compared to 10–20 million for smallRNASeq and mRNASeq), in order to identify and classify differentially expressed genes. The expected decrease in sequencing costs however, will compensate for the required sequencing depth.
Current methods based on RNA selection cannot quantitatively determine transcript level ratios across RNA classes. While RNAomeSeq detects most, if not all, RNA classes besides rRNA, it is conceivable that the technical procedures of RNAomeSeq introduce detection biases toward or against specific RNA species and classes. Therefore, it remains a question to what extent RNAomeSeq can be used to quantitatively determine transcript level ratios between RNA classes or map a complete quantitative RNAome from a sample. Qualitative analysis, i.e., comparisons between experimental groups, is not hampered by biases. Two putative biases could be identified. Small RNAs are favored over longer RNAs in NGS methods and therefore overrepresented. Secondly, RNA fragmentation by sonication could result in a break at the hydroxyl or the phosphate group at the 3′ end. The 3′ adapter used in the NGS protocol is specifically modified to ligate to RNAs with a 3′ hydroxyl group, such as microRNAs, resulting from enzymatic cleavage by Dicer or other RNA processing enzymes. However, the detection of numerous isomiRs, to which specialized enzymes add additional nucleotides at the 3′ end after Dicer cleavage, would suggest that the 3′ adapter has tolerance for other 3′ ends as well. Furthermore, if we assume that breakage by sonication occurs randomly, we would expect that only 1 in 2 fragments could be used in sequence adapter ligation and subsequent cDNA formation, which could translate into a 2-fold underrepresentation of non-enzymatically processed small and longer RNAs in RNAomeSeq.
To estimate an underrepresentation or overrepresentation of specific RNA classes, it is essential to know the ratio between specific RNA classes. Single cell sequencing experiments and subsequent follow up studies have provided an estimate for the total number of mRNAs33 and microRNAs34 in a single mES cell. These data indicate that for every mRNA molecule 5 microRNA molecules are present in mES cells.33,34 Since the smallRNASeq adapter ligation kit for RNAomeSeq was used, we assume that microRNAs and isomiRs are very efficient labeled and sequenced in which 1 microRNA translates to 1 sequence read. 2.2% of the detected reads in RNAomeSeq aligned to coding transcripts and 1.6% to microRNA transcripts. (Fig. 3) Coding transcript with a mean length of 3300 nucleotides (Fig. 4A) are likely to break evenly during fragmentation with an average fragment size of 300 nucleotides (Fig. S1). Thus, we expect approximately 11 fragments per transcript. Subsequent calculations estimate the presence of 1 mRNA molecule per 8 microRNA molecules in RNAomeSeq, suggesting a ∼1.6 fold overrepresentation of microRNA or underrepresentation of mRNA molecules.
While exact RNA content in a single cell or sample is difficult to assess, several observations allow us to provide a rough estimate of the expected number of mRNA sequencing reads in RNAomeSeq. The poly(A) content of a typical cell is 1% of the total RNA,35 implicating an underrepresentation of the poly(A) content in RNAomeSeq, since our experiments indicate ∼0.3% poly(A) content and an estimated 0.2 – 025% mRNAs in total RNA from mES cells (Fig. 3A and Fig. S4). Compared to other cell types however, mES cells have fewer mRNA molecules per cell (20-fold reduction) as well as lower total RNA content per cell (5.5-fold reduction). 33 This suggests a relative ∼3.6-fold lower mRNA content in mES cells. The standard mRNASeq data indicates that ∼71% of all poly(A) RNA refers to coding transcripts (Fig. 3B). Extrapolating these estimations, one would expect ∼2% of the reads in RNAomeSeq to refer to coding transcripts, which is in agreement with our observations. These calculations suggest an overrepresentation of microRNAs rather than underrepresentation of mRNA molecules in RNAomeSeq.
Transcripts from intergenic and intronic regions were abundantly present among small and large RNA classes, among which we could also identify differential expressed RNAs, suggesting functional roles in the cellular cisplatin response (intronic, Supplemental Dataset 2; intergenic, Supplemental Dataset 3). In particular the large content of intronic transcripts was intriguing for both cisplatin- and mock-treated samples. We found in RNAomeSeq that on average 37.4% of the reads originated from intronic regions (note that the intronic DNA content of the genome is ∼12.8 times larger than the exonic DNA content). It has long been thought that the splicing process is very fast and spliced intronic RNA is rapidly degraded, but it is becoming clear that some introns have additional functions and escape the rapid degradation process.36 Evidently, a part of the intronic RNA content in RNAomeSeq can be explained by the presence of precursor-mRNAs (pre-mRNAs). Secondly, some introns could be more stable than anticipated or, thirdly, functional non-coding RNAs could originate from intronic regions. If co-transcriptional splicing is very fast and spliced introns are directly degraded, one would predominantly expect sequence reads overlapping 5′ exon – intron 3′ boundaries as pre-mRNA is captured during isolation. RNAomeSeq indeed detected sequence reads that overlap exon-intron boundaries, but also equal numbers of reads that map to intron-exon boundaries. In total, these reads were much less than sequence reads that span exon-exon boundaries (Table S6). Remarkably, standard mRNASeq, which requires a double selection for poly(A)+ RNAs, also detects sequence reads that overlap exon-intron or intron-exon boundaries, suggesting that a significant part of reads spanning exon-intron or intron-exon boundaries originate from introns that are maintained in mature mRNA and are thus very stable (Table S6). Based on RNaomeSeq data, we suggest that a significant part of all intronic transcripts are likely bona fide non-coding RNAs that is consistent with our results in which snoRNAs are present in intronic regions (Fig. 4C) and previous reports indicating the presence of intron-derived non-coding RNAs.37-40 Finally, RNAomeSeq is highly strand specific (99.8% correct orientation; Table S5). Using strand-specificity sequence information intrinsic to the RNAomeSeq method, we regularly observed intronic reads expressed from the opposite strand compared to gene orientation, strongly indicating the presence of a non-coding RNA encoded in the intron. Further RNAomeSeq studies will allow more in depth analysis of intron biology.
In addition, we also noted numerous intergenic RNAs upstream of gene promoters, which were not present in mRNASeq or smallRNASeq. Their location and size were reminiscent of a recently identified class of non-poly(A) non-coding RNAs, named eRNAs. These are detected as sequence peaks upstream of the promoter. Since only very few eRNAs have been experimentally verified, we did not systematically categorize them in a distinct RNA class as seen in Figure 3. The widespread occurrence of non-poly(A) RNAs in close proximity of highly expressed genes (examples see Figure S6) suggests that RNAomeSeq can also detect eRNAs, exemplifying that RNAomeSeq (but not mRNASeq) can be used to study relationships between different RNA classes in an unbiased manner.
One of RNAomeSeq's strengths is monitoring global upregulation or repression of complete RNA classes since it quantitatively preserves all RNA species in a single sample. This allows for monitoring/identifying pathways that control the expression of complete RNA classes. A prime example is repression of the microRNA biogenesis pathway during tumorigenesis, leading to reduced numbers of mature microRNAs in human cancer.41 We observed a specific global repression of the microRNA and isomiR classes after cisplatin treatment (Fig. 6), demonstrating that RNAomeSeq can be used to study behavior of complete RNA classes.
In summary, we show that RNAomeSeq quantitatively preserves global and differential RNA expression patterns of RNA classes. Besides novel RNA species identification, RNAomeSeq can identify relationships between different RNA classes, allowing the elucidation of RNA networks in much greater detail. For example, mRNA expression levels are determined by transcriptional activity, but also by microRNA expression. It is becoming clear that eRNAs, generated upstream of the gene locus, are needed for transcriptional activity 16 and therefore can serve as marks for active transcription. MicroRNAs predominantly act via mRNA degradation, which can be visualized by RNA sequencing methods.42 Analyzing mRNAs, microRNAs and eRNAs simultaneously could indicate which mechanism controls observed gene expression changes. In toto, the described characteristics of RNAomeSeq will significantly improve expression analysis as well as studies on RNA biology not covered by existing methods.
Material and Methods
Total RNA isolation
Mouse embryonic stem (mES) cells (HM1) were cultured as described.21 One vial of mES cells was thawed and grown for 2 passages on feeder-coated plates followed by one passage on gelatin-coated plates before beginning the experiment. The mES cells in experiment were treated with 2.7 μM cisplatin (75% survival; Platosin) or mock-treated (equal volume dimethylsulfoxide (DMSO)). After 8 h continuous exposure total RNA was isolated using Qiazol Lysis Reagent (Qiagen) and total RNA was purified with the miRNeasy kit (Qiagen), according to manufacturer's protocols. The integrity (scores >9.0) of the RNA was determined on the Agilent 2100 Bioanalyzer (Agilent) according to manufacturer's protocol. This procedure was repeated 4 times to obtain 4 independent biological replicates. Subsequent sequencing and array protocols were performed on the total RNA from the same biological samples.
Microarray sample preparation
The poly(A) RNA enrichment for Affymetrix GeneTitan® array was performed by ServiceXS, following their standard protocol. In short, 100 ng of total RNA was labeled with the Affymetrix 3′ IVT-Express Labeling Kit (containing oligo dT primers), amplified and fragmented before hybridizing to Affymetrix HT Mouse Genome 430 PM Array.
mRNASeq sample preparation
Total RNA enrichment for sequencing poly(A) RNAs was performed with the TruSeq mRNA sample preparation kit (Illumina) according to the manufacturer's protocols. In short, 1 μg of total RNA for each sample was used for poly(A) RNA selection using magnetic beads coated with poly-dT, followed by thermal fragmentation. The fragmented poly(A) RNA enriched samples were subjected to cDNA synthesis using Illumina TruSeq preperation kit according to the manufacturer's protocol. Briefly, cDNA was synthesized by reverse transcriptase (Super-Script II) using poly-dT and random hexamer primers. The cDNA fragments were then blunt-ended through an end-repair reaction, followed by dA-tailing. Subsequently, specific double-stranded bar-coded adapters were ligated and library amplification for 15 cycles was performed.
smallRNASeq sample preparation
The cDNA library for smallRNASeq was generated by the small RNASeq kit (Illumina TruSeq smallRNA v1.5) according to the manufacturer's protocol. In short, specific bar-coded adapters were ligated to 1 μg of total RNA followed by reverse transcriptase and amplification for 11 cycles. Small RNAs were enriched by fractionation on a 15% Tris-borate-EDTA gel, excising the RNAs of 15–30 nucleotide of length.
RNAomeSeq sample preparation
rRNA (rRNA) depletion was performed using RiboMinus Eukaryote Kit (Life Science), according to the manufacturer's protocol. 10 μg of total RNA was incubated with biotin-labeled LNA probes (2 for each of the 4 rRNA species, i.e. 5 S, 5.8 S, 18 S and 28 S) and hybridized to streptavidin-coated magnetic beads. The rRNA-depleted samples were concentrated using the RiboMinus Concentration Module, according to manufacturer's protocols. The concentrated rRNA-depleted samples were fragmented by sonication (Covaris s200, duty cycle 5% and 200burst/cycle for 210 sec), to fragments smaller than 500 nucleotides. The cDNA library preparation was performed according to the smallRNASeq sample preparation. Adapter dimers, approx. 145 nucleotides in length, were removed by excising RNAs ranging 160– 645 nucleotide of length from the gel, corresponding to RNAs 15–500 nt in length. The excised gel containing the adapter-ligated cDNA fragments were extracted from the gel using the gel breaker kit (IST Engineering). Finally, the cDNA was pooled after extraction and further prepared for sequencing.
Sequencing
The pooled cDNA libraries all consisted of equal concentration bar-coded samples, i.e., 3 mock- and 3 cisplatin-treated samples. The mRNASeq and smallRNASeq pooled libraries were sequenced in one lane each and the RNAomeSeq pooled library was sequenced in 2 lanes, all 36 bp single read on the HiSeq2000 (Illumina).
Total RNA analysis pipeline
The analysis of the sequencing datasets was performed with TRAP, which stands for Total RNA Analysis Pipeline. The analysis was performed on a quad-core CPU desktop with 64-bits windows system and 16 gigabyte RAM. Per sample, the analysis takes around 5 minutes for mRNASeq and 20 minutes for smallRNASeq.
The RNAomeSeq and smallRNASeq reads were, prior to the analysis with TRAP, trimmed for adapter sequences with a custom script. Reads from RNAomeSeq and mRNASeq were aligned to the mouse mm9 reference genome using Tophat (version 1.3.1.Linux_x86_64, –coverage-search, -butterfly-search, –segment-mismatches 1,–segment-length 18) via the NARWHAL automation software.22 We have developed NARWHAL to automate sequence data processing using pre-existing open-source tools. TRAP makes use of several R Bioconductor43 packages, e.g., Biostrings (version 2.26.3), Rsamtools (version 1.10.2), IRanges (version 1.16.6), GenomicRanges (version 1.10.7), Limma44 and EdgeR.26 Reads that aligned within and between RefSeq transcripts were extracted from the resulting BAM files using Scripts 1 and 2 in module I. RefSeq can be replaced in TRAP by other annotations such as GENCODE depending on the users preference. Exonic reads were summed per transcript. In module II, a specific transcript or region was referred to as expressed, when a predefined threshold was reached (1 read per million). The threshold was defined as a minimum number of reads that could be aligned to a transcript or non-exonic region across all biological replicates in at least one of the experimental groups. In module III, expressed transcripts were divided by RefSeq identifiers into coding and non-coding transcripts. The non-exonic regions were divided by location into an intergenic or intronic category. Statistical analysis of the transcripts and regions can be performed with several published statistical algorithms for mRNASeq that are all compatible with TRAP. We used in our analysis EdgeR,26 since this was the best performing statistical algorithm.
Next, we used TRAP to analyze reads smaller than 36 nucleotides from smallRNASeq and RNAomeSeq. In module I, trimmed sequence reads were discarded if smaller than 14 nucleotides of length. Reads were referred to as expressed when the threshold was reached, which was defined as a predefined minimal reads being present in all biological replicates in at least one experimental group. In Module II, the expressed reads were first aligned to rRNA sequences (5 s and 5.8 s), tRNA sequences, the miRBase23 database (v19) (using vmatchPattern from the Biostrings package) or the genome (using NARWHAL,22 using only bowtie; –best, −l 32, −n 2, −M 1). In module III, statistical analysis of the tRNA aligned reads and miRBase23 aligned reads (microRNAs) was performed with EdgeR.26 The reads aligned to the genome (small RNAs) were further processed as long RNAs in Script 1 and 2 in TRAP. Threshold in TRAP can be manually set and adjusted according to needs.
Statistics and pathway analysis
Differentially expressed (DE) transcripts were identified in the mRNASeq data set with EdgeR,26 assuming negative binomial distribution of the reads. DE transcripts were identified in the Affymetrix dataset by computing a linear model using Limma.44 For both platforms cut-offs were used for DE transcripts detection (fold change > 1.5 and FDR < 0.05). Pathway analysis was performed with Ingenuity Pathway Analysis Software.
Proportion of precursor-mRNA
The presence of precursor mRNA (pre-mRNA) was defined as the number of reads spanning exon-intron and intron-exon borders compared to reads spanning exon-exon borders. Exon-exon reads were reads with a start position 10 - 5 bp to the 5′ side of the 5′ part of the exon-intron junction which span to the next exon. Exon-intron and intron-exon reads were reads that do not have a span (“N” in cigar string). The reads aligning to exon-intron reads run into the intron and had a start position 10 - 5 bp to the 5′ side of the 5′ part of the exon-intron junction. Intron-exon reads have a start position 10 - 5 bp to the 5′ side of the 3′ part of the junction and run into the exon. Pre-mRNA reads were reads aligning to exon-intron and intron-exon borders.
Strand specificity
To determine the strand specificity of a gene, we divided the number of reads aligning to the gene to either the plus or minus strand by the total number of reads aligning to the gene. We determined the percentage of reads aligning to genes orientated on the plus or minus strand and calculated the percentage of reads with the correct orientation over all genes. Reads aligning to the genome were visualized using integrative genomics viewer (IGV).45
Proportion of RNA species
The proportion of RNA species was defined by the number of reads that primary aligned to the genome (script 1 and 2 proportion). Only reads used to align to the genome were 36 nucleotides of length or did not align to miRBase,23 tRNA or rRNA sequences. The proportion of small RNA reads (<36 nucleotides) was defined by being uniquely aligned to miRBase,23 rRNA or tRNA. The proportion of protein-coding RNAs found in the RNAomeSeq data set was validated using a gel-analysis of poly(A) RNA enriched by poly-dT beads. We added magnetic beads coated with poly-dT, from the mRNASeq protocol (Illumina TruSeq), to 1 ug of total RNA. The bound poly(A) RNA was subsequently analyzed on an RNA pico-chip Agilent 2100 Bioanalyzer (Agilent), using manufacturer's protocols.
Availability of Supporting Data
Data has been deposited in the GEO database under the number GSE48084.
Competing Interests
The authors declare that they have no competing interests.
Author Contributions
K.W.J.D. and J.P. conceived the project and designed concept. K.W.J.D., W.IJ. and J.P. invented the method. K.W.J.D. developed the scripts. K.W.J.D. performed all experimental procedures. K.W.J.D. and C.E.M.K. performed all RNA sequencing experiments. B.M. and R.B. and M.H. examined and commented on the scripts. C.P.G. performed the microarray data analysis. K.W.J.D. and J.P. wrote the paper. W.IJ., H.V. and J.H.J.H. provided valuable input during the course of the project and commented on the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Z. Ozgur and A. van der Sloot for technical assistance.
Funding
This work was supported by the Netherlands Toxicogenomics Center (NTC), Cancer Genomics Center (CGC) and National Institute of Health (NIH)/National Institute of Aging (NIA) (1PO1 AG-17242-02), NIEHS (1UO1 ES011044) and an ERC advanced grant DamAge.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009; 136:215-33; PMID:19167326; http://dx.doi.org/ 10.1016/j.cell.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mattick JS. Long noncoding RNAs in cell and developmental biology. Semin Cell Dev Biol 2011; 22:327; PMID:21621631; http://dx.doi.org/ 10.1016/j.semcdb.2011.05.002 [DOI] [PubMed] [Google Scholar]
- 3. Hermeking H. MicroRNAs in the p53 network: micromanagement of tumour suppression. Nat Rev Cancer 2012; 12:613-26; PMID:22898542; http://dx.doi.org/ 10.1038/nrc3318 [DOI] [PubMed] [Google Scholar]
- 4. Leung AK, Sharp PA. MicroRNA functions in stress responses. Molecular cell 2010; 40:205-15; PMID:20965416; http://dx.doi.org/ 10.1016/j.molcel.2010.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 2013; 498:516-20; PMID:23728302; http://dx.doi.org/ 10.1038/nature12210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature 2012; 482:339-46; PMID:22337053; http://dx.doi.org/ 10.1038/nature10887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nallar SC, Kalvakolanu DV. Regulation of snoRNAs in cancer: close encounters with interferon. J Interferon Cytokine Res 2013; 33:189-98; PMID:23570385; http://dx.doi.org/ 10.1089/jir.2012.0106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cawley K, Logue SE, Gorman AM, Zeng Q, Patterson J, Gupta S, Samali A. Disruption of microRNA biogenesis confers resistance to ER stress-induced cell death upstream of the mitochondrion. PLoS One 2013; 8:e73870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shen J, Xia W, Khotskaya YB, Huo L, Nakanishi K, Lim SO, Du Y, Wang Y, Chang WC, Chen CH, et al. EGFR modulates microRNA maturation in response to hypoxia through phosphorylation of AGO2. Nature 2013; 497:383-7; PMID:23636329; http://dx.doi.org/ 10.1038/nature12080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van Kouwenhove M, Kedde M, Agami R. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat Rev Cancer 2011; 11:644-56; PMID:21822212; http://dx.doi.org/ 10.1038/nrc3107 [DOI] [PubMed] [Google Scholar]
- 11. Pothof J, Verkaik NS, van IW, Wiemer EA, Ta VT, van der Horst GT, Jaspers NG, van Gent DC, Hoeijmakers JH, Persengiev SP. MicroRNA-mediated gene silencing modulates the UV-induced DNA-damage response. Embo J 2009; 28:2090-9; PMID:19536137; http://dx.doi.org/ 10.1038/emboj.2009.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010; 142:409-19; PMID:20673990; http://dx.doi.org/ 10.1016/j.cell.2010.06.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G, Young G, Lucas AB, Ach R, Bruhn L, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011; 477:295-300; PMID:21874018; http://dx.doi.org/ 10.1038/nature10398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genetics 2009; 10:155-9; PMID:19188922; http://dx.doi.org/ 10.1038/nrg2521 [DOI] [PubMed] [Google Scholar]
- 15. Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010; 465:182-7; PMID:20393465; http://dx.doi.org/ 10.1038/nature09033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 2011; 474:390-4; PMID:21572438; http://dx.doi.org/ 10.1038/nature10006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Melo CA, Drost J, Wijchers PJ, van de Werken H, de Wit E, Oude Vrielink JA, Elkon R, Melo SA, Leveille N, Kalluri R, et al. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol Cell 2013; 49:524-35; PMID:23273978; http://dx.doi.org/ 10.1016/j.molcel.2012.11.021 [DOI] [PubMed] [Google Scholar]
- 18. Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. Landscape of transcription in human cells. Nature 2012; 489:101-8; PMID:22955620; http://dx.doi.org/ 10.1038/nature11233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cui P, Lin Q, Ding F, Xin C, Gong W, Zhang L, Geng J, Zhang B, Yu X, Yang J, et al. A comparison between ribo-minus RNA-sequencing and polyA-selected RNA-sequencing. Genomics 2010; 96:259-65; PMID:20688152; http://dx.doi.org/ 10.1016/j.ygeno.2010.07.010 [DOI] [PubMed] [Google Scholar]
- 20. Yang L, Duff MO, Graveley BR, Carmichael GG, Chen LL. Genomewide characterization of non-polyadenylated RNAs. Genome Biol 2011; 12:R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kruse JJ, Svensson JP, Huigsloot M, Giphart-Gassler M, Schoonen WG, Polman JE, Jean Horbach G, van de Water B, Vrieling H. A portrait of cisplatin-induced transcriptional changes in mouse embryonic stem cells reveals a dominant p53-like response. Mutat Res 2007; 617:58-70; PMID:17327130; http://dx.doi.org/ 10.1016/j.mrfmmm.2006.12.004 [DOI] [PubMed] [Google Scholar]
- 22. Brouwer RW, van den Hout MC, Grosveld FG, van Ijcken WF. NARWHAL, a primary analysis pipeline for NGS data. Bioinformatics 2012; 28:284-5; PMID:22072383; http://dx.doi.org/ 10.1093/bioinfor-matics/btr613 [DOI] [PubMed] [Google Scholar]
- 23. Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic acids research 2006; 34:D140-4; PMID:16381832; http://dx.doi.org/ 10.1093/nar/gkj112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li J, Tibshirani R. Finding consistent patterns: a nonparametric approach for identifying differential expression in RNA-Seq data. Statistic Methods Med Res 2013; 22:519-36; PMID:22127579; http://dx.doi.org/ 10.1177/0962280211428386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010; 11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010; 26:139-40; PMID:19910308; http://dx.doi.org/ 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Auer PL, Doerge RW. A Two-Stage Poisson Model for Testing RNA-Seq Data. Stat Appl Genet Mol Biol 2011; 10 [Google Scholar]
- 28. Kapranov P, Laurent G, St, Raz T, Ozsolak F, Reynolds CP, Sorensen PH, Reaman G, Milos P, Arceci RJ, Thompson JF, et al. The majority of total nuclear-encoded non-ribosomal RNA in a human cell is 'dark matter' un-annotated RNA. BMC biology 2010; 8:149; PMID:21176148; http://dx.doi.org/ 10.1186/1741-7007-8-149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gong M, Chen Y, Senturia R, Ulgherait M, Faller M, Guo F. Caspases cleave and inhibit the microRNA processing protein DiGeorge Critical Region 8. Protein Sci 2012; 21:797-808; PMID:22434730; http://dx.doi.org/ 10.1002/pro.2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ghodgaonkar MM, Shah RG, Kandan-Kulangara F, Affar EB, Qi HH, Wiemer E, Shah GM. Abrogation of DNA vector-based RNAi during apoptosis in mammalian cells due to caspase-mediated cleavage and inactivation of Dicer-1. Cell Death Differ 2009; 16:858-68; PMID:19229243; http://dx.doi.org/ 10.1038/cdd.2009.15 [DOI] [PubMed] [Google Scholar]
- 31. Neilsen CT, Goodall GJ, Bracken CP. IsomiRs–the overlooked repertoire in the dynamic microRNAome. Trends Genet 2012; 28:544-9; PMID:22883467; http://dx.doi.org/ 10.1016/j.tig.2012.07.005 [DOI] [PubMed] [Google Scholar]
- 32. Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007; 129:1401-14; PMID:17604727; http://dx.doi.org/ 10.1016/j.cell.2007.04.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Islam S, Kjallquist U, Moliner A, Zajac P, Fan JB, Lonnerberg P, Linnarsson S. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res 2011; 21:1160-7; PMID:21543516; http://dx.doi.org/ 10.1101/gr.110882.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Calabrese JM, Seila AC, Yeo GW, Sharp PA. RNA sequence analysis defines Dicer's role in mouse embryonic stem cells. Proc Natl Acad Sci U S A 2007; 104:18097-102; PMID:17989215; http://dx.doi.org/ 10.1073/pnas.0709193104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sasagawa Y, Nikaido I, Hayashi T, Danno H, Uno KD, Imai T, Ueda HR. Quartz-Seq: a highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol 2013; 14:R31; PMID:23594475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hesselberth JR. Lives that introns lead after splicing. Wiley Interdisciplinary Rev RNA 2013; 4:677-91; PMID:23881603 [DOI] [PubMed] [Google Scholar]
- 37. Yin QF, Yang L, Zhang Y, Xiang JF, Wu YW, Carmichael GG, Chen LL. Long noncoding RNAs with snoRNA ends. Mol Cell 2012; 48:219-30; PMID:22959273; http://dx.doi.org/ 10.1016/j.molcel.2012.07.033 [DOI] [PubMed] [Google Scholar]
- 38. Livyatan I, Harikumar A, Nissim-Rafinia M, Duttagupta R, Gingeras TR, Meshorer E. Non-polyadenylated transcription in embryonic stem cells reveals novel non-coding RNA related to pluripotency and differentiation. Nucl Acids Res 2013; PMID:23630323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chorev M, Carmel L. Computational identification of functional introns: high positional conservation of introns that harbor RNA genes. Nucl Acids Res 2013; PMID:23605046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Eswaran J, Horvath A, Godbole S, Reddy SD, Mudvari P, Ohshiro K, Cyanam D, Nair S, Fuqua SA, Polyak K, et al. RNA sequencing of cancer reveals novel splicing alterations. Sci Rep 2013; 3:1689; PMID:23604310; http://dx.doi.org/ 10.1038/srep01689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet 2007; 39:673-7; PMID:17401365; http://dx.doi.org/ 10.1038/ng2003 [DOI] [PubMed] [Google Scholar]
- 42. Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010; 466:835-40; PMID:20703300; http://dx.doi.org/ 10.1038/nature09267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004; 5:R80; PMID:15461798; http://dx.doi.org/ 10.1186/gb-2004-5-10-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004; 3:Article3; PMID:16646809 [DOI] [PubMed] [Google Scholar]
- 45. Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings Bioinformat 2013; 14:178-92; PMID:22517427; http://dx.doi.org/ 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.