

Abstract
Background
APOL1 genotype is associated with advanced kidney disease in African-Americans, but the pathogenic mechanisms are unclear. Here, associations of APOL1 genotype with urine biomarkers of glomerular and tubular injury, and with kidney function decline, were evaluated.
Study Design
Observational study.
Setting & Participants
431 HIV-infected African-American women enrolled in Women's Interagency HIV Study (WIHS).
Predictor
APOL1 genotype.
Outcomes
Albumin-creatinine ratio (ACR), four tubular injury biomarkers (interleukin 18 [IL-18], kidney injury molecule 1 [KIM-1], neutrophil gelatinase-associated lipocalin [NGAL], and α1-microglobulin [α1m]), and kidney function estimated using the CKD-EPI cystatin C equation.
Measurements
Participants were genotyped for APOL1 single-nucleotide polymorphisms rs73885319 (G1 allele) and rs71785313 (G2 allele). Urine biomarker levels were measured using stored samples from 1999-2000. Cystatin C was measured using serum collected at baseline and 4- and 8-year follow-up.
Results
At baseline, ACR levels were higher among 47 women with 2 APOL1 risk alleles versus 384 women with 0/1 risk allele (median, 24 vs. 11 mg/g; p < 0.001). Compared to women with 0/1 risk allele, women with 2 risk alleles had 104% higher ACR (95% CI, 29-223 mg/g) and 2-fold greater risk of ACR > 30 mg/g (95% CI, 1.17-3.44) after multivariable adjustment. APOL1 genotype showed little association with urine IL-18:Cr, KIM-1:Cr, and NGAL:Cr (estimates of -5% [95% CI, -24% to 18%], -20% [95% CI, -36% to 1%], and 10% [95% CI, -26% to 64%], respectively), or detectable urine α1m (prevalence ratio, 1.13; 95% CI, 0.65-1.97) in adjusted analyses. Compared to women with 0/1 allele, women with 2 risk alleles had faster eGFR decline, by 1.2 (95% CI, -2.2 to -0.2) ml/min/1.73 m2 per year, and had 1.7- and 3.4-fold greater rates of incident chronic kidney disease (95% CI, 1.1-2.5) and 10% annual eGFR decline (95% CI, 1.7-6.7), respectively, with minimal attenuation after adjustment for glomerular and tubular injury biomarkers.
Limitations
Results may not be generalizable to men.
Conclusions
Among HIV-infected African-American women, APOL1-associated kidney injury appears to localize to the glomerulus, rather than the tubules.
Index Words: APOL1 genotype, risk variant, risk allele, G1 allele, G2 allele, single-nucleotide polymorphism (SNP), albumin-creatinine ratio (ACR), proteinuria, tubular injury biomarker, apolipoprotein L1, kidney disease, renal function, glomerular injury, African American, Women's Interagency HIV Study (WIHS)
African-Americans have a 3- to 4-fold greater risk of end-stage renal disease (ESRD) compared to Caucasians in HIV-infected and uninfected populations.1-4 Two prior studies revealed strong associations between chromosome 22q and ESRD without diabetes among individuals of African ancestry.5,6 Subsequent studies identified two risk variants on the APOL1 (apolipoprotein L1) gene which account for these associations.7,8 The G1 allele comprises two SNPs (reference single nucleotide polymorphism [SNP] identification number [rs]73885319 and rs60910145) encoding 2 amino acid substitutions and the G2 allele encodes a two amino-acid deletion (rs71785313).7 Heterozygosity for the risk alleles appears sufficient to confer resistance to Trypanosoma brucei rhodesiense,9 while homozygosity or compound heterozygosity leads to substantially elevated kidney disease risk.7
The pathogenesis of APOL1-associated kidney disease, however, is poorly understood. Madhavan et al. demonstrated that APOL1 localizes to podocytes, proximal tubules, and endothelium in the normal kidney, and that glomerular and proximal tubular APOL1 staining is reduced among individuals with FSGS and HIVAN.10 Ma et al. subsequently reported that APOL1 protein and mRNA were detectable in podocytes and renal tubular cells of cryosections from individuals with normal kidney function.11 However, the specific effects of APOL1 risk alleles on glomerular and proximal tubular function were not evaluated.
Biomarkers of tubular injury and dysfunction may be useful in earlier detection and localization of pathology within the nephron. In the Women's Interagency HIV Study (WIHS), we previously found that African-American race was a strong and independent risk factor for albuminuria, and was associated with higher levels of three urinary markers of tubular injury and dysfunction: interleukin 18 (IL-18), neutrophil gelatinase-associated lipocalin (NGAL), and α1-microglobulin (α1m).12,13 Furthermore, albuminuria, IL-18 and α1m were each independently associated with longitudinal kidney function decline and mortality.13-15 These findings suggest that the higher risk of kidney disease progression among HIV-infected African-Americans may be mediated through their more extensive glomerular and tubular injury. However, the specific contributions of the APOL1 risk alleles to these observations have not been ascertained.
In this study of HIV-infected African-Americans enrolled in the WIHS, we first evaluated the cross-sectional associations of APOL1 risk alleles with glomerular injury, quantified by albumin-creatinine ratio (ACR), as well as four biomarkers of tubular injury and dysfunction: IL-18, kidney injury molecule 1 (KIM-1), NGAL, and α1m. We then assessed the associations of APOL1 risk alleles with longitudinal kidney function decline, adjusting for baseline levels of the injury markers.
Methods
Study Population
The WIHS is a prospective cohort study that enrolled 3,067 HIV-infected and 1,070 uninfected women from six US locations (Bronx, Brooklyn, Chicago, Los Angeles, San Francisco, and Washington, DC) in 1994-1995, 2001-2002, or 2011-2012. Details of the study design and data collection methods were published previously.16,17 Participants underwent semiannual visits that included a standardized questionnaire, physical examination, and collection of laboratory specimens. Among the 795 African-American HIV-infected women who were evaluated at the WIHS clinical visits in 1999-2000, we chose a priori to include all 431 women who had the following: 1) consent for DNA testing; 2) available DNA for APOL1 genotyping, and 3) stored urine available from the 1999-2000 visits for biomarker measurements.
The institutional review boards of participating institutions approved the study protocol at all WIHS study sites, and informed consent was obtained from all study participants. This cross-sectional study was also approved by the Institutional Review Board at the Johns Hopkins School of Medicine.
Exposure Variable
Participants were genotyped for the APOL1 SNPs rs73885319 (G1 allele) and rs71785313 (G2 allele) using TaqMan assays (Applied Biosystems, Foster City, CA)18 and categorized as having zero, one or two risk alleles. The G1 allele rs60910145 was not tested because rs73885319 and rs60910145 are in near perfect linkage disequilibrium.7 Genotyping for ancestry informative markers (AIMs) was performed using the Illumina GoldenGate platform (Illumina, San Diego, CA). Selection of AIMs in the WIHS was described previously.19 Briefly, West African, European, East Asian and Native American populations were distinguished using a subset of markers published by Smith et al.18 Of the larger panel of 384 AIMs, 185 were selected, and 168 passed quality control measures. Genetic ancestry components were evaluated by principal components analysis using EIGENSTRAT software.20,21
Outcome Variables
All biomarkers were measured at the Cincinnati Children's Hospital Medical Center Biomarker Laboratory, using random urine samples. Urine specimen storage and coefficients of variation for the biomarker assays are described in Item S1 (provided as online supplementary material). Urine albumin and creatinine were measured by immunoturbidimetry and colorimetric enzyme assay, respectively, using a Siemens Dimension Xpand plus HM clinical analyzer (Siemens, Munich, Germany). Urine IL-18 was measured using a commercially available ELISA kit (Medical & Biological Laboratories Co., Nagoya, Japan). The urine KIM-1 ELISA was constructed using commercially available reagents (R&D Systems Inc., Minneapolis, MN).22 Urine NGAL was assayed using a human-specific commercially available ELISA (BioPorto, Grusbakken, Denmark).23 Urine α1m was measured by a commercially available assay (Siemens BN II Nephelometer, Munich, Germany).
Serum for cystatin C measurement was collected at baseline and two follow-up visits, occurring approximately 4 and 8 years after the initial visit. Kidney function was estimated using the CKD-EPI equation for serum cystatin C, which is less susceptible to bias by muscle mass and health status than serum creatinine.24 Cystatin C was measured centrally at the University of California, Los Angeles, Clinical Immunology Research Laboratory using a particle-enhanced immunoturbidimetric assay (Gentian, Moss, Norway), which has been calibrated against the new World Standard Reference material ERM-DA471/IFCC (International Federation of Clinical Chemistry and Laboratory Medicine).25 Additional details are described in Item S1.
Continuous outcomes included baseline and annual change in eGFR in ml/min/1.73m2 over approximately 8 years of follow-up. Additionally, two dichotomous kidney outcomes were analyzed: 1) incident chronic kidney disease (CKD), defined as eGFR <60 ml/min/1.73m2 at either of two follow-up visits among participants with baseline eGFR ≥60 ml/min/1.73m2, and 2) rapid decline, defined as 10% or greater annual decline in eGFR.
Covariates
Demographic and clinical characteristics were ascertained at the time of urine collection (1999-2000). Hypertension was defined by self-reported use of anti-hypertensive medications, systolic blood pressure ≥ 140mmHg, diastolic blood pressure ≥ 90mmHg, or self-reported diagnosis of hypertension confirmed by any of the previous criteria within 2 years. Diabetes mellitus was defined by self-reported use of oral hypoglycemic medications or insulin, fasting blood glucose ≥126 mg/dl, or self-reported history of diabetes diagnosis confirmed by either of the former criteria within 2 years. Hepatitis C virus (HCV) co-infection was defined by detectable HCV RNA and positive HCV antibody result. The HIV-related characteristics included current CD4 lymphocyte count, history of clinical AIDS diagnosis,26 HIV RNA level, and current highly active antiretroviral therapy (HAART) use. Historical suppression of HIV RNA was defined as the percentage of historical HIV RNA values <400 copies/mL.
Statistical Analysis
We stratified women by number of APOL1 risk alleles and compared demographic and clinical characteristics using the chi-square and Kruskal-Wallis tests for categorical and continuous variables, respectively. We then evaluated the associations of APOL1 risk alleles with creatinine-standardized and unstandardized ACR, IL-18, KIM-1, and NGAL, using linear regression with robust Huber-White standard errors27,28 (to correct for heteroscedasticity). Due to their right-skewed distributions, the continuous outcomes were log-transformed to normalize their distributions; results were back-transformed to produce estimated percentage differences.
Consistent with our previous WIHS publication,13 α1m was handled as a dichotomous variable, detectable or undetectable. Because α1m is fully reabsorbed by proximal tubular cells after filtration at the glomerulus,29 its presence in the urine indicates proximal tubular dysfunction.30 We used Poisson relative risk regression with a robust variance estimator31 to assess the associations of APOL1 risk alleles with detectable α1m, and with the clinically established threshold for ACR >30mg/g.
Primary analyses were performed using a recessive model of inheritance (2 vs 0/1 risk alleles). In secondary analyses, we evaluated dominant (2/1 vs 0 risk alleles) and additive (2 vs 1 vs 0 risk alleles) models of inheritance.
We then constructed multivariable regression models to determine whether APOL1 risk genotype was independently associated with each biomarker. Model covariates were selected on the basis of known biological associations with the development and progression of kidney disease, and included age, hypertension, diabetes mellitus, body mass index, HCV infection, HIV viral load, CD4 lymphocyte count, HAART use, and baseline eGFR. Additionally, we adjusted for genetic ancestry data by including in our models the principal components that were associated with the biomarkers in univariate analyses (components 1, 3 and 5). We assessed for effect modification between APOL1 risk genotype and historical suppression of HIV RNA for the outcome of albuminuria, by evaluating an interaction term in the overall model. To determine whether APOL1-associated kidney injury precedes overt CKD, we also performed subgroup analyses restricted to individuals with eGFR >60 ml/min/1.73m2.
Finally, we evaluated the associations of APOL1 risk genotype with kidney function at baseline and during follow-up, using a recessive model of inheritance. We used linear mixed models to evaluate the associations of APOL1 risk alleles with annual eGFR change, with random intercepts and slopes across time to account for the correlation between repeated measures at baseline and at years 4 and 8. We then used Poisson regression with a robust variance estimator to assess the associations of APOL1 risk alleles with incident CKD and rapid decline, expressing the results as incident rate ratios for each outcome. Women with eGFR <60 ml/min/1.73m2 at baseline were excluded from incident CKD analyses. Multivariable models sequentially adjusted for demographics, traditional risk factors for kidney disease progression, ACR, and the tubular injury markers. To account for losses to follow-up in kidney decline analyses, we adjusted estimates using an inverse probability weighting approach by modeling the participant's probability of having a non-missing outcome, with separate weights calculated at each visit.32 The inverse of this probability was then used as a weight applied to persons with known outcomes in the multivariable regression analyses of kidney decline.
Multiple imputation with the Markov chain Monte Carlo method was used to impute missing covariates, with 10 imputations to yield ∼95% relative efficiency.33 The percentage of missing observations for each covariate ranged from less than 1% to 31% (Table S1). Right-skewed variables were log-transformed in the imputation model. Estimates obtained from imputed data were combined using the MIANALYZE procedure to reflect the uncertainty due to missing data.
Compared to the women included in this substudy, women who were excluded (n=364) due to missing APOL1 genotype or urine biomarker measurements had higher prevalence of diabetes mellitus, lower CD4 lymphocyte count, and lower prevalence of HAART use (Table S2). As an alternate approach, we used multiple imputation to include all 795 HIV-infected African-American women in the analyses (Tables S3 and S4).
Results
Study Population by APOL1 Genotype
Among the 431 HIV-infected African-American women included in this study, 11% (n=47) and 44% (n=191) had 2 and 1 APOL1 risk alleles, respectively, which are similar to the frequencies reported among the general African-American population in the United States.34 Hypertension and HCV infection were each present in 31% and 35% of the participants, respectively, whereas diabetes was present in 10% of the cohort. There were no statistically significant differences in the prevalence of hypertension, diabetes mellitus, or HCV infection by number of APOL1 risk alleles (Table 1). Median CD4 cell count and HIV RNA levels were similar across APOL1 risk allele groups.
Table 1. Baseline characteristics of HIV-infected African-American women, stratified by number of APOL1 risk alleles.
| Characteristic | 0 allele (n = 193) | 1 allele (n = 191) | 2 alleles (n = 47) | P-value |
|---|---|---|---|---|
| Baseline Age (y) | 41 [35-46] | 42 [37-47] | 37 [35-47] | 0.1 |
| Hypertension | 50 (26%) | 67 (35%) | 17 (36%) | 0.1 |
| Diabetes mellitus | 13 (7%) | 26 (14%) | 6 (13%) | 0.08 |
| HCV infection | 66 (35%) | 67 (35%) | 16 (34%) | 0.9 |
| Current CD4+ cell count (cells/mm3) | 380 [246-563] | 388 [242-583] | 474 [318-673] | 0.09 |
| History of AIDS | 86 (45%) | 96 (50%) | 25 (53%) | 0.4 |
| HIV RNA load | 0.8 | |||
| ≤80 copies/mL | 52 (28%) | 56 (29%) | 13 (28%) | |
| 81-1999 copies/mL | 42 (22%) | 47 (25%) | 10 (22%) | |
| 2000-9999 copies/mL | 30 (16%) | 29 (15%) | 11 (24%) | |
| ≥10,000 copies/mL | 65 (34%) | 59 (31%) | 12 (26%) | |
| Historical HIV suppression1 | 68 (35%) | 73 (38%) | 15 (32%) | 0.7 |
| Current cART use | 117 (61%) | 116 (61%) | 23 (49%) | 0.3 |
Note: Values for categorical variables are given as number (percentage); values for continuous variables, as median [interquartile range]. P-values from chi-square and Kruskal-Wallis tests for categorical and continuous variables, respectively.
Defined as percentage of historical HIV RNA values less than 400 copies/mL for each study participant.
Abbreviations: cART, combination antiretroviral therapy; HCV, hepatitis C virus
Associations of APOL1 Genotype With Glomerular and Tubular Injury Markers
The distribution of ACR among women with two APOL1 risk alleles was shifted to the right relative to those with 0/1 risk allele, indicating higher ACR levels (Figure 1A). By contrast, the distributions of IL-18, KIM-1, NGAL, and α 1m did not appear to differ by number of APOL1 risk alleles (Figure 1B, Figure S1A-C).
Figure 1.
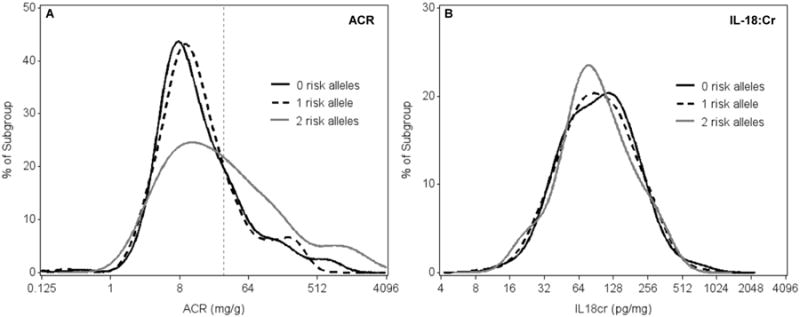
Distribution of albumin-creatinine ratio (A) and interleukin 18–creatinine ratio (B) in HIV-infected African-American women, stratified by number of APOL1 risk alleles. Dashed vertical line in panel A indicates ACR threshold of 30 mg/g. Abbreviations: ACR, albumin-creatinine ratio; IL-18:Cr, interleukin 18–creatinine ratio.
Using a recessive model of inheritance (Table 2), the presence of 2 APOL1 risk alleles compared with 0/1 risk allele was associated with higher levels of ACR (median, 24 vs. 11 mg/g; p < 0.001) and a doubling in the prevalence of ACR >30 mg/g (47% vs. 21%; p=0.001). After multivariable adjustment for traditional kidney risk factors and HIV-related risk factors (Table 3), the presence of 2 APOL1 risk alleles remained associated with 104% higher ACR (p=0.002) and 2-fold greater prevalence of ACR >30 mg/g (p=0.02). There was no significant interaction between historical suppression of HIV RNA and APOL1 genotype for the outcome of ACR (p=0.4).
Table 2. Urine biomarker levels in HIV-infected African-American women, stratified by number of APOL1 risk alleles.
| 0 allele (n = 193) | 1 allele (n = 191) | 2 alleles (n = 47) | P-value | |||
|---|---|---|---|---|---|---|
| Recessive1 | Dominant2 | Additive3 | ||||
| Glomerular Injury | ||||||
| Cr-standardized | ||||||
| ACR (mg/g) | 10 (7-29) | 11 (7-24) | 24 (8-88) | <0.001 | 0.02 | <0.001 |
| ACR >30 mg/g | 44 (23%) | 38 (20%) | 22 (47%) | 0.001 | 0.1 | 0.005 |
| Unstandardized | ||||||
| Urine Albumin (mg/mL) | 0.16 (0.08-0.37) | 0.16 (0.09-0.37) | 0.40 (0.11-1.10) | <0.001 | 0.009 | <0.001 |
| Tubular Injury | ||||||
| Cr-standardized | ||||||
| IL-18:Cr (pg/mg) | 97 (55-149) | 94 (56-150) | 79 (59-148) | 0.3 | 0.4 | 0.3 |
| KIM-1:Cr (pg/mg) | 323 (228-493) | 325 (219-568) | 265 (171-465) | 0.007 | 0.06 | 0.01 |
| NGAL:Cr (ng/mg) | 29 (15-65) | 34 (16-58) | 27 (14-75) | 0.9 | 0.7 | 0.9 |
| Unstandardized | ||||||
| IL-18 (pg/mL) | 122 (66-247) | 131 (70-253) | 119 (83-215) | 0.9 | 0.9 | 0.9 |
| KIM-1 (pg/mL) | 459 (241-874) | 530 (310-866) | 415 (252-845) | 0.3 | 0.6 | 0.3 |
| NGAL (ng/mL) | 39 (19-86) | 44 (21-91) | 38 (21-159) | 0.5 | 0.3 | 0.4 |
| Tubular Dysfunction | ||||||
| Detectable A1M | 118 (61%) | 130 (68%) | 30 (64%) | 0.7 | 0.8 | 0.9 |
| Urine Cr (mg/mL) | 1.50 (0.91-2.10) | 1.54 (0.96-2.08) | 1.51 (0.98-2.27) | 0.5 | 0.4 | 0.4 |
Note: Values for categorical variables are given as number (percentage); values for continuous variables, as median [interquartile range].
P-value for 2 risk alleles vs 0/1 risk alleles, adjusted for age and ACR.
P-value for 2/1 risk alleles vs 0 risk alleles, adjusted for age and ACR.
P-value for trend (2 vs 1 vs 0 risk alleles), adjusted for age and ACR.
Abbreviations: ACR, albumin-creatinine ratio; A1M, α1-microglobulin; Cr, creatinine; IL-18, interleukin 18; KIM-1, kidney injury molecule 1; NGAL, neutrophil gelatinase-associated lipocalin.
Table 3. Adjusted associations of APOL1 risk alleles with biomarkers in HIV-infected African-American women.
| % Estimate5 (95% CI) | Prevalence Ratio6 (95% CI) | P Values by Model of inheritance | |||
|---|---|---|---|---|---|
| Recessive2 | Dominant3 | Additive4 | |||
| Continuous Outcomes | |||||
| ACR (mg/g) | 104 (29, 223) | -- | 0.002 | 0.6 | 0.01 |
| IL-18:Cr (pg/mg) | -5 (-24, 18) | -- | 0.6 | 0.5 | 0.5 |
| KIM-1:Cr (pg/mg) | -20 (-36, -1) | -- | 0.06 | 0.5 | 0.06 |
| NGAL:Cr (ng/mg) | 10 (-26, 64) | -- | 0.6 | 0.9 | 0.6 |
| Dichotomous Outcomes | |||||
| ACR > 30 mg/g | -- | 2.00 (1.17, 3.44) | 0.02 | 0.8 | 0.08 |
| Detectable A1M | -- | 1.13 (0.65, 1.97) | 0.8 | 0.7 | 0.9 |
Note: Multivariable models adjust for age, hypertension, diabetes mellitus, body mass index, hepatitis C virus infection, HIV viral load, CD4 cell count, current combination antiretroviral therapy, creatinine-based estimated glomerular filtration rate, and principal components 1, 3, and 5.
P-value compares 2 vs 0/1 APOL1 risk alleles
P-value compares 2/1 vs 0 APOL1 risk alleles
P-value for trend (2 vs 1 vs 0 APOL1 risk alleles)
Estimated percentage difference attributable to having 2 vs 0/1 APOL1 risk alleles.
Adjusted prevalence ratio among individuals with 2 vs 0/1 APOL1 risk alleles.
Abbreviations: ACR, albumin-creatinine ratio; A1M, α1-microglobulin; CI, confidence interval; Cr, creatinine; IL-18, interleukin 18; KIM-1, kidney injury molecule 1; NGAL, neutrophil gelatinase-associated lipocalin.
APOL1 genotype was associated with higher ACR levels when analyzed using dominant (p=0.02) and additive (p < 0.001) models of inheritance (Table 2). After multivariable adjustment, the observed associations remained robust using the additive (p=0.01) but not dominant (p=0.6) model (Table 3).
APOL1 risk genotype was not associated with higher levels of urine IL-18, NGAL, or detectable α1m, by recessive, dominant, or additive models of inheritance (Tables 2 and 3). In unadjusted analyses, APOL1 risk genotype was associated with lower urine levels of creatinine-standardized but not unstandardized KIM-1, in recessive and additive models. However, after multivariable adjustment (Table 3), the association between APOL1 risk genotype and creatinine-standardized KIM-1 levels no longer met statistical significance. Urine creatinine levels were similar across APOL1 risk allele groups (Table 2).
Findings were similar when we restricted analyses to participants with baseline eGFR >60 ml/min/1.73m2 (n=408; Tables S5 and S6), and when we used multiple imputation to include all participants with missing APOL1 genotype or urine biomarker measurements (n=795; Table S3).
Associations of APOL1 Genotype With Longitudinal Kidney Function Decline
Mean baseline eGFR was 83.5 ml/min/1.73m2 in women with 2 APOL1 risk alleles and 89.1 ml/min/1.73m2 in women with 0/1 risk allele (p=0.2). Over approximately 8 years of follow-up, women with 2 APOL1 risk alleles experienced faster kidney function decline, as compared to women with 0/1 risk allele (Table 4). The unadjusted rates of annual eGFR decline were -2.6 (95% confidence interval [CI], -3.5 to -1.6) ml/min/1.73 m2 among women with 2 risk alleles, and -1.3 (95% CI, -1.6 to -1.1) ml/min/1.73m2 among women with 0/1 risk allele (p=0.02). Adjustment for common risk factors, ACR, and the tubular injury markers minimally affected the magnitude of difference in annual kidney function decline by APOL1 risk genotype.
Table 4. Associations of APOL1 risk alleles with kidney function outcomes in HIV-infected African-American women.
| Outcome | 0 allele (n = 193) | 1 allele (n = 191) | 2 alleles (n = 47) |
|---|---|---|---|
| Baseline eGFR (ml/min/1.73 m2) | |||
| Mean (95% CI) | 90.8 (88.0, 93.5) | 87.3 (84.4, 90.3) | 83.5 (76.1, 91.0) |
| Estimated difference, 2 vs 0/1 alleles (95% CI)1 | |||
| Unadjusted | Reference | Reference | -5.5 (-13.0, 1.9) |
| Adjusted + ACR | --- | --- | -4.7 (-10.4, 1.0) |
| Adjusted + ACR, IL-18, KIM-1, NGAL, A1M | --- | --- | -4.1 (-9.4, 1.2) |
| Annual change in eGFR (ml/min/1.73 m2) | |||
| Mean (95% CI) | -1.4 (-1.8, -1.1) | -1.2 (-1.6, -0.9) | -2.6 (-3.5, -1.6) |
| Estimated difference, 2 vs 0/1 alleles (95% CI)2 | |||
| Unadjusted | Reference | Reference | -1.2 (-2.2, -0.2) |
| Adjusted + ACR | --- | --- | -1.1 (-2.1, -0.1) |
| Adjusted + ACR, IL-18, KIM-1, NGAL, A1M | --- | --- | -1.1 (-2.1, -0.1) |
| Incident CKD | |||
| No. of Events | 42 | 42 | 16 |
| No. at risk | 181 | 174 | 41 |
| Incident Rate Ratio, 2 vs 0/1 alleles (95% CI)3 | |||
| Unadjusted | Reference | Reference | 1.65 (1.08, 2.52) |
| Adjusted + ACR | --- | --- | 1.74 (1.15, 2.64) |
| Adjusted + ACR, IL-18, KIM-1, NGAL, A1M | --- | --- | 1.68 (1.15, 2.46) |
| 10% annual eGFR decline | |||
| No. of Events | 13 | 11 | 10 |
| No. at risk | 193 | 191 | 47 |
| Incident Rate Ratio, 2 vs 0/1 alleles (95% CI)3 | |||
| Unadjusted | Reference | Reference | 3.40 (1.74, 6.67) |
| Adjusted + ACR | --- | --- | 3.20 (1.48, 6.91) |
| Adjusted + ACR, IL-18, KIM-1, NGAL, A1M | --- | --- | 2.82 (1.21, 6.54) |
Note: Adjusted models control for age, hypertension, diabetes mellitus, hepatitis C virus infection, HIV viral load, CD4 lymphocyte count, combination antiretroviral therapy use, and kidney injury markers listed.
Abbreviations: CI, confidence interval; CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate. A1M, α1-microglobulin; ACR, albumin-creatinine ratio; IL-18, interleukin 18; KIM-1, kidney injury molecule 1; NGAL, neutrophil gelatinase-associated lipocalin
Estimated difference in baseline eGFR attributable to having 2 vs 0/1 APOL1 risk alleles
Estimated difference in annual eGFR change attributable to having 2 vs 0/1 APOL1 risk alleles
Incident rate ratio for 2 vs 0/1 APOL1 risk alleles
During follow-up, 100 (25%) and 34 (8%) cases of incident CKD and rapid decline occurred, respectively. Compared to women with 0/1 risk allele, women with 2 APOL1 risk alleles experienced higher rates of incident CKD and rapid decline by 1.7- and 3.4 fold, respectively, with minimal attenuation after adjustment for the kidney injury markers (Table 4).
Findings were similar when we performed the longitudinal analyses in all 795 HIV-infected African-American participants, using multiple imputation to include individuals with missing APOL1 genotype or urine biomarker measurements (Table S4).
Discussion
Among HIV-infected African-American women, we found that the presence of 2 APOL1 risk alleles is associated with albuminuria, a traditional marker of glomerular injury, but not with higher levels of tubular injury markers, including IL-18, KIM-1, NGAL, and α1m. In longitudinal analyses, women with 2 APOL1 risk alleles experienced faster kidney function decline, and higher rates of incident CKD and 10% annual eGFR decline, compared with women having 0/1 risk allele. Notably, adjustment for baseline levels of glomerular and tubular injury markers did not attenuate the associations of APOL1 risk genotype with longitudinal kidney outcomes. To our knowledge, this is the first study to utilize these urine biomarkers to distinguish associations of the APOL1 risk genotype with glomerular and tubular injury.
The strong association of the APOL1 risk genotype with albuminuria is consistent with results of prior investigations of individuals with mild kidney disease. Among non-diabetic African-Americans in the Dallas Heart Study, individuals with 2 APOL1 risk alleles were observed to be 2.8-fold more likely to have microalbuminuria, compared to those with 0/1 risk allele.34 The APOL1 risk genotype has also been reported to be associated with macroalbuminuria in first-degree relatives of African-Americans with nondiabetic ESRD.35 Finally, among HIV-infected African-Americans without prior AIDS, we previously found that the presence of 2 APOL1 risk alleles was independently associated with 69% higher (95% CI, 36%-108%) urine protein excretion, compared with 0/1 risk allele.36 The present study builds upon our prior work by localizing APOL1-associated kidney injury to the glomerulus, rather than the tubules.
Contrary to our hypothesis, albuminuria did not appreciably attenuate the associations between APOL1 genotype and longitudinal kidney function decline. Because we measured albuminuria only at baseline, we cannot exclude the possibility that individuals with 2 APOL1 risk alleles developed albuminuria prior to ascertainment of eGFR at the 4- or 8-year follow-up visits. Second, the relatively small sample size of individuals with 2 APOL1 risk alleles may have reduced power to detect a smaller effect size of albuminuria on the longitudinal kidney outcomes. Finally, glomerular injury may be one of several pathways linking APOL1 genotype with the development of overt kidney disease among HIV-infected African-Americans.
Although the pathophysiology underlying APOL1-associated kidney disease remains speculative, the presence of APOLI protein in podocytes in healthy kidney samples suggests a role for APOL1 in normal glomerular health.10,11 APOL1 is a 43 kDa member of the Bcl-2 family of proteins, which are key regulators of programmed cell death.37 When overexpressed in cancer cell lines, APOL1 induces autophagic cell death.38 Recent studies suggest an important role for autophagy in the maintenance of glomerular homeostasis. Hartleben et al. observed that podocytes display unusually high levels of autophagic activity compared to tubular cells, and that mice lacking autophagic capability exhibit increased susceptibility to glomerular disease.39 Furthermore, Lan et al. demonstrated that APOL1 overexpression from G1 and G2 APOL1 genetic variants in cultured human podocytes results in podocyte necrosis, and that APOL1 variant–induced podocyte injury is augmented by the presence of HIV infection.40 Podocyte dysregulation has been implicated in the development of HIV–associated nephropathy (HIVAN),41 although gene-environment interactions appear to be necessary for the development of overt kidney disease. Papeta et al. found that the introduction of HIV to mice with genetic susceptibility to HIVAN led to alterations in the expression of specific proteins important in maintaining podocyte structure and function.42 Collectively, these in vitro and animal studies suggest that host APOL1 genotype may interact synergistically with HIV to modulate pathways involved in podocyte integrity.
In humans, the contributions of the APOL1 genotype and HIV to the development and progression of glomerular disease have been studied in kidney biopsy series. In HIV-infected African Americans with non-HIVAN pathology, Fine et al. found that number of APOL1 risk alleles correlated with the type of non-HIVAN histopathology, and with risk for end-stage renal disease (ESRD).43 However, among individuals with biopsy-confirmed HIVAN, Atta et al. reported no differences in renal pathology or risk for ESRD by APOL1 genotype. The discordant findings of these studies may reflect the heterogeneity of HIV-related kidney disease in African-Americans, and highlight the need for larger studies that evaluate pathophysiologic mechanisms mediating APOL1-associated kidney risk.
Of note, because APOL1 has been detected in vascular endothelium,10 albuminuria among individuals with APOL1 risk variants could signify the presence of subclinical vascular disease. Albuminuria is an established risk factor for cardiovascular disease, even among individuals with preserved eGFR.44 Recent studies evaluating the associations of the APOL1 genotype with cardiovascular outcomes have yielded conflicting results.45 Among African-Americans in the Jackson Heart Study and the Women's Health Initiative, 2 APOL1 risk alleles were associated with a 2-fold greater risk of atherosclerotic cardiovascular disease.46 By contrast, Langefeld et al. found no association between APOL1 risk variants and prevalent cardiovascular disease, in African-Americans enrolled in the Systolic Blood Pressure Intervention Trial,47 while Freedman et al. reported lower risk for subclinical atherosclerosis and death in association with the APOL1 risk genotype, among African-Americans in the Diabetes Heart Study.48 Understanding the interplay between APOL1 risk genotype, HIV suppression, kidney injury, and the development of cardiovascular disease through future studies may have substantial implications for the aging HIV-infected population.
The findings of the present study do not support a strong pathogenic role for APOL1 risk variants in tubular injury and dysfunction among HIV-infected African-Americans with well-preserved kidney function. However, tubular injury may play a more prominent role in the absence of HIV infection, or at more advanced stages of APOL1-associated kidney disease. For instance, in the Focal Segmental Glomerulosclerosis Clinical Trial, in which persons with HIV were excluded, Kopp et al. found that individuals with the high-risk APOL1 genotype had more extensive tubular atrophy and fibrosis on kidney biopsy.49 Furthermore, in African-Americans with “hypertension-associated” nephropathy, Larsen et al. reported higher prevalence of tubular atrophy and microcystic dilatation on kidney biopsy samples of individuals with 2 vs 0/1 APOL1 risk alleles.50 Notably, participants of that study had advanced kidney disease, with a mean serum creatinine of 4.3 mg/dl among individuals with 2 APOL1 risk alleles. Additional studies are needed to validate these findings, and to investigate the contributions of tubulointerstitial damage to prognosis at later stages of APOL1-associated kidney disease.
Our study has several limitations. First, as a study of women, the results are not generalizable to men, although there is currently no evidence to suggest a gender-based interaction between APOL1 genotype and kidney disease risk. Second, as a cross-sectional study nested within a cohort, our study is subject to selection bias due to study dropout or competing events, and we cannot make conclusions regarding causality. Third, almost half of the HIV-infected African-American women in the WIHS were excluded from initial analyses, due to missing APOL1 genotype or urine biomarker measurements, which could reduce power or bias the results. However, our findings remained robust when we repeated the analyses, using multiple imputation to include the participants with missing data. Fourth, urine samples were collected over a decade prior to biomarker measurement, so degradation over time may have contributed to the null associations between APOL1 genotype and tubular biomarkers. Alternatively, the tubular injury markers may lack sensitivity for detection of tubular dysfunction at very early stages. However, these urine biomarkers had strong associations with subsequent eGFR decline and mortality risk.14,15 Finally, despite our adjusting for multiple potential confounders, the possibility of residual confounding remains.
In summary, among HIV-infected women of African ancestry, APOL1-associated kidney injury appears to localize to the glomerulus, rather than the tubules. Additionally, the APOL1 risk genotype was strongly predictive of kidney function decline. Future studies should evaluate the pathophysiologic mechanisms by which variant forms of APOL1 result in glomerular injury, as well as targeted therapies to slow progression of APOL1-mediated kidney disease.
Supplementary Material
Figure S1: Distributions of urine KIM-1, NGAL, and A1M, stratified by number of APOL1 risk alleles.
Table S1: Demographic and clinical characteristics, stratified by inclusion/exclusion in APOL1 substudy.
Table S2: Proportion of missing data for variables included in multiple imputation model.
Table S3: Adjusted associations of APOL1 risk alleles with biomarkers using multiple imputation.
Table S4: Associations of APOL1 risk alleles with kidney function outcomes using multiple imputation.
Table S5: Urine biomarker levels in women with eGFR > 60, stratified by number of APOL1 risk alleles.
Table S6: Adjusted associations of APOL1 risk alleles with biomarkers among women with eGFR > 60.
Acknowledgments
This paper is dedicated to the memory of our colleague, Dr WH Linda Kao, and her pioneering work on the genetic basis of CKD.
Support: This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (1 K23DK08131) and through a mini-grant provided by the National Kidney Foundation of Maryland (both to Dr Estrella). Data in this manuscript were collected by the WIHS. The WIHS is funded primarily by the National Institute of Allergy and Infectious Diseases, with additional co-funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development, the National Cancer Institute (NCI), the National Institute on Drug Abuse, and the National Institute on Mental Health (NIMH). Targeted supplemental funding for specific projects is also provided by the National Institute of Dental and Craniofacial Research, the National Institute on Alcohol Abuse and Alcoholism, the National Institute on Deafness and other Communication Disorders, and the NIH Office of Research on Women's Health. The contents of this publication are solely the responsibility of the authors and do not represent the official views of the National Institutes of Health (NIH). WIHS data collection is also supported by UL1-TR000004 (University of California, San Francisco, Clinical and Translational Science Award [CTSA]) and UL1-TR000454 (Atlanta CTSA). The WIHS Principal Investigators are as follows: UAB-MS WIHS (Michael Saag, Mirjam-Colette Kempf, Deborah Konkle-Parker), U01-AI-103401; Atlanta WIHS (Ighovwerha Ofotokun, Gina Wingood), U01-AI-103408; Bronx WIHS (Kathryn Anastos), U01-AI-035004; Brooklyn WIHS (Howard Minkoff, Deborah Gustafson), U01-AI-031834; Chicago WIHS (Mardge Cohen), U01-AI-034993; Metropolitan Washington WIHS (Mary Young), U01-AI-034994; Miami WIHS (Margaret Fischl, Lisa Metsch), U01-AI-103397; UNC WIHS (Adaora Adimora), U01-AI-103390; Connie Wofsy Women's HIV Study, Northern California (Ruth Greenblatt, Bradley Aouizerat, Phyllis Tien), U01-AI-034989; WIHS Data Management and Analysis Center (Stephen Gange, Elizabeth Golub), U01-AI-042590; Southern California WIHS (Alexandra Levine, Marek Nowicki), U01-HD-032632 (WIHS I–WIHS IV).
Footnotes
N SECTION: Because the Editor-in-Chief recused himself from consideration of this article, the Deputy Editor (Daniel E. Weiner, MD, MS) served as Acting Editor-in-Chief. Details of the journal's procedures for potential editor conflicts are given in the Information for Authors & Editorial Policies.
Contributions: research idea and study design: VJ, RS, MGS, MME; data acquisition: MGS, MB, MHC, MN, AS, MY, PCT, CRP, MME; data analysis/interpretation: VJ, RS, CRP, RSP, MGS, MME; statistical analysis: RS; supervision or mentorship: MGS, RSP, CRP, MME. WHLK died before the manuscript was submitted; MME affirms that she contributed to data acquisition and vouches for her coauthorship status; all other authors approved the final author list. Except as noted, each author contributed important intellectual content during manuscript drafting or revision and accepts accountability for the overall work by ensuring that questions pertaining to the accuracy or integrity of any portion of the work are appropriately investigated and resolved. VJ takes responsibility that this study has been reported honestly, accurately, and transparently; that no important aspects of the study have been omitted, and that any discrepancies from the study as planned have been explained.
Financial Disclosure: The authors declare that they have no other relevant financial interests.
Supplementary Material: Note: The supplementary material accompanying this article (doi:_________) is available at www.ajkd.org
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Choi AI, Rodriguez RA, Bacchetti P, Bertenthal D, Volberding PA, O'Hare AM. The impact of HIV on chronic kidney disease outcomes. Kidney international. 2007;72(11):1380–1387. doi: 10.1038/sj.ki.5002541. [DOI] [PubMed] [Google Scholar]
- 2.Jotwani V, Li Y, Grunfeld C, Choi AI, Shlipak M. Risk factors for end-stage renal disease in HIV-infected individuals: traditional and HIV-related factors. AJKD. 2012;59(5):628–35. doi: 10.1053/j.ajkd.2011.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lucas GM, Lau B, Atta MG, Fine DM, Keruly J, Moore RD. Chronic kidney disease incidence, and progression to end-stage renal disease, in HIV-infected individuals: a tale of two races. The Journal of infectious diseases. 2008;197(11):1548–1557. doi: 10.1086/587994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsu CY, Lin F, Vittinghoff E, Shlipak MG. Racial differences in the progression from chronic renal insufficiency to end-stage renal disease in the United States. Journal of the American Society of Nephrology: JASN. 2003;14(11):2902–2907. doi: 10.1097/01.asn.0000091586.46532.b4. [DOI] [PubMed] [Google Scholar]
- 5.Kopp JB, Smith MW, Nelson GW, et al. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet. 2008;40(10):1175–1184. doi: 10.1038/ng.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kao WH, Klag MJ, Meoni LA, et al. MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat Genet. 2008;40(10):1185–1192. doi: 10.1038/ng.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329(5993):841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tzur S, Rosset S, Shemer R, et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Human genetics. 2010;128(3):345–350. doi: 10.1007/s00439-010-0861-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F, et al. Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science. 2005;309(5733):469–472. doi: 10.1126/science.1114566. [DOI] [PubMed] [Google Scholar]
- 10.Madhavan SM, O'Toole JF, Konieczkowski M, Ganesan S, Bruggeman LA, Sedor JR. APOL1 localization in normal kidney and nondiabetic kidney disease. Journal of the American Society of Nephrology : JASN. 2011;22(11):2119–2128. doi: 10.1681/ASN.2011010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma L, Shelness GS, Snipes JA, et al. Localization of APOL1 Protein and mRNA in the Human Kidney: Nondiseased Tissue, Primary Cells, and Immortalized Cell Lines. Journal of the American Society of Nephrology : JASN. 2014 doi: 10.1681/ASN.2013091017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jotwani V, Scherzer R, Abraham A, et al. Does HIV infection promote early kidney injury in women? Antiviral therapy. 2014;19(1):79–87. doi: 10.3851/IMP2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jotwani V, Scherzer R, Abraham A, et al. Association of Urine alpha1-Microglobulin with Kidney Function Decline and Mortality in HIV-Infected Women. Clin J Am Soc Nephrol. 2015;10(1):63–73. doi: 10.2215/CJN.03220314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shlipak MG, Scherzer R, Abraham A, et al. Urinary Markers of Kidney Injury and Kidney Function Decline in HIV-Infected Women: Biomarkers and Kidney Decline. Journal of acquired immune deficiency syndromes. 2012;61(5):565–73. doi: 10.1097/QAI.0b013e3182737706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peralta C, Scherzer R, Grunfeld C, et al. Urinary biomarkers of kidney injury are associated with all-cause mortality in the Women's Interagency HIV Study (WIHS) HIV Med. 2014 May;15(5):291–300. doi: 10.1111/hiv.12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barkan SE, Melnick SL, Preston-Martin S, et al. The Women's Interagency HIV Study. WIHS Collaborative Study Group. Epidemiology. 1998;9(2):117–125. [PubMed] [Google Scholar]
- 17.Bacon MC, von Wyl V, Alden C, et al. The Women's Interagency HIV Study: an observational cohort brings clinical sciences to the bench. Clin Diagn Lab Immunol. 2005;12(9):1013–1019. doi: 10.1128/CDLI.12.9.1013-1019.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith MW, Patterson N, Lautenberger JA, et al. A high-density admixture map for disease gene discovery in african americans. American journal of human genetics. 2004;74(5):1001–1013. doi: 10.1086/420856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frasco MA, Mack WJ, Van Den Berg D, et al. Underlying genetic structure impacts the association between CYP2B6 polymorphisms and response to efavirenz and nevirapine. Aids. 2012;26(16):2097–2106. doi: 10.1097/QAD.0b013e3283593602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 21.Price AL, Zaitlen NA, Reich D, Patterson N. New approaches to population stratification in genome-wide association studies. Nat Rev Genet. 2010;11(7):459–463. doi: 10.1038/nrg2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chaturvedi S, Farmer T, Kapke GF. Assay validation for KIM-1: human urinary renal dysfunction biomarker. Int J Biol Sci. 2009;5(2):128–134. doi: 10.7150/ijbs.5.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bennett M, Dent CL, Ma Q, et al. Urine NGAL predicts severity of acute kidney injury after cardiac surgery: a prospective study. Clinical journal of the American Society of Nephrology : CJASN. 2008;3(3):665–673. doi: 10.2215/CJN.04010907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Odden MC, Scherzer R, Bacchetti P, et al. Cystatin C level as a marker of kidney function in human immunodeficiency virus infection: the FRAM study. Archives of internal medicine. 2007;167:2213–2219. doi: 10.1001/archinte.167.20.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inker LA, Eckfeldt J, Levey AS, et al. Expressing the CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) cystatin C equations for estimating GFR with standardized serum cystatin C values. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2011;58(4):682–684. doi: 10.1053/j.ajkd.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castro KG, W J, Slutsker L, et al. 1993 Revised Classification System for HIV Infection and Expanded Surveillance Case Definition for AIDS Among Adolescents and Adults: Center for Disease Control and Prevention. 1992 [PubMed] [Google Scholar]
- 27.Huber P. The behavior of maximum likelihood estimates under nonstandard conditions. Proceedings of the Fifth Berkeley Symposium on Mathematical Statistics and Probability. 1967;1:221–233. [Google Scholar]
- 28.White H. A heteroskedasticity-consistent covariance matrix estimator and a direct test for heteroskedasticity. Econometrica. 1980;48:817–830. [Google Scholar]
- 29.Akerstrom B, Logdberg L, Berggard T, Osmark P, Lindqvist A. alpha(1)-Microglobulin: a yellow-brown lipocalin. Biochimica et biophysica acta. 2000;1482(1-2):172–184. doi: 10.1016/s0167-4838(00)00157-6. [DOI] [PubMed] [Google Scholar]
- 30.Weber MH, Verwiebe R. Alpha 1-microglobulin (protein HC): features of a promising indicator of proximal tubular dysfunction. Eur J Clin Chem Clin Biochem. 1992;30(10):683–691. [PubMed] [Google Scholar]
- 31.Zou G. A modified poisson regression approach to prospective studies with binary data. American journal of epidemiology. 2004;159(7):702–706. doi: 10.1093/aje/kwh090. [DOI] [PubMed] [Google Scholar]
- 32.Robins JM, Finkelstein DM. Correcting for noncompliance and dependent censoring in an AIDS Clinical Trial with inverse probability of censoring weighted (IPCW) log-rank tests. Biometrics. 2000;56(3):779–788. doi: 10.1111/j.0006-341x.2000.00779.x. [DOI] [PubMed] [Google Scholar]
- 33.Gilks WR, Richardson S, Spiegehalter DJ. Markov chain Monte Carlo in practice. London: Chapman & Hall; 1996. [Google Scholar]
- 34.Friedman DJ, Kozlitina J, Genovese G, Jog P, Pollak MR. Population-based risk assessment of APOL1 on renal disease. Journal of the American Society of Nephrology : JASN. 2011;22(11):2098–2105. doi: 10.1681/ASN.2011050519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Freedman BI, Langefeld CD, Turner J, et al. Association of APOL1 variants with mild kidney disease in the first-degree relatives of African American patients with non-diabetic end-stage renal disease. Kidney international. 2012;82(7):805–811. doi: 10.1038/ki.2012.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Estrella MM, Wyatt CM, Pearce CL, et al. Host APOL1 genotype is independently associated with proteinuria in HIV infection. Kidney international. 2013;84(4):834–840. doi: 10.1038/ki.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vanhollebeke B, Pays E. The function of apolipoproteins L. Cell Mol Life Sci. 2006;63(17):1937–1944. doi: 10.1007/s00018-006-6091-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wan G, Zhaorigetu S, Liu Z, Kaini R, Jiang Z, Hu CA. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. The Journal of biological chemistry. 2008;283(31):21540–21549. doi: 10.1074/jbc.M800214200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartleben B, Godel M, Meyer-Schwesinger C, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. The Journal of clinical investigation. 2010;120(4):1084–1096. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lan X, Jhaveri A, Cheng K, et al. APOL1 Risk Variants Enhance Podocyte Necrosis through Compromising Lysosomal Membrane Permeability. American journal of physiology Renal physiology. 2014;307(3):F326–36. doi: 10.1152/ajprenal.00647.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Husain M, D'Agati VD, He JC, Klotman ME, Klotman PE. HIV-1 Nef induces dedifferentiation of podocytes in vivo: a characteristic feature of HIVAN. Aids. 2005;19(17):1975–1980. doi: 10.1097/01.aids.0000191918.42110.27. [DOI] [PubMed] [Google Scholar]
- 42.Papeta N, Chan KT, Prakash S, et al. Susceptibility loci for murine HIV-associated nephropathy encode trans-regulators of podocyte gene expression. The Journal of clinical investigation. 2009;119(5):1178–1188. doi: 10.1172/JCI37131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fine DM, Wasser WG, Estrella MM, et al. APOL1 risk variants predict histopathology and progression to ESRD in HIV-related kidney disease. Journal of the American Society of Nephrology : JASN. 2012;23(2):343–350. doi: 10.1681/ASN.2011060562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chronic Kidney Disease Prognosis C. Matsushita K, van der Velde M, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet. 2010;375(9731):2073–2081. doi: 10.1016/S0140-6736(10)60674-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lipkowitz MS. Apolipoprotein L1: from obscurity to consistency to controversy. Kidney international. 2015;87(1):14–17. doi: 10.1038/ki.2014.319. [DOI] [PubMed] [Google Scholar]
- 46.Ito K, Bick AG, Flannick J, et al. Increased burden of cardiovascular disease in carriers of APOL1 genetic variants. Circulation research. 2014;114(5):845–850. doi: 10.1161/CIRCRESAHA.114.302347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langefeld CD, Divers J, Pajewski NM, et al. Apolipoprotein L1 gene variants associate with prevalent kidney but not prevalent cardiovascular disease in the Systolic Blood Pressure Intervention Trial. Kidney international. 2015;87(1):169–175. doi: 10.1038/ki.2014.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Freedman BI, Langefeld CD, Lu L, et al. APOL1 associations with nephropathy, atherosclerosis, and all-cause mortality in African Americans with type 2 diabetes. Kidney international. 2015;87(1):176–181. doi: 10.1038/ki.2014.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kopp JB, Winkler CA, Zhao X, et al. Clinical Features and Histology of Apolipoprotein L1-Associated Nephropathy in the FSGS Clinical Trial. J Am Soc Nephrol. 2015 doi: 10.1681/ASN.2013111242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Larsen CP, Beggs ML, Saeed M, et al. Histopathologic findings associated with APOL1 risk variants in chronic kidney disease. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2015;28(1):95–102. doi: 10.1038/modpathol.2014.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Distributions of urine KIM-1, NGAL, and A1M, stratified by number of APOL1 risk alleles.
Table S1: Demographic and clinical characteristics, stratified by inclusion/exclusion in APOL1 substudy.
Table S2: Proportion of missing data for variables included in multiple imputation model.
Table S3: Adjusted associations of APOL1 risk alleles with biomarkers using multiple imputation.
Table S4: Associations of APOL1 risk alleles with kidney function outcomes using multiple imputation.
Table S5: Urine biomarker levels in women with eGFR > 60, stratified by number of APOL1 risk alleles.
Table S6: Adjusted associations of APOL1 risk alleles with biomarkers among women with eGFR > 60.