

Abstract
Generation of a strong electrical potential in the cochlea is uniquely mammalian and may reflect recent evolutionary advances in cellular voltage-dependent amplifiers. This endocochlear potential is hypothesized to dramatically improve hearing sensitivity, a concept that is difficult to explore experimentally, because manipulating cochlear function frequently causes rapid degenerative changes early in development. Here, we examine the deafness phenotype in adult Claudin 11-null mice, which lack the basal cell tight junctions that give rise to the intrastrial compartment and find little evidence of cochlear pathology. Potassium ion recycling is normal in these mutants, but endocochlear potentials were below 30 mV and hearing thresholds were elevated 50 dB sound pressure level across the frequency spectrum. Together, these data demonstrate the central importance of basal cell tight junctions in the stria vascularis and directly verify the two-cell hypothesis for generation of endocochlear potential. Furthermore, these data indicate that endocochlear potential is an essential component of the power source for the mammalian cochlear amplifier.
Keywords: evoked potentials, targeted deletion, oligodendrocyte-specific protein, β-galactosidase, freeze fracture, homologous recombination
Introduction
Advances in the identification of deafness genes have provided extraordinary insight into, and have shaped debate about, the cellular and molecular machinery underlying auditory function (Petit, 1996; Steel and Kros, 2001; Ahituv and Avraham, 2002). Although consensus has not been achieved, evidence is converging for key functions of this machinery, which serves to: (1) maintain a network of transcellular conduits that constantly provide K+ to the stria vascularis (SV), (2) generate and maintain high extracellular K+ concentrations in endolymph (120-150 mm), (3) generate and maintain strong endocochlear potentials (EPs; 80-100 mV), and (4) maximize detection of low intensity sounds by amplifying small deflections of stereocilia bundles (Salt et al., 1987; Kikuchi et al., 2000; Marcus et al., 2002; Wangemann, 2002).
Several of these functions were affected within the SV, which is a secretory epithelium that maintains K+ homeostasis in the endolymph (Wangemann, 2002). The tripartite structure of this epithelium comprises polarized marginal cells covering the lateral surface of the cochlear duct, polarized basal cells juxtaposed to fibrocytes in the underlying spiral ligament, and neural crest-derived intermediate cells within the intrastrial space. Electrogenic machinery that generates EP is localized in the SV, and recent evidence suggests that intermediate cells are the predominant cellular component. However, marginal and basal cells are also major components because of the tight junctions (TJs) they elaborate.
Basal and marginal cell TJs preclude paracellular diffusion into and out of the intrastrial space and isolate this compartment. Concomitantly, basal and intermediate cells secrete into this space K+ derived from fibrocytes through gap junctions (Kikuchi et al., 2000; Takeuchi et al., 2000; Marcus et al., 2002). Marginal cell Na+/K+ pumps, Na+/Cl-/K+ cotransporters, and ion channels efficiently transfer this K+ into the endolymph, and close appositions between marginal, intermediate, and basal cells are key. A model proposed by Salt et al. (1987) to describe this process, the two-cell hypothesis, posits that marginal and basal cell layers are necessary for generating EP and, in particular, that basal cell TJs are an essential component of the electrogenic machinery (Salt et al., 1987). However, this notion has not been verified experimentally in vivo.
The apparent importance of basal cell TJs in the generation of EP prompted us to examine mutant mice that cannot generate this paracellular barrier. From auditory brainstem responses (ABRs), otoacoustic emissions, and other physiological measures, we demonstrate that adult Claudin 11-null mice exhibit severe deafness associated with low EP. In the absence of degenerative changes in the cochlea, our data directly establish the importance of basal cell TJs and validate the two-cell hypothesis (Salt et al., 1987). Importantly, inner hair cells from Claudin 11-null mice are functional; thus, elevated hearing thresholds in these animals between 47 and 51 dB sound pressure level (SPL) may represent a loss of gain in the cochlear amplifier (Davis, 1983; Dallos and Evans, 1995). Indeed, similar losses in amplifier gain occur after outer hair cell (OHC) degeneration and, in the absence of the OHC motor protein, prestin (Ryan and Dallos, 1975; Liberman et al., 2002; Liu et al., 2003).
Materials and Methods
Genotyping Claudin 11-null mice. Disruption of the Claudin 11 gene was generated previously by replacing the coding region in exon 1 and the 5′-end of intron 1 with the lacZ coding region (Gow et al., 1999). Genotypes of Claudin 11-null mice and littermates are determined by multiplex PCR from phenol-purified tail DNA using primers and amplification protocols.
Cytochemistry. Primary antibodies to claudin 1, occludin, and zonaoccludens-1 (ZO-1) were obtained from Zymed (San Francisco, CA). The monoclonal antibody, 37E3, raised against claudin 11 was generated by the Hybridoma Shared Research Facility (Mount Sinai School of Medicine, New York, NY). Secondary antibodies were obtained from Southern Biotech (Bermingham, AL) and Sigma (St. Louis, MO). Secondary-only controls were included in all Western blotting and tissue staining experiments to verify the specificity of primary antibodies. In addition, the 37E3 monoclonal does not label brain or inner ear sections from Claudin 11-null mice (data not shown).
To detect β-galactosidase expression, inner ears were dissected into PBS, pH 7.4, and fixed in 2% paraformaldehyde, 2 mm MgCl2, 1.25 mm EGTA, 0.1 m PIPES, pH 6.9, for 2 hr before processing (Gow et al., 1992). To render the bony labyrinth transparent, inner ears were incubated in benzyl benzoate:benzyl alcohol (1:1). For immunocytochemistry, inner ears were dissected from mice perfused with 4% paraformaldehyde in 0.1 m sodium phosphate buffer, pH 7.4. After decalcification in 0.5 M ETDA for 12-96 hr, inner ears were dehydrated through alcohol over 24 hr and embedded in paraffin. Sections (7 μm) were dewaxed in xylene, rehydrated through alcohol into PBS, treated for antigen recovery (target unmasking fluid; Zymed), and labeled with antibodies (Gow et al., 1998). For whole-mount immunocytochemistry, temporal bones from CO2-anesthetized rats were dissected into modified Leibowitz medium (Invitrogen, Rockville, MD) and perfused through the round and oval windows with 4% paraformaldehyde, PBS, pH 7.4, for 2 hr. The stria vascularis was dissected, permeabilized in 0.5% Triton X-100, PBS, pH 7.4, for 30 min, and incubated overnight in 5% goat serum, 2% BSA, PBS, pH 7.4. Samples were labeled with antibodies for 90 min and mounted for confocal microscopy (LSM510; Zeiss, Oberkochen, Germany). For phalloidin labeling, 4% paraformaldehyde-perfused inner ears were dissected, and organs of Corti incubated in 1 μg/ml phalloidin-fluorescein conjugate (Rzadzinska et al., 2004). For immunoelectron microscopy, we used a postembedding protocol (Matsubara et al., 1996). Rat cochleas were dissected and perfused with 4% paraformaldehyde, 0.5% glutaraldehyde, PBS, pH 7.4, for 2 hr, cryoprotected in glycerol, and stria vascularis dissected and frozen in liquid propane. Frozen sections were immersed in 1.5% uranyl acetate and methanol at -90°C for freeze substitution and then infiltrated with Lowicryl HM20 resin at -45°C and polymerized with ultraviolet light. Thin sections were incubated in 0.1% sodium-borohydrate, 50 mm glycine, TBS, 0.1% Triton X-100 (TBST), followed by 10% goat serum, TBST, primary antibody in 1% goat serum, TBST, immunogold (Amersham Biosciences, Arlington Heights, IL), 1% NGS, and 0.5% polyethylene glycol in TBST. Finally, sections were stained with 1% uranyl acetate for microscopy. For freeze fracture microscopy, mice were perfused with 4% glutaraldehyde in 0.15 m cacodylate buffer, pH 7.2, and the stria vascularis was dissected and processed (Gow et al., 1999).
Horseradish peroxidase tracer experiments in cochlea. Mice were anesthetized with carbon dioxide, and temporal bones were dissected into Leibowitz medium. Cochleas were perfused through the round and oval windows with 1% horseradish peroxidase (HRP) (grade VI; Sigma) in Leibowitz for 5 min at 4°C and then flushed for 1 min and fixed with 2.5% paraformaldehyde, 2% glutaraldehyde, HEPES, pH 7.4, for 2 hr. After an overnight wash with buffer, cochleas were incubated in DAB (SK-4100 kit; Vector Laboratories, Burlingame, CA), postfixed in 1% osmium tetroxide for 1 hr, and dehydrated in an ethanol series for embedding in Epon 812. Thick sections were stained with toluidine blue for light microscopy.
Auditory brainstem responses, distortion-product otoacoustic emissions, EPs, and K+ measurements. The ABR protocol was modified from the study by Zheng et al. (1999). Mice were anesthetized with fresh 2,2,2-tribromoethanol (375 μg/gm; Sigma) in PBS and placed on a 39°C pad. Subdermal electrodes were placed lateroventrally to the outer meatus of each ear, at the vertex (reference), and at the midline over the cervical region (ground). Mice were presented with 1024 pure-tone stimuli (102 μsec; Blackman envelope) at stimulus rates of 19.1/sec, although 9.1 and 29.1/sec yield indistinguishable data. The left and right ears were tested alternately, and ipsilateral EEGs (amplified × 200 K) were bandpass filtered (300-3000 Hz), parsed with 15 μV artifact rejection, and recorded for 12.5 msec (Intelligent Hearing Systems, Miami, FL). Distortion-product otoacoustic emissions (DPOAEs) between 3 and 16 kHz were averaged from eight blocks of eight sweeps using an ER-10C probe (Etymotic Research, Elk Grove Village, IL) at eight intervals per octave, f2:f1 = 1.2, and L1 = L2 = 60 dB SPL controlled with DP2000 software (Starkey Laboratories, Eden Prairie, MN).
To measure EPs and K+ in perilymph and endolymph, inactin (140 mg/kg, i.p.) was used to induce deep anesthesia for an extended period, and succinylcholine chloride (20-40 mg/kg) was used for artificially respiring the animal in the absence of muscle movements. Mice were ventilated with a Harvard Apparatus MiniVent Type 845 (Harvard Apparatus, Holliston, MA) ventilator. Measurements were made in the second cochlear turn with calibrated, double-barreled microelectrodes (Marcus et al., 2002) using the K+-selective ion exchanger Fluka 60398, K+ ionophore I-Cocktail B (Fluka, Buchs, Switzerland).
Northern and Western blotting. For Northern blotting, inner ears were dissected into PBS, and extraneous tissue was removed. Semicircular canals were removed with a scalpel, cut at the oval window, and RNA from pooled cochleas (∼7) was purified over CsCl (Southwood et al., 2002). For Western blotting, whole mouse brains or single cochleas (prepared as above) are homogenized in 1× SDS-PAGE buffer in an Ependorf tube (Eppendorf Scientific, Westbury, NY), centrifuged at 14,000 × g, and run on 12% Laemmli gels (Laemmli, 1970) and then electroblotted for 1 hr at 4°C, as recommended by the manufacturer (Hoefer Scientific, San Francisco, CA).
Results
Members of the claudin family of transmembrane proteins are likely to be the dominant structural component comprising the physical paracellular barrier in most epithelial TJ networks. Importantly, individual family members give rise to intramembranous fibrils in the plasmalemma with distinct ultrastructural morphologies and electrophysiological properties. Indeed, we have demonstrated that the subset of parallel stranded, rarely anastomosing fibrils characteristic of CNS myelin sheaths and Sertoli cells in the testis are comprised entirely of claudin 11 (Gow et al., 1999). Here, we demonstrate that the same holds true for intramembranous fibrils between basal cells of the stria vascularis, and we characterize the pathophysiology associated with the loss of these junctions in Claudin 11-null mice.
Claudin 11 is expressed by basal cells of the stria vascularis
Intramembranous fibrils with similar morphology to CNS myelin and Sertoli cell TJs are elaborated by a number of cell types, including epithelial cells in the lateral wall of the cochlear duct (Luciano et al., 1995). This observation, together with our previous demonstration that Claudin 11 is expressed in the vestibulocochlear apparatus of mouse embryos (Gow et al., 1999), prompted us to examine expression of this gene in the inner ear of postnatal mice. To establish the pattern of Claudin 11 expression, we harvested inner ears from wild-type and Claudin 11-null mice at several ages for 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) histochemistry (Gow et al., 1999). Figure 1Aa demonstrates that β-galactosidase is expressed in Claudin 11-null mice in epithelial cells lining the vestibulocochlear apparatus, including the semicircular canals, ampulas, utricle and saccule, suprastrial zone, lateral wall of the cochlear duct, and CNS myelin in the eighth cranial nerve. Cross sections through the cochlea of X-gal-stained inner ears show that β-galactosidase expression is localized primarily to the basal cell layer of the strial vascularis (Southwood and Gow, 2001). Finally, X-gal reaction product surrounding blood vessels in the stria vascularis (Fig. 1, inset, arrow) indicates that claudin 11 TJs occlude vasculature exit points from this epithelium.
Figure 1.

Localization of TJ proteins in the inner ear of rodents. A, Claudin 11 gene expression is widespread in mouse inner ear. a, X-gal histochemistry reveals expression of lacZ regulated by the endogenous Claudin 11 promoter. 1, Semicircular canals; 2, ampulla; 3, utricle and saccule; 4, suprastrial zone; 5, lateral wall of the cochlear duct; 6, CNS myelin in the eighth cranial nerve. Inset, Strial capillaries (arrow) express Claudin 11. b, Paraffin section of wild-type cochlea labeled with anti-claudin 11 antibodies (37E3) shows expression in the stria vascularis. Inset, Antibodies label the basal cell layer (arrows) but not the marginal cell layer (arrowheads). c, Secondary antibodies do not label a section adjacent to that in b. B, Specificity of anti-claudin 11 antibodies. Antibodies used in Ab label a 24 kDa band on a Western blot of whole brain from wild-type (+/+, lane 1) but not Claudin 11-null (-/-, lane 2) mice. Secondary antibodies do not label the Western blot (+/+, lane 3). Mr, relative mobility (in kilodaltons). C, Claudin 11 expression in rat stria vascularis. The schematic shows intimate relationships between different cells in the stria vascularis. a-h, Confocal micrographs reveal labeling of strial marginal cells (a-d) and basal cells (e-h) using antibodies against TJ proteins claudins 11 and 1, occludin, and ZO-1. D, Immunoelectron micrographs of TJ proteins in basal cells. Colocalization of claudin 11 and occludin (a) ZO-1 and occludin (b) but not claudin 1 (c) at membrane “kiss” sites in basal cell TJs is shown. Cldn 11, claudin 11; Occ, occludin; Cldn 1, claudin 1.
Labeling sections of cochlea (Fig. 1Ab) and semicircular canals (data not shown) from wild-type mice with a monoclonal antibody (37E3) raised against the unique carboxyl terminus of mouse claudin 11 demonstrates that X-gal histochemistry in Claudin 11-null mice accurately reflects expression of Claudin 11 throughout the inner ear. Claudin 11 is strongly expressed in the lateral wall of the cochlear duct and is localized to the basal cell layer of the stria vascularis (inset, arrows), whereas the marginal cell layer is unstained (arrowheads). Labeling sections with secondary antibody alone does not yield significant staining (Fig. 1Ac), and the specificity of 37E3 for claudin 11 is demonstrated on Western blots (Fig. 1B) where a single band at 24 kDa is detected in brain homogenates from wild-type (lane 1) but not Claudin 11-null mice (lane 2) or wild-type brain homogenate incubated only with secondary antibody (lane 3).
Claudins 1 and 11 are incorporated into canonical TJs in the stria vascularis
The schematic in Figure 1C depicts salient features of the trilaminar structure of the stria vascularis (adapted from Krstic, 1991) including the simple polarized marginal cell epithelium (Fig. 1C, top), the intermediate cell layer (central), and the stratified polarized basal cell epithelium (bottom). Tight junction networks are represented as thick dashes. Cells shaded in gray reflect the close association between basolateral infoldings of marginal cells and the apical processes of basal cells. Intermediate cell processes also interdigitate with marginal cell infoldings. A major purpose of such close cell appositions in the stria vascularis is likely to be facilitated ion transfer, particularly K+, between these cell types.
Whole-mount confocal microscopy of dissected rat stria vascularis labeled with antibodies raised against several TJ proteins shows that marginal cell TJs contain claudin 1 (Fig. 1Cb), occludin (Fig. 1Cc), and ZO-1 (Fig. 1Cd). Viewed en face, the “chicken wire” staining pattern of these proteins is typical of TJ networks in most simple polarized epithelia. Note that marginal cells do not express claudin 11 (Fig. 1Ca). Unlike the archetypal single-cell layer organization of simple polarized epithelia, the lateral edges of basal cells overlap extensively, and TJs localize to the juxtaposed surfaces of these cells (Fig. 1C, schematic). Viewed en face, claudin 11 staining of the basal cell epithelium does not delineate individual cells but rather broad regions of TJ strands (Fig. 1Ce). Nonetheless, occludin and ZO-1 are also expressed by basal cells, and labeling with antibodies is similar to that of claudin 11 (Fig. 1Cg,Ch). Figure 1Cf demonstrates that claudin 1 is not expressed by basal cells. Together, these data indicate that claudin 11, occludin, and ZO-1 are likely colocalized to basal cell TJs.
To confirm the confocal data in Figure 1Ce-Ch, we examined the ultrastructure of basal cell TJs in adult mice and rats. Double immunoelectron microscopy labeling of basal cells (Fig. 1D) using antibodies against claudin 11 (10 nm gold particles) (Fig. 1Da) and occludin (5 nm particles) demonstrates the colocalization of these proteins at cell membranes (double arrows) in TJ strands (arrowheads). Similarly, ZO-1 (10 nm particles) and occludin (5 nm particles) are colocalized in Figure 1Db. In contrast, anti-claudin 1 antibodies do not label basal cell TJs (Fig. 1Dc).
Absence of basal cell TJs in Claudin 11-null mice
Studies in several mammalian species suggest that basal cell TJs are important for the generation and maintenance of EPs between 80 and 100 mV (Salt et al., 1987; Wangemann, 2002). In light of the claudin 11 expression data in Figure 1 and the ultrastructural abnormalities in brain and testis of Claudin 11-null mice (Gow et al., 1999), where claudin 11 TJs in CNS myelin and between Sertoli cells are completely absent, we examined basal cell intramembranous fibrils by freeze-fracture electron microscopy.
The replica in Figure 2a from a 3-month-old wild-type mouse shows the typical morphology and organization of intramembranous fibrils in basal cell membranes (white arrowheads). These parallel, rarely anastomosing TJ strands are reminiscent of CNS myelin and Sertoli cell TJs, consistent with the notion that TJs of similar morphology in different tissues are likely comprised of the same claudin family members (Gow et al., 1999; Southwood and Gow, 2001). A striking example of linear gap junction arrays (Fig. 2a, black arrowheads) interposed between the TJ strands is also shown in Figure 2a. Such intercalation is commonplace in strial basal cell membranes from several species (Souter and Forge, 1998) as well as in Sertoli cell junctions (B. Kachar, unpublished observation) but not in other epithelia.
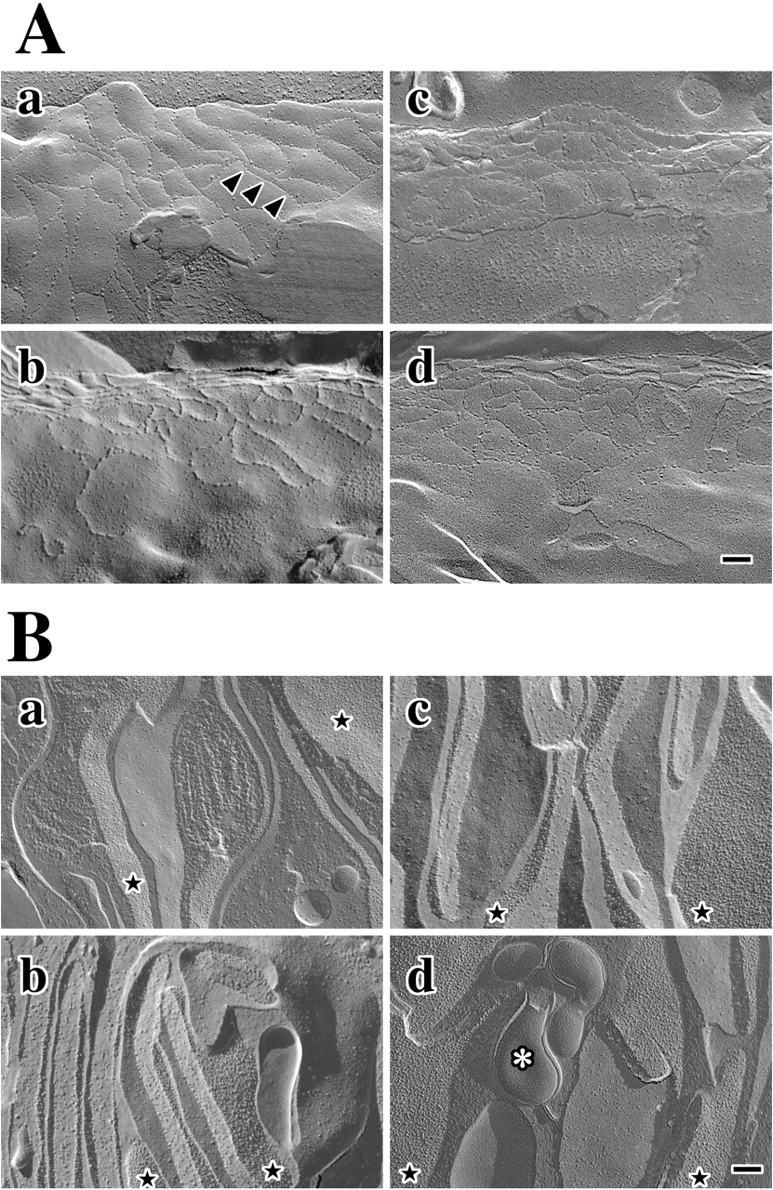
Figure 2.

Freeze fracture showing intercellular junctions in wild-type and Claudin 11-null mice. a, Parallel intramembranous fibrils of claudin 11 TJs (white arrowheads) separate small gap junction domains (black arrowheads) in wild-type P 90 mice. b, Tight junction strands are absent in P90 Claudin 11-null mice, and gap junctions form large domains with defined borders. c, Tight and gap junctions are well developed in P30 wild-type mice and are similar to adults. d, Tight junction domains are absent in null mutant mice but gap junctions are present. Scale bar, 0.15 μm.
Freeze-fracture micrographs from 3-month-old Claudin 11- null mice reveal two prominent abnormalities in strial basal cells (Fig. 2b). First, intramembranous fibrils are completely absent, which indicates that other claudin family members do not normally copolymerize with claudin 11 or compensate for its absence in the mutants. Second, extensive gap junction domains with well defined perimeters are prominent features (Fig. 2b, black arrowheads). Importantly, the presence of these domains demonstrates that plasma membranes of adjacent basal cells are closely apposed, which indicates that the overall organization of the epithelium is essentially unperturbed by the absence of TJ strands. Nevertheless, the spatial organization of the gap junctions in the mutants is not regulated by the basal cell cytoplasmic TJ plaque, thereby demonstrating that intramembranous fibrils in this epithelium normally impede gap junction coalescence in the plane of the bilayer.
Intermingling of gap junctions and TJs is also apparent in basal cell membranes from young mice shortly after the onset of auditory function (Fig. 2c). The similarity of this morphology with adults indicates that, once established, the relationship between these intercellular junctions is stable into adulthood. In a similar manner to adult Claudin 11-null mice, TJ strands in young mutants are absent, and gap junctions form large domains in closely apposed basal cell membranes (Fig. 2d). Again, the lack of intramembranous strands indicates that compensation by other claudin family members does not occur, and it is likely that the paracellular space between basal cells remains open throughout the life of these mutants.
Absence of basal cell paracellular diffusion barrier in Claudin 11-null mice
In view of the close apposition of basal cell membranes in the mutant mice, we sought to determine whether paracellular diffusion of macromolecules into the stria vascularis of these animals is restricted in the absence of claudin 11. Accordingly, we injected the tracer protein HRP into the oval and round windows of cochleas dissected from Claudin 11-null mice. In 1 μm plastic sections of the lateral wall of the cochleas from wild-type and Claudin 11-null mice, the triple-layered epithelial organization of the stria vascularis is easily distinguished from the underlying fibrocytes of the spiral ligament (Fig. 3a,b, see below the double arrowheads). Within the stria vascularis, marginal and basal cell layers are denoted by single and double arrowheads, respectively. Intermediate cells (i) are apparent between these epithelial layers along with capillaries (Fig. 3, asterisks); thus, the organization and dominant cell types of the stria vascularis are essentially normal in the mutants. The intrastrial compartment in these animals appears to be more loosely structured than controls and may reflect changes in ion composition of the intrastrial space stemming from infusion of perilymph from surrounding regions. Alternatively, this feature may signify a processing artifact because it is not a consistent feature of all strial preparations from the mutants (see below). The cross-sectional thickness of the stria vascularis in the second turn of cochleas from postnatal day 90 (P90) null animals (25.0 ± 1.6 μm; n = 4 mice) is similar to controls (24.6 ± 0.6 μm; n = 4 mice), which indicates that the absence of basal cell TJs does not cause significant cell loss.
Figure 3.
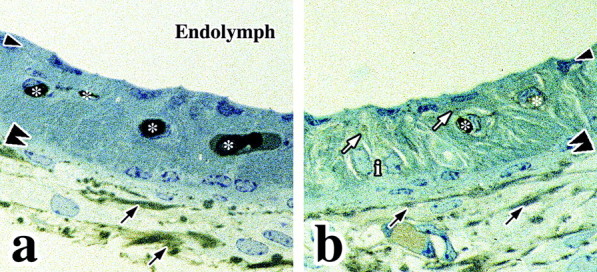
HRP infiltration into the stria vascular is of Claudin 11-null mice. a, HRP is present in the spiral ligament but does not penetrate into the intrastrial space of wild-type cochlea. b, HRP is present in the spiral ligament and stria vascularis from null mutants. Arrowheads, Marginal cell layer; double arrowheads, basal cell layer; black arrows, exogenous HRP tracer activity in the spiral ligament; white arrows, exogenous HRP tracer activity in the stria vascularis; asterisks, endogenous HRP activity in blood vessels; i, intermediate cell nuclei.
The extent of diffusion of HRP from the cochlear duct into the lateral wall tissue is determined histochemically by relying on the enzymatic activity of HRP to convert diaminobenzidine into its insoluble brown product. In wild-type cochleas, HRP is evident between fibrocytes in the spiral ligament (Fig. 3a, arrows). Paracellular barriers between marginal and basal cells of these animals are intact, and HRP does not reach the intermediate cell layer. Note that the HRP activity observed in the vasculature of the stria vascularis (Fig. 3, asterisks) reflects endogenous peroxidase activity in red blood cells.
HRP activity is detected between fibrocytes in the spiral ligament of Claudin 11-null mutants in a similar manner to controls (Fig. 3b, black arrows). In addition, HRP is evident between marginal cell infoldings throughout the intermediate compartment of the stria vascularis (Fig. 3b, white arrows). Because marginal cell TJs are intact in the null mutants (see below), HRP must enter the stria vascularis through paracellular spaces between basal cells in the absence of claudin 11 TJs. Thus, we conclude that the close apposition of basal cells in Claudin 11-null mice (Fig. 2) cannot supplant the diffusion barrier function of TJs and that the paracellular space of this epithelium is open.
Hearing loss in Claudin 11-null mice
To identify physiological consequences stemming from the absence of basal cell TJs, we measured ABRs in adult Claudin 11- null mice to determine sound level thresholds for hearing. In wild-type mice, auditory response thresholds using click and pure-tone stimuli at 8, 16, and 32 kHz are between 15 and 40 dB SPL (Fig. 4A, +/+). These thresholds are invariant between 1 and 6 months of age (data not shown). Furthermore, ABR thresholds measured in heterozygous mice (+/-) are similar to controls at all ages examined. In contrast, ABRs from Claudin 11-null mice (-/-) are increased dramatically between 60 and 85 dB SPL and indicate severe hearing impairment in these animals. Together, these data indicate that thresholds are elevated between 46.6 and 50.5 dB SPL across the frequency spectrum and demonstrate that deafness is not associated with a specific region of the cochlear duct but, rather, is manifested along its entire length. These data accord with expectations when considering the expression of claudin 11 along the stria vascularis.
Figure 4.
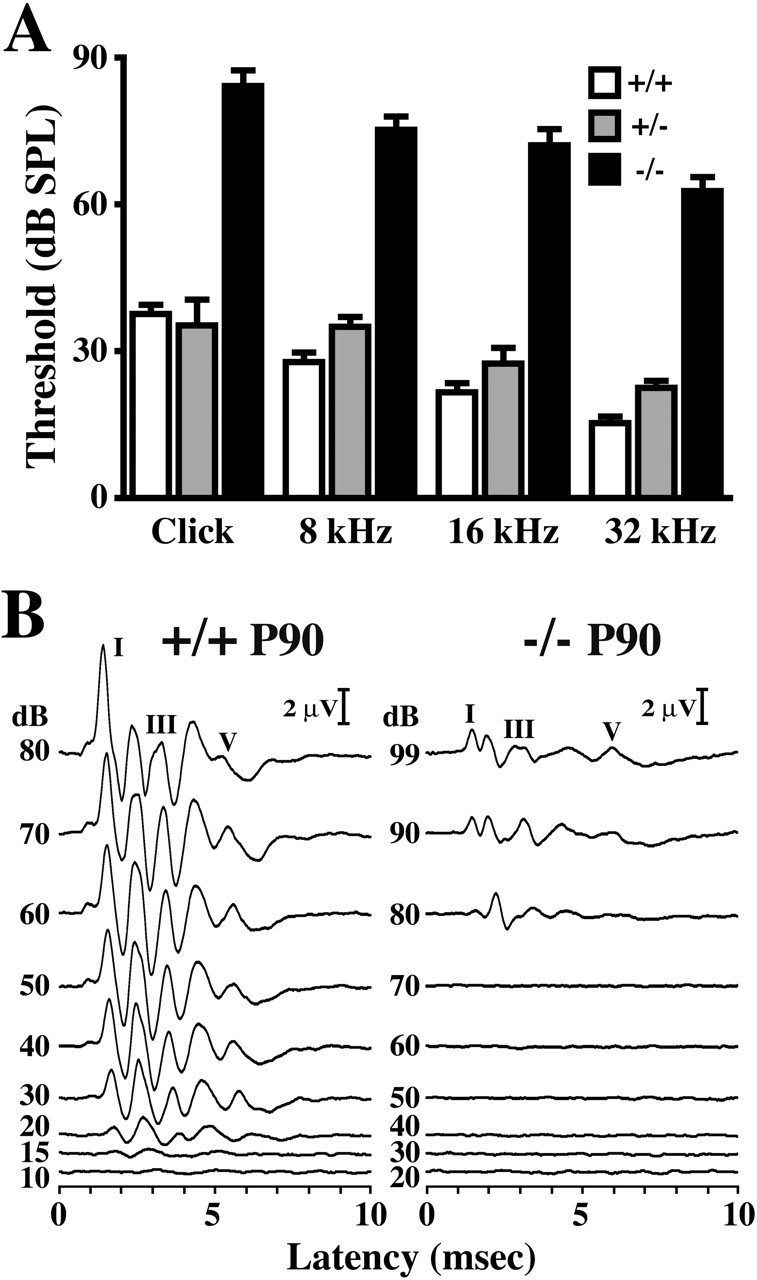
Auditory brainstem responses in Claudin 11-null mice. A, Hearing thresholds for adult wild-type, heterozygous, and null mutant mice using click, 8, 16, and 32 kHz tone pips. B, Auditory brainstem responses from P90 wild-type (+/+) and null (-/-) mice for 32 kHz stimuli at different intensities (99-10 dB SPL). The major ABR Peak I, III, and V for each animal are labeled.
A detailed examination of ABRs from 32 kHz pure-tone stimuli is shown for adult wild-type and Claudin 11-null mice in Figure 4B. For wild-type mice (Fig. 4B, left panel), five well-defined maxima are observed between 1 and 6 msec after stimulus. The major Peaks, I, III, and V, are labeled. Recordings from lower intensity stimuli also exhibit five peaks down to 20 dB SPL, although amplitudes progressively decrease. At 15 dB SPL, ABR waveforms are barely distinguishable from baseline, which signifies the threshold of hearing for this animal. Auditory brainstem responses from adult Claudin 11-null mice (Fig. 4B, right panel) are similar to littermate controls in terms of peak resolution (five peaks) and latencies for Peaks I-III at 80 dB SPL. Indeed, average latencies for these peaks from null mutant mice (n = 5) are 104, 107, and 106%, respectively, compared with controls (n = 4). Nonetheless, important differences are evident. Most notably, the threshold of hearing is increased dramatically (∼80 dB SPL) and peak amplitudes are decreased several-fold. Furthermore, the latency for Peak V from the mutant is longer than that from the control; however, this latency change likely reflects differences in CNS rather than cochlear dysfunction.
A cochlear defect accounts for hearing loss in Claudin 11-null mice
Although ABR measurements are convenient for large-scale screening to reveal auditory impairment, this technique frequently fails to shed light on causes of dysfunction that may range from structural defects in the cochlear duct to disruption of sensory input to the brainstem. Thus, to further characterize the hearing defect in Claudin 11-null mice, we measured cubic DPOAE, which originates in the cochlear duct after auditory stimuli at different frequencies.
Figure 5A shows DPOAEs as a function of f2 stimulus frequencies between 3 and 16 kHz for wild-type mice and Claudin 11-null mice between P80 and P90. Wild-type mice (Fig. 5A, open symbols) are relatively insensitive to stimuli below ∼4 kHz, but DPOAEs rise sharply above this frequency and remain markedly above the noise floor up to 16 kHz. The DPOAEs from adult mutant mice exhibit considerable deficiencies compared with controls with an attenuation >30 dB near f2 = 14 kHz. These data indicate that hearing loss in Claudin 11-null mice stems from a global defect in the cochlear duct that is most likely attributable to the absence of basal cell TJs.
Figure 5.
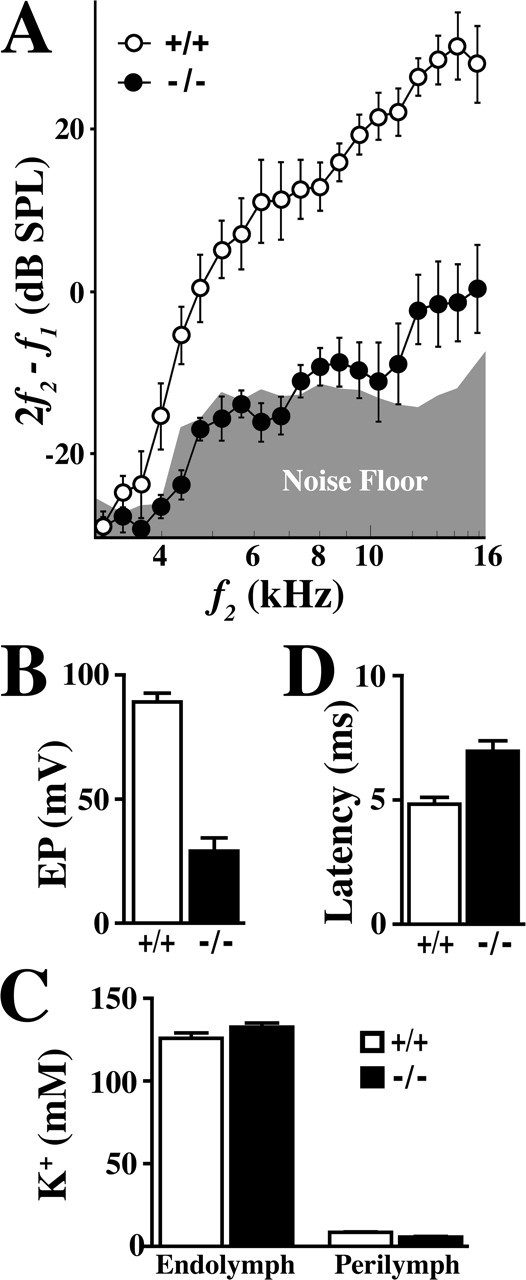
DPOAE, EP, and ABR latency measurements in Claudin 11-null mice. A, DPOAEs from adult wild-type (+/+; open symbols) and Claudin 11-null mice (-/-; closed symbols). The average noise floor is represented by the gray region along the abscissa. B, Endocochlear potentials from adult wild-type (+/+) and mutant (-/-) mice. C, K+ concentrations in endolymph and perilymph from adult wild-type (+/+) and mutant (-/-) mice. D, Latencies of ABR Peak V from adult wild-type (+/+) and mutant (-/-) mice.
Low EPs in Claudin 11-null mice
Electrophysiological studies in several rodent species treated with ototoxic drugs or exhibiting age- and noise-induced hearing loss have led to a commonly held view that EP plays a critical role in maintaining normal hearing sensitivity. In view of our structural and functional data obtained from Claudin 11-null mice (Figs. 2, 3, 4, 5A), we measured EPs in adult mutants and wild-type controls in a blinded study with respect to genotype. Endocochlear potentials measured in adult wild-type mice are ∼89 mV (n = 16) and are similar to published values (Fig. 5B). In contrast, EPs in Claudin 11-null mice are ∼29 mV (n = 15), which directly demonstrates the importance of basal cell TJs to the electrogenic machinery for establishing a high electrical potential in the scala media.
Considering the magnitude of the total electrical potential across mechanotransducer channels in outer hair cell apical membranes, the 60 mV decrease in EP measured in the null mutants causes a surprisingly large change in ABR thresholds (Fig. 4). Total electrical potential is ∼160 mV in wild-type mice (90 mV derived from the EP and assuming a hair cell resting potential of -70 mV) and 100 mV in Claudin 11-null mice. Thus, reducing the potential in the mutants to 63% of that in controls causes a 50 dB SPL increase in hearing threshold, which represents more than a 250-fold decrease in hearing sensitivity. These data demonstrate an extremely nonlinear relationship between electrical driving force and hearing sensitivity.
Normal K+ levels in endolymph of Claudin 11-null mice
Because of the central involvement of K+ in maintaining osmolality of the endolymph, sustaining hair cell function, and carrying most of the charge to drive the cochlear amplifier, we determined the concentrations of this critical electrolyte in perilymph and endolymph of Claudin 11-null mice (Fig. 5C). Measurements in wild-type cochleas are consistent with recent published literature (Marcus et al., 2002) and demonstrate that K+ levels in perilymph and endolymph are 8.5 and 126 mm (n = 10), respectively. In cochleas from Claudin 11-null mice, K+ levels are indistinguishable from littermate controls (5.7 and 133 mm, respectively; n = 11). These data are important for several reasons. First, it is likely that the influx of K+ from fibrocytes into basal and intermediate cells is unperturbed in the mutants and that secretion of this ion through Kir4.1 channels into the intrastrial space and uptake by marginal cells is efficient. Second, secretion of K+ through Isk channels into endolymph is normal, which accounts for the normal size of scala media (Fig. 1Ab). Third, the likelihood of electrolyte imbalance in endolymph, which could cause hair cell dysfunction by loading these cells with Na+ or Ca2+, is minimal. Finally, our data are consistent with previous indications of a low extracellular K+ concentration in the intrastrial space (Salt et al., 1987; Takeuchi et al., 2000), because ion diffusion between basal cells in Claudin 11-null mice does not cause degenerative changes in strial cells. Thus, the absence of basal cell TJs appears to cause minimal disruption to ion homeostasis in the cochlea, despite dramatic declines in EP to one-third of control values.
Increased latency of evoked potentials in Claudin 11-null mice
In a previous study, we measured visual evoked potentials (VEPs) in Claudin 11-null mice and found that the absence of TJs in CNS myelin increases the latency of nerve impulses traveling from the retina to the visual cortex. These data are consistent with slowed conduction velocity along the optic tract, although reduced performance of nerve synapses cannot be formally ruled out (Gow et al., 1999). In the current study, ABRs from null mutants and controls are used to estimate the latency for nerve impulses traveling from the cochlear nuclei to other regions of the brain. In this regard, Peak V arises from nerve transmission between the brainstem and the inferior colliculus, and the latency of this peak affords an estimate of nerve conduction velocity in the afferent auditory tract (for review, see Parham et al., 2001). In wild-type mice, the average latency of Peak V is 4.9 ± 0.3 msec (Fig. 5C), and this value is invariant with age (data not shown). The average latency of Peak V in Claudin 11-null mice is 7.0 ± 0.4 msec, which represents a 40% increase over controls. These data are consistent with our previous evoked potential measurements along the optic tract (Gow et al., 1999), for which we estimate an increased latency of ∼120% in mutants compared with controls.
Hair cells and spiral ganglion neurons survive in Claudin 11-null mice
In light of DPOAE and EP data (Fig. 5) suggesting that hearing loss in Claudin 11-null mice is localized to the cochlea, we focused on several cell types in this tissue that might be adversely affected by the absence of basal cell TJs. First, we examined the morphology of outer and inner hair cells to determine whether the loss of these cells might account for increased ABR thresholds (Fig. 4). Inner ears isolated from P90 mutant mice were fixed and dissected to expose the organ of Corti at each cochlear turn. Phalloidin labeling of the basal turn from a wild-type organ of Corti reveals the actin cytoskeleton in stereocilia of outer and inner hair cells (Fig. 6Aa). Three rows of OHCs (Fig. 6Aa, top) and a single row of inner hair cells (Fig. 6Aa, bottom) are apparent. Equal spacing in each row indicates that there is little cell loss. Cell loss is not observed in the second or basal cochlear turns from these animals (data not shown).
Figure 6.
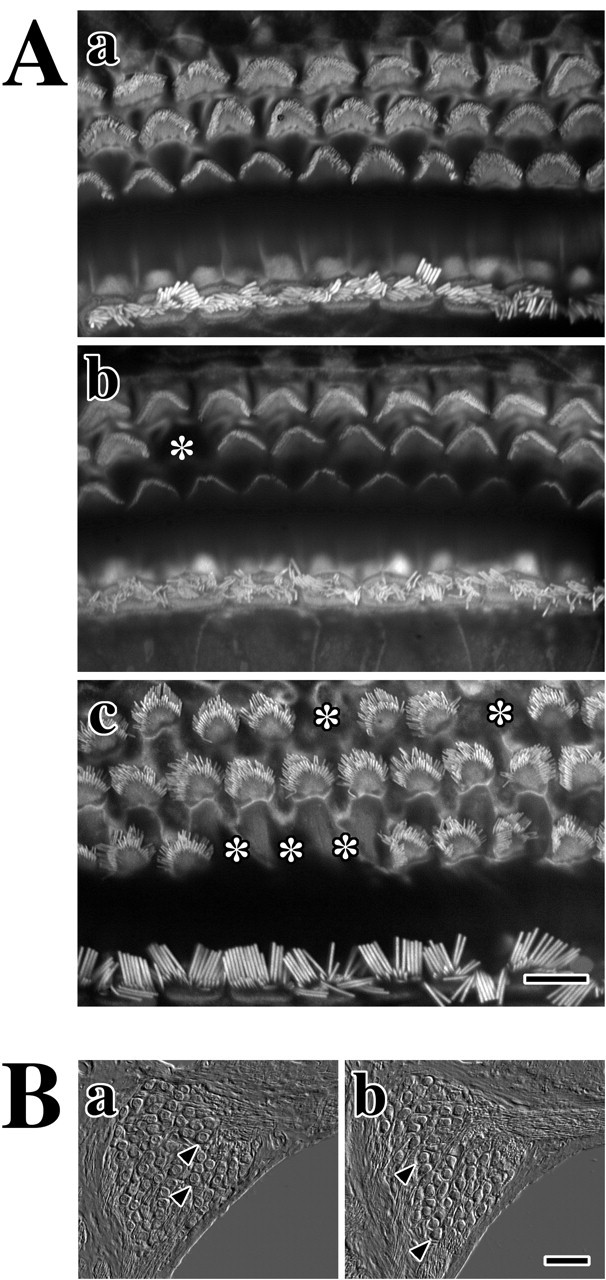
Normal morphologies of hair cells and spiral ganglion neurons in adult Claudin 11-null mice. A, Fluorescence microscopy from phalloidin-labeled hair cell stereocilia in organ of Corti from adult mice. a, Outer and inner hair cells from the basal cochlear turn of a P90 wild-type mouse. b, c, Outer and inner hair cells from the basal cochlear turn (b) and the apical turn (c) of a P90 Claudin 11-null mouse. Asterisks mark sites of individual OHC loss. Scale bar, 10 μm. B, Differential interference contrast microscopy showing cross sections of spiral ganglia in the midcochlear region reveal neuron cell bodies (arrowheads) from P90 wild-type (a) and Claudin 11-null (b) mice. Scale bar, 40 μm.
Outer and inner hair cells in basal and apical regions of cochleas from adult Claudin 11-null mice (Fig. 6Ab,Ac) exhibit similar overall organization and morphology to controls. Degenerative changes are apparent by the absence of individual OHCs in basal and apical regions from the mutants (Fig. 6, asterisks), and similar changes occur in the second turn. However, OHC loss is relatively low (<20% in two animals) and is unlikely to account for increased ABR thresholds observed in mutants (Fig. 4). Moreover, we do not observe inner hair cell loss in these mice. Thus, we conclude that the limited extent of hair cell loss in Claudin 11-null mice is not associated with the sensorineural deafness phenotype.
Excluding hair cell degeneration as a cause of deafness in Claudin 11-null mice, we examined the morphology of spiral ganglion cells for evidence of nerve cell loss. The micrograph in Figure 6Ba shows a cross section through the spiral ganglion at the second cochlear turn from an adult wild-type mouse. Round neuron cell bodies (Fig. 6, arrowheads) are tightly packed inside the bony labyrinth and number 70-80 per section in accord with published data (Vetter et al., 1996). Dendrites exit the spiral ganglion to the right, and axons exit at the bottom. Figure 6Bb shows the spiral ganglion in an age-matched null mutant from a comparable region in the cochlea, which is morphologically similar to the control. Thus, we conclude that hearing loss in the mutants does not reflect neurodegenerative changes in the spiral ganglion.
Normal marginal cell morphology in Claudin 11-null mice
Another cell type in the cochlea of Claudin 11-null mice to which damage could account for the deafness phenotype is the marginal cell. Dysfunction or loss of these cells would lead to diminished K+ recycling and endolymph secretion into the cochlear duct from the stria vascularis and cause the collapse of the scala media compartment. Such pathology has been observed in humans and mutant animals with mutations in the SLC12A2, KCNQ1, and KCNE1 genes (for review, see Wangemann, 2002). We do not observe collapse or bulging of Reissner's membrane, which indicates that marginal cell function is normal; nevertheless, we examined the morphology of these cells in the null mutants by freeze-fracture electron microscopy (Fig. 7). As indicated in Figure 1C, marginal cells elaborate a claudin 1 TJ network. These junctions are extensive and well developed by P30 (Fig. 7Aa, arrowheads) and have a morphology that is indistinguishable from those at P90 (Fig. 7Ac). Likewise, the TJ network is well established in Claudin 11-null mice at P30 and persists into adulthood. Thus, these data indicate that marginal cells in the null mutants function appropriately to form a polarized epithelial layer using an extensive claudin 1 TJ network.
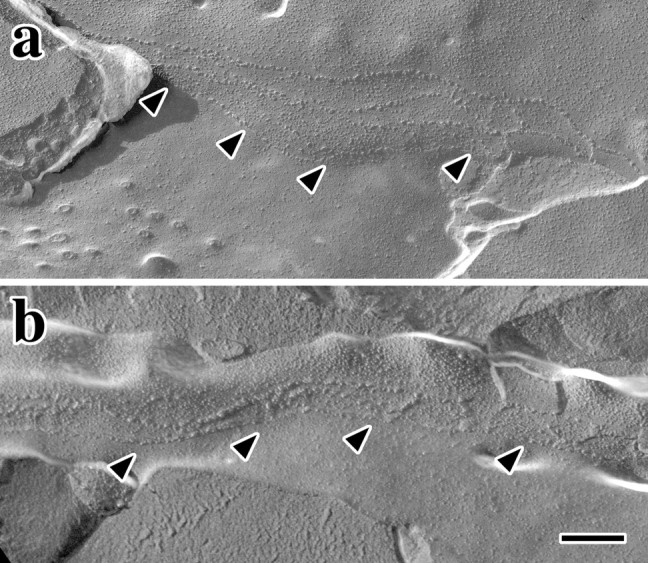
Figure 7.
Freeze fracture showing normal marginal cell TJs and basolateral infoldings in Claudin 11-null mice. A, Claudin 1 TJ strands (arrowheads) in marginal cells from wild-type (a, c) and null mutant (b, d) mice at P90 (a, b) and P30 (c, d). Scale bar, 100 nm. B, Basolateral infoldings in marginal cells from wild-type (a, c) and null mutant (b, d) mice at P90 (a, b) and P30 (c, d). Characteristic packing of intramembranous particles reflects the presence of Na+/K+ ATPases and other integral proteins (star). Asterisk in d marks atypical morphology that may be a sign of localized edema in the stria vascularis of an adult mutant. Scale bar, 100 nm.
The extensively infolded basolateral membrane of marginal cells is laden with Na+/K+ ATP-dependent pumps to facilitate rapid uptake of K+ from the intrastrial space. These infoldings are discernable in Figure 7B by vertically oriented alternating fracture surfaces in each panel and are similar to previous reports illustrating marginal cell freeze-fracture morphology (Souter and Forge, 1998). Presumptive Na+/K+ pumps, and probably other transmembrane proteins, are clearly visible in wild-type tissue at P30 as intramembranous particles with a characteristic high density in several fracture surfaces across the field (Fig. 7Ba, stars). Furthermore, this high particle density is similar to adults (Fig. 7Bc). Marginal cell basolateral membranes in Claudin 11-null mice are extensively infolded in a similar manner to controls, as are the appearance and density of intramembranous particles. An unusual feature observed in freeze-fracture replicas from P90 null mutant mice is shown in Figure 7Bd (asterisk). This pathology may represent local regions within the stria vascularis where marginal cell membranes have separated from other cell types. Presumably, these changes stem from edema that may be caused by the buildup of Na+ that has infiltrated the stria vascularis between basal cells. We anticipate that such changes do not contribute significantly to the progressive hearing loss observed in the mutants, because they represent a minor aspect of the pathology.
Normal strial endothelial cell TJs in Claudin 11-null mice
Expression of β-galactosidase by endothelial cells in the stria vascularis of Claudin 11-null mice (Fig. 1Aa) raises the possibility that TJs in strial endothelial cells are abnormal or missing, which could lead to electrolyte imbalance in the stria vascularis. To investigate this possibility, we examined the morphology of strial microvasculature intramembranous fibrils in freeze-fracture replicas for evidence of pathology.
Tight junction strands elaborated by endothelial cells in the stria vascularis from adult wild-type mice (Fig. 8a, arrowheads) exhibit an anastomosing morphology similar to capillaries from other organs (Bowman et al., 1992). Gap junctions are rare. These morphological characteristics are also evident in freeze-fracture replicas from adult Claudin 11-null mice and are indistinguishable from controls (Fig. 8b, arrowheads), which demonstrates that the absence of claudin 11 is without major consequence to endothelial membrane ultrastructure. Indeed, strial capillaries from the mutants also appear normal using scanning and transmission electron microscopy (data not shown). In addition to morphology, endothelial TJs in the mutants are functionally intact, as demonstrated by the K+ concentration data shown in Figure 5C and by the absence of extravasated immunoglobulins in the intrastrial space (data not shown). Thus, we find no evidence of endothelial cell pathology in the stria vascularis of Claudin 11-null mice. The persistence of intramembranous fibrils in these cells indicates that multiple claudin family members contribute to the TJ strands, as has been demonstrated for other endothelia (Nitta et al., 2003).
Figure 8.
Freeze fracture showing normal endothelial cell TJs in Claudin 11-null mice. Intramembranous fibrils (arrowheads) in strial endothelial cell membranes from adult wild-type (a) and Claudin 11-null (b) mice are morphologically similar. Scale bar, 200 nm.
Normal expression of intercellular junctions, ion channels, and pumps in Claudin 11-null mice
In the absence of major morphological changes in the cochlea of Claudin 11-null mice that might account for hearing loss revealed by our electrophysiological measurements, we turned to a temporal analysis of genes known to be important for auditory function that are expressed by different cell types and might provide evidence of cell dysfunction. These genes are grouped into three classes: intercellular junctions, ion channels, and ion pumps. The cDNAs from each gene were used to probe Northern blots of RNA from pooled cochleas at P6.5, P14.5, P30, and P100. The data show that Claudin 11 is expressed in wild-type (+/+) (Fig. 9) but not null mutant (-/-) cochleas. This expression commences well before the onset of hearing at approximately P14, although the role played by claudin 11 during this period is not known. Indeed, β-galactosidase histochemistry indicates that Claudin 11 is broadly and continuously expressed in cochlea from late embryogenesis (data not shown).
Figure 9.
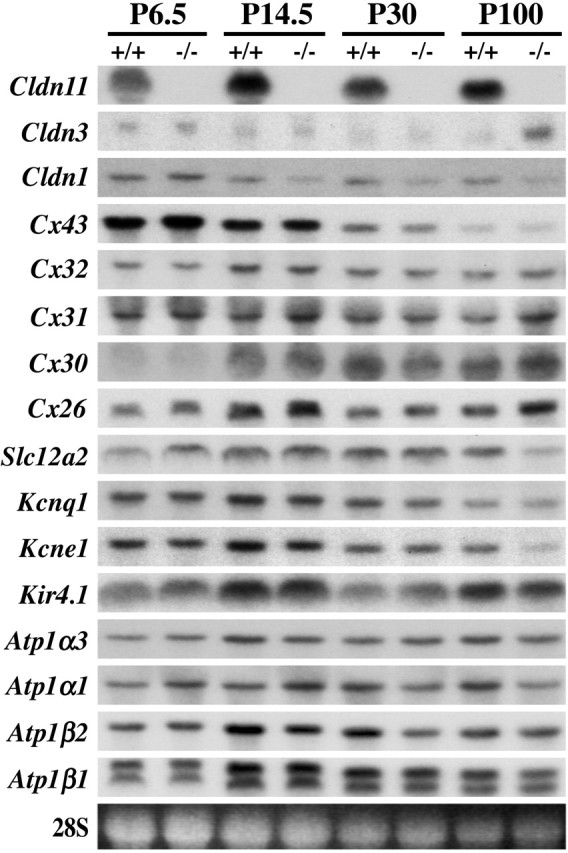
Northern blotting of cochlear RNA shows temporal expression profiles of intercellular junction, ion channel, and ATPase genes. Total RNA obtained from pooled cochleas of P6.5, P14.5, P30, and P100 wild-type (+/+) and Claudin 11-null mice (-/-) probed with cDNAs indicated on the left. 28S rRNA is included as a loading control.
In addition to Claudin 11 expression, we examined expression of Claudins 1, 3, 6, 9, and 14 in the cochlea. Interestingly, we find that expression of Claudin 3 is induced in the mutants approximately fivefold compared with littermate controls, suggesting that some form of compensation may be manifested in adults. However, we are unable to visualize increases in claudin 3 by immunofluorescence microscopy and cannot ascertain the purpose for induction of this gene. Clearly, compensation does not result in TJ assembly between basal cells. Claudin 1 expression may be slightly diminished in the mutants from P14.5 to P100; however, we view changes in signal intensity of twofold or less with skepticism because of limitations inherent with this technique. Moreover, the anastomosing intramembranous fibrils between marginal cells in replicas from Claudin 11-null mice are indistinguishable from controls, and we conclude that even if claudin 1 is expressed at slightly lower levels, the marginal cell epithelium is polarized.
Of the remaining genes examined by Northern blot analysis, the expression of several may be aberrantly regulated in Claudin 11-null mice with changes less than threefold compared with controls. We observe increases in expression of Connexins 31 and 26 and decreases in the Slc12a2 Na+/2Cl-/K+ cotransporter Kcne1 and Atp1α1 at P100. Such reductions may be of some interest because these genes are all expressed by marginal cells and may signify altered behavior. Nonetheless, it is unlikely that major marginal cell functions, particularly secretion of the endolymph, are significantly perturbed, because neither collapse of the membranous labyrinth nor endolymphatic hydrops are features of pathology in these animals. Furthermore, it is doubtful that the altered expression profiles portend a diminution in marginal cell population because expression of other genes by this cell type, such as Atp1β2 and Kcnq1, are normal. Finally, we examined the expression patterns of a number of the genes in Figure 9 using commercial antibodies (when available) to label paraffin sections of cochlea. We find no alterations in these patterns for the claudins, connexins, and ATPase in the stria vascularis.
Discussion
Previous studies have determined that claudin 11 TJs are assembled in several tissues, including CNS myelin and Sertoli cells (Gow et al., 1999; Morita et al., 1999). Here, we characterize expression of Claudin 11 in the inner ear and examine consequences to the auditory system associated with the absence of this TJ network. Our task is simplified because claudin 11 typically forms “pure” TJs in the sense that other claudins are not present in these junctions (Gow et al., 1999), and because degenerative changes in the cochlear duct of Claudin 11-null mice are minimal despite the dramatic functional changes that we observe.
A major finding in the current study is a direct demonstration of the dependence on basal cell TJs for the generation of EP. Early experiments in the cochlea demonstrate a strong voltage potential at the surface of the stria vascularis (Tasaki et al., 1952; von Bekesy, 1952). Subsequent electrophysiological studies indicate that a compartment within this lateral wall epithelium exhibits an electrochemical potential with respect to surrounding tissue and a low K+ concentration compared with those of an intracellular milieu and endolymph. Accordingly, these data point to the extracellular compartment of the intrastrial space as the site of high-voltage potential and identify basal and intermediate cells as the electrogenic cell population (Salt et al., 1987; Hoshino et al., 2000; Takeuchi et al., 2000; Marcus et al., 2002). Furthermore, the model posits that basal cell TJs are necessary to electrically isolate the intrastrial compartment. Here, we characterize a mouse mutant that cannot generate a paracellular barrier between basal cells, which renders the intrastrial space open to perilymph and abolishes its electrical isolation. In Claudin 11-null mice, EP is very low and the absence of degenerative changes in the cochlea directly demonstrates that claudin 11 TJs comprise a critical component of the electrogenic machinery in the stria vascularis.
Models distinct from the two-cell hypothesis developed by Salt et al. (1987) have been proposed to account for EP (for review, see Wangemann, 1995). These models stipulate that a single polarized epithelium, the marginal cell layer, is sufficient to generate a strong voltage potential across the stria vascularis and posit that ion pumps in the apical and/or basolateral surfaces of marginal cells serve as electrogenic machinery. The current study does not support these models and, rather, adds considerable weight to mounting evidence against such mechanisms. For example, these models predict normal EP in the absence of basal cell TJs. Clearly, this prediction contrasts with our experimental data, except in scenarios where the absence of claudin 11 damages other cells in the cochlea. However, there is little evidence to support such an explanation. Normal appearance of the scala media and an absence of degeneration in the organ of Corti indicate that marginal cells are fully functional in Claudin 11-null mice. Furthermore, normal TJ morphologies for marginal cells and strial capillaries, normal basolateral surfaces of marginal cells, and at most minor expression changes in genes associated with K+ homeostasis strengthen our conclusion.
A second finding to emerge from this study is a frequency-independent elevation of ABR thresholds in Claudin 11-null mice, which affords us an opportunity for comment about the dependence of the cochlear amplifier on EP. Importantly, null mutants exhibit good resolution of ABR Peaks I-V at all frequencies tested with normal latencies for Peaks I-III. In mammalian cochleas, the presence of a mechanism to maximize detection of low-intensity sound through active resonance amplification has been hypothesized for more than a decade. Cellular and molecular machinery associated with this amplifier have come to light and include the OHC, in which the force-generating mechanism for amplification is localized, and a transmembrane motor protein, prestin, that effects amplification by somatic “electromotility” in direct response to voltage changes across the OHC lateral membrane (Zheng et al., 2000). Genetic studies in which the cochlear amplifier has been inactivated indicate that the resulting loss of sensitivity is 40-60 dB SPL across the frequency spectrum (Liberman et al., 2002), which accords with data from cochleas that are devoid of OHCs (Ryan and Dallos, 1975). In this regard, it is tempting to speculate that the ∼50 dB SPL ABR threshold elevation observed in Claudin 11-null mice stems from a failure to efficiently power the cochlear amplifier. If so, might our data suggest that EP is an essential component of the power source for the amplification machinery? Clearly, this is too bold a conclusion from our current understanding of the Claudin 11-null phenotype. However, it is a tantalizing assessment from an evolutionary perspective because of the possibility that electromotility in OHC and high EP coemerged in mammals ∼240 million years ago (Santos-Sacchi, 2003).
Several groups have used loop diuretics such as furosemide and bumetanide to transiently lower EPs and have measured evoked responses as a function of EP. These data indicate that ABR thresholds increase as EP decreases and suggest an inverse correlation between EP and cochlear amplifier function. However, a major technical deficiency with these data is continuously variable (i.e., unstable) EPs characterized by a precipitous fall within 2 min after administration (to negative values with larger drug doses) and an immediate, nonlinear rebound over 1-2 hr. Moreover, and concomitant with the initial decline in EP, dramatic morphological changes in intermediate and marginal cells reflect widespread edema in the intrastrial space and separation of intermediate cell processes from marginal cell infoldings (Pike and Bosher, 1980; Santi and Lakhani, 1983). In addition, the K+ concentration in endolymph declines, probably in response to strial pathology, which may cause hair cell damage and reduce the driving force for transduction (Forge and Brown, 1982; Rybak and Morizono, 1982; Comis et al., 1990; Santos-Sacchi et al., 2001). In light of such pathological changes, observed ABR threshold shifts with loop diuretics could equally reflect correlations between thresholds and damage to the stria vascularis, damage to hair cells, or both. In contrast, Claudin 11-null mice exhibit very little pathology in the cochlea and have normal levels of K+ in endolymph; thus, the data in the current study present a clear view of the relationship between EP and the function of the cochlear amplifier.
Previously, we found that the latencies of VEPs in adult null mice were increased 15-20% compared with littermates (Gow et al., 1999). In the current study, we find that Peak V latencies in the mutants are increased by 40%. Although claudin 11 comprises TJs of CNS myelin sheaths, we reason that conduction velocity changes are unlikely to account for the progressive deterioration of ABRs and DPOAEs in Claudin 11-null mice for several reasons. First, hypomyelination and neuronal loss, which could cause abnormal auditory function, are not features of the Claudin 11-null phenotype. Furthermore, we detect no ultrastructural abnormalities in CNS myelin sheaths at nodes of Ranvier or in the internodes, beyond the absence of claudin 11 intramembranous fibrils (Gow et al., 1999). Second, we do not observe atrophy of the spiral ganglion neurons from these animals at P90 (Fig. 6B), which indicates that atrophy of the eighth nerve is minimal. Finally, for genetic mutant mice exhibiting moderate to severe CNS or PNS hypomyelination, ABRs are distinct from those observed in Claudin 11-null mice. For example, three myelin mutants examined by other groups exhibit significantly increased latencies for Peak I (Zhou et al., 1995a,b) or Peaks I-V (Fujiyoshi et al., 1994), whereas Peak V latency is longer in Claudin 11-null mice and Peaks I-III are indistinguishable from controls.
In view of the expression of claudin 11 in the vestibular system (Fig. 1Aa, 1-3), which we have confirmed with antibodies on paraffin sections to be confined to the squamous epithelium of the membranous labyrinth (A. Gow, unpublished data), we are curious as to why Claudin 11-null mice do not exhibit circling or waltzing behavior. Many mice expressing mutated genes in the inner ear, particularly those involved with ion homeostasis, exhibit overt vestibular dysfunction. For example Isk-null mice lacking functional K+ channels at the apical surface of marginal cells fail to secrete K+ into the endolymph and display a shaker/waltzer phenotype (Vetter et al., 1996). Presumably, claudin 11 TJs in the vestibule perform similar functions to those in the basal cell layer to occlude the paracellular space of an epithelium separating two compartments with differing K+ and Na+ compositions, and we speculate that the absence of this barrier could disrupt K+ homeostasis within the vestibular system. Indeed, Claudin 11-null mice perform poorly in a rotarod analysis from an early age (Gow et al., 1999), which may reflect mild dysfunction. In contrast, persistence of hair cells in the cristae ampullares in adult null mice (Gow, unpublished data) are consistent with relatively normal K+ homeostasis (Vetter et al., 1996), and other claudin family members may compensate for the absence of claudin 11. In this regard, immunocytochemical analyses are ongoing and indicate that claudin 3 is one candidate.
Footnotes
This work was supported by National Institutes of Health (NIH)-National Institute on Deafness and Other Communication Disorders (NIDCD) Grant RO1 DC006262, NIH-National Institute of Neurological Disorders and Stroke Grant RO1 NS43783 (A.G.), and NIH-NIDCD Grant RO1 DC00212 (D.C.M.). We thank Dr. T. Moran (Director; Hybridoma Core, Mount Sinai School of Medicine, NewYork, NY) and HelenPark, who generated the claudin 11 monoclonal antibodies (37E3); Dr. G. Manley (Institut fur Zoologie, Technischen Universitat Munchen, Garching, Germany) for critical reading; Dr. D. Forrest (Genetics, Mount Sinai School of Medicine, New York, NY) for his support and advice; Dr. A. Rzadzinska for her help processing organs of Corti (NIH-NIDCD, Bethesda, MD); and Brunella Bertucci, Cunli Xiang, Tabitha Doci, and Jarrod MacFarlane (Center for Molecular Medicine and Genetics, Wayne State University, Detroit, MI) for technical assistance.
Correspondence should be addressed to either of the following: Alexander Gow, Molecular Medicine and Genetics, Wayne State University School of Medicine, 3216 Scott Hall, 541 East Canfield, Detroit, MI 48201, E-mail: agow@genetics.wayne.edu; or Bechara Kachar, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Building 50, Room 4249, Bethesda, MD 20892, E-mail: kacharb@nidcd.nih.gov.
Copyright © 2004 Society for Neuroscience 0270-6474/04/247051-11$15.00/0
References
- Ahituv N, Avraham KB (2002) Mouse models for human deafness: current tools for new fashions. Trends Mol Med 8: 447-451. [DOI] [PubMed] [Google Scholar]
- Bowman PD, do Bois M, Shivers RR, Dorovini-Zis K (1992) Endothelial tight junctions. In: Tight junctions, Ed 1 (Cereijido M, ed), pp 305-320. Boca Raton, FL: CRC.
- Comis SD, Osborne MP, Jeffries DJ (1990) Effect of furosemide upon morphology of hair bundles in guinea pig cochlear hair cells. Acta Otolaryngol 109: 49-56. [DOI] [PubMed] [Google Scholar]
- Dallos P, Evans BN (1995) High-frequency motility of outer hair cells and the cochlear amplifier. Science 267: 2006-2009. [DOI] [PubMed] [Google Scholar]
- Davis H (1983) An active process in cochlear mechanics. Hear Res 9: 79-90. [DOI] [PubMed] [Google Scholar]
- Forge A, Brown AM (1982) Ultrastructural and electrophysiological studies of acute ototoxic effects of furosemide. Br J Audiol 16: 109-116. [DOI] [PubMed] [Google Scholar]
- Fujiyoshi T, Hood L, Yoo TJ (1994) Restoration of brain stem auditory-evoked potentials by gene transfer in shiverer mice. Ann Otol Rhinol Laryngol 103: 449-456. [DOI] [PubMed] [Google Scholar]
- Gow A, Friedrich VL, Lazzarini RA (1992) Myelin basic protein gene contains separate enhancers for oligodendrocytes and Schwann cell expression. J Cell Biol 119: 605-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow A, Southwood CM, Lazzarini RA (1998) Disrupted proteolipid protein trafficking results in oligodendrocyte apoptosis in an animal model of Pelizaeus-Merzbacher disease. J Cell Biol 140: 925-934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow A, Southwood CM, Li JS, Pariali M, Riordan GP, Brodie SE, Danias J, Bronstein JM, Kachar B, Lazzarini RA (1999) CNS myelin and sertoli cell tight junction strands are absent in Osp/Claudin 11-null mice. Cell 99: 649-659. [DOI] [PubMed] [Google Scholar]
- Hoshino T, Mizuta K, Gao J, Araki S, Araki K, Takeshita T, Wu R, Morita H (2000) Cochlear findings in the white spotting (Ws) rat. Hear Res 140: 145-156. [DOI] [PubMed] [Google Scholar]
- Kikuchi T, Kimura RS, Paul DL, Takasaka T, Adams JC (2000) Gap junction systems in the mammalian cochlea. Brain Res Brain Res Rev 32: 163-166. [DOI] [PubMed] [Google Scholar]
- Krstic R (1991) Human microscopic anatomy, pp 564-565. New York: Springer.
- Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680-685. [DOI] [PubMed] [Google Scholar]
- Liberman MC, Gao J, He DZ, Wu X, Jia S, Zuo J (2002) Prestin is required for electromotility of the outer hair cell and for the cochlear amplifier. Nature 419: 300-304. [DOI] [PubMed] [Google Scholar]
- Liu XZ, Ouyang XM, Xia XJ, Zheng J, Pandya A, Li F, Du LL, Welch KO, Petit C, Smith RJ, Webb BT, Yan D, Arnos KS, Corey D, Dallos P, Nance WE, Chen ZY (2003) Prestin, a cochlear motor protein, is defective in nonsyndromic hearing loss. Hum Mol Genet 12: 1155-1162. [DOI] [PubMed] [Google Scholar]
- Luciano L, Reiss G, Iurato S, Reale E (1995) The junctions of the spindle-shaped cells of the stria vascularis: a link that completes the barrier between perilymph and endolymph. Hear Res 85: 199-209. [DOI] [PubMed] [Google Scholar]
- Marcus DC, Wu T, Wangemann P, Kofuji P (2002) KCNJ10 (Kir4.1) potassium channel knockout abolishes endocochlear potential. Am J Physiol Cell Physiol 282: C403-C407. [DOI] [PubMed] [Google Scholar]
- Matsubara A, Laake JH, Davanger S, Usami S, Ottersen OP (1996) Organization of AMPA receptor subunits at a glutamate synapse: a quantitative immunogold analysis of hair cell synapses in the rat organ of Corti. J Neurosci 16: 4457-4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K, Sasaki H, Fujimoto K, Furuse M, Tsukita S (1999) Claudin-11/OSP-based tight junctions of myelin sheaths in brain and Sertoli cells in testis. J Cell Biol 145: 579-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S (2003) Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol 161: 653-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham K, Sun X-M, Kim DO (2001) Noninvasive assessment of auditory function in mice: auditory brainstem response and distortion product otoacoustic emissions. In: Handbook of mouse auditory research. From behavior to molecular biology (Willott JF, ed), pp 37-58. Boca Raton, FL: CRC.
- Petit C (1996) Genes responsible for human hereditary deafness: symphony of a thousand. Nat Genet 14: 385-391. [DOI] [PubMed] [Google Scholar]
- Pike DA, Bosher SK (1980) The time course of the strial changes produced by intravenous furosemide. Hear Res 3: 79-89. [DOI] [PubMed] [Google Scholar]
- Ryan A, Dallos P (1975) Effect of absence of cochlear outer hair cells on behavioural auditory threshold. Nature 253: 44-46. [DOI] [PubMed] [Google Scholar]
- Rybak LP, Morizono T (1982) Effect of furosemide upon endolymph potassium concentration. Hear Res 7: 223-231. [DOI] [PubMed] [Google Scholar]
- Rzadzinska AK, Schneider ME, Davies C, Riordan GP, Kachar B (2004) An actin molecular treadmill and myosins maintain stereocilia functional architecture and self-renewal. J Cell Biol 164: 887-897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salt AN, Melichar I, Thalmann R (1987) Mechanisms of endocochlear potential generation by stria vascularis. Laryngoscope 97: 984-991. [PubMed] [Google Scholar]
- Santi PA, Lakhani BN (1983) The effect of bumetanide on the stria vascularis: a stereological analysis of cell volume density. Hear Res 12: 151-165. [DOI] [PubMed] [Google Scholar]
- Santos-Sacchi J (2003) New tunes from Corti's organ: the outer hair cell boogie rules. Curr Opin Neurobiol 13: 459-468. [DOI] [PubMed] [Google Scholar]
- Santos-Sacchi J, Wu M, Kakehata S (2001) Furosemide alters nonlinear capacitance in isolated outer hair cells. Hear Res 159: 69-73. [DOI] [PubMed] [Google Scholar]
- Souter M, Forge A (1998) Intercellular junctional maturation in the stria vascularis: possible association with onset and rise of endocochlear potential. Hear Res 119: 81-95. [DOI] [PubMed] [Google Scholar]
- Southwood CM, Gow A (2001) Functions of OSP/claudin 11-containing parallel tight junctions: implications from the knockout mouse. In: Tight junctions, Ed 2 (Anderson JM, Cereijido M, eds), pp 719-741. New York: CRC.
- Southwood CM, Garbern J, Jiang W, Gow A (2002) The unfolded protein response modulates disease severity in Pelizaeus-Merzbacher disease. Neuron 36: 585-596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel KP, Kros CJ (2001) A genetic approach to understanding auditory function. Nat Genet 27: 143-149. [DOI] [PubMed] [Google Scholar]
- Takeuchi S, Ando M, Kakigi A (2000) Mechanism generating endocochlear potential: role played by intermediate cells in stria vascularis. Biophys J 79: 2572-2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki I, Davis H, Eldredge DH (1952) Exploration of cochlear potentials in guinea pig with a microelectrode. J Acoust Soc Am 26: 765-773. [Google Scholar]
- Vetter DE, Mann JR, Wangemann P, Liu J, McLaughlin KJ, Lesage F, Marcus DC, Lazdunski M, Heinemann SF, Barhanin J (1996) Inner ear defects induced by null mutation of the isk gene. Neuron 17: 1251-1264. [DOI] [PubMed] [Google Scholar]
- von Bekesy G (1952) DC resting potentials inside the cochlear partition. J Acoust Soc Am 24: 72-76. [DOI] [PubMed] [Google Scholar]
- Wangemann P (1995) Comparison of ion transport mechanisms between vestibular dark cells and strial marginal cells. Hear Res 90: 149-157. [DOI] [PubMed] [Google Scholar]
- Wangemann P (2002) K+ cycling and the endocochlear potential. Hear Res 165: 1-9. [DOI] [PubMed] [Google Scholar]
- Zheng J, Shen W, He DZ, Long KB, Madison LD, Dallos P (2000) Prestin is the motor protein of cochlear outer hair cells. Nature 405: 149-155. [DOI] [PubMed] [Google Scholar]
- Zheng QY, Johnson KR, Erway LC (1999) Assessment of hearing in 80 in-bred strains of mice by ABR threshold analyses. Hear Res 130: 94-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Abbas PJ, Assouline JG (1995a) Electrically evoked auditory brainstem response in peripherally myelin-deficient mice. Hear Res 88: 98-106. [DOI] [PubMed] [Google Scholar]
- Zhou R, Assouline JG, Abbas PJ, Messing A, Gantz BJ (1995b) Anatomical and physiological measures of auditory system in mice with peripheral myelin deficiency. Hear Res 88: 87-97. [DOI] [PubMed] [Google Scholar]