

Abstract
Toxicology is the highly interdisciplinary field studying the adverse effects of chemicals on living organisms. It requires sensitive tools to detect such effects. After their initial implementation during the 1990s, single-molecule fluorescence detection tools were quickly recognized for their potential to contribute greatly to many different areas of scientific inquiry. In the intervening time, technical advances in the field have generated ever-improving spatial and temporal resolution, and have enabled the application of single-molecule fluorescence to increasingly complex systems, such as live cells. In this review, we give an overview of the optical components necessary to implement the most common versions of single-molecule fluorescence detection. We then discuss current applications to enzymology and structural studies, systems biology, and nanotechnology, presenting the technical considerations that are unique to each area of study, along with noteworthy recent results. We also highlight future directions that have the potential to revolutionize these areas of study by further exploiting the capabilities of single-molecule fluorescence microscopy.
Keywords: Fluorescence, single-molecule, microscopy, enzymology, systems biology, nanotechnology
Introduction
In late 1959, American physicist and Nobel laureate Richard Feynman envisioned observing and arranging atoms one by one (Feynmann 1961), a vision that became reality when optical single molecule detection came into being in the 1990s. Inspired by the adage that “seeing is believing,” single-molecule microscopy tools were maturely developed in the late 1990s to allow visualization of single molecules with a high degree of precision. As we highlighted in a previous review (Walter et al. 2008), single-molecule study is particularly relevant as it can provide detailed information about molecular structure, localization, assembly and reaction mechanisms. Specifically, single-molecule microscopy reveals the heterogeneity of the sample (McDowell et al. 2010), affords precise localization and counting of molecules (Pitchiaya et al. 2012), allows for the sensitive detection of low numbers of molecules in cells without the need for enrichment (Pitchiaya et al. 2014), enables quantitative measurement of kinetics (Krishnan et al. 2013), reveals rare or intermediate species, and enables miniaturization and multiplexing of biological assays. In the last 30 years, the field of fluorescence microscopy has been revolutionized by techniques such as single-molecule fluorescence resonance energy transfer (smFRET), a wide array of super-resolution microscopy approaches, and intracellular single-molecule detection. As a result, single-molecule microscopy is contributing at ever-increasing rates to fields as diverse as enzymology, structural biology, systems biology and nanotechnology (Banerjee and Deniz 2014; Joo et al. 2008; Li and Xie 2011; Wildenberg et al. 2011; Xia et al. 2013). Each of these fields is relevant to toxicology as the discipline that broadly studies the adverse effects of chemicals on living organisms. Toxicology requires sensitive tools to detect such effects, and single molecule microscopy offers the ultimate level of chemical sensitivity.
This review will focus on fluorescence-based single-molecule detection, which offers numerous advantages beyond the ability to visualize individual molecules directly. The use of multiple fluorophores with different excitation and emission properties allows multiplexing of signals within one sample and optical setup, while the ability to observe hundreds of molecules in a single field of view allows statistically robust datasets to be accumulated rapidly. In addition, the set of single molecule fluorescence-based observables is constantly expanding. For example, recent work has demonstrated single-molecule fluorescence detected linear dichroism (Phelps et al. 2013), which is sensitive to the orientation of the absorbing and emitting probe, and has extended single-molecule detection into the ultraviolet range of the spectrum (Alemán et al. 2014), allowing powerful probes such as fluorescent nucleic acid base analogues to be utilized. With such a powerful toolkit, scientific questions that are of immense scope and depth can be addressed through single-molecule fluorescence. The layout of this review is as follows. First, we discuss the optical setups necessary for single-molecule fluorescence detection, focusing on those aspects that are shared by the most common approaches rather than on the details of any single approach. Second, we discuss current applications of single molecule fluorescence to enzymology and structural biology, systems biology, and nanotechnology, focusing on technical considerations unique to these applications as well as on recent results in these fields empowered by single-molecule fluorescence. We end with a preview of advances to come.
Single-molecule fluorescence optical setups
The five basic components of single-molecule optical detection are the microscope, light source(s), optical detector(s), probe(s) and sample (Lackowickz 2007; Walter et al. 2008). The requirements for the probes and sample have been reviewed in detail elsewhere (Ha and Tinnefeld 2012; Pitchiaya et al. 2014; Roy et al. 2008; Walter et al. 2008; Wang et al. 2014; Xia et al. 2013), so here we focus on the requirements of the optical system.
The light source and associated optics
While single-molecule detection can be accomplished using a broadband light source such as a halogen lamp as the excitation source, continuous-wave (CW) lasers are preferred because their high powers and narrow bandwidths allow specific fluorophores to be efficiently and selectively excited. Diode lasers and diode-pumped solid-state lasers are particularly popular because of their compact size and the wide range of wavelengths available. One of the most common lasers used in single-molecule fluorescence measurements is diode-pumped Nd:YVO4, which lases at 1064 nm, typically followed by frequency doubling to 532 nm. Pulsed lasers are used for specialized varieties of single-molecule detection, such as fluorescence lifetime imaging and two-photon absorption microscopy. Continuous variation of the laser power can be achieved either with a combination of a half-wave plate and polarizer or with a continuously variable neutral density filter. A challenge common to all single-molecule microscopy is the elimination of background signal, which is usually accomplished by confining the excitation volume to a small region of the sample. While countless variations exist, one of the most common solutions to this problem is total internal reflection fluorescence microscopy (TIRFM) (Fig. 1a, b). In TIRFM, a laser beam is coupled into an object of high refractive index above the critical angle for total internal reflection. In the sample, which resides on the far side of the interface at which TIR occurs, an evanescent field is generated that penetrates only ~50–150 nm, depending on the incidence angle and the relative refractive indices at the interface (Axelrod et al. 1984; Walter et al. 2008). Thus, only molecules residing close to the interface can be excited, avoiding background fluorescence from the bulk of the sample. TIRFM is typically accomplished by coupling the laser beam into either a prism (Fig. 1a) or a microscope objective (Fig. 1b). In the former, the prism sits on a microscope slide and the objective contacts the opposite face of a sample chamber that is constructed on the slide. In the latter, both excitation and detection occur through the same objective, yielding the advantage that the opposite face of the sample chamber has no requirements for shape, size or optical properties.
Fig. 1.

Single-molecule fluorescence optical setups and data collection. a Prism-based TIRF optical setup. b Objective-based TIRF optical setup. c Example experimental setup and fluorescence intensity trace for an experiment investigating binding and unbinding of fluorescently-labeled probes to an immobilized oligonucleotide. d Example experimental setup and data for an intracellular single-molecule high resolution localization and counting (iSHiRLoC) experiment investigating microRNA diffusion in live cells (Johnson-Buck and Walter 2014; Pitchiaya et al. 2013)
The detector and associated optics
Single-molecule fluorescence detection is most often accomplished by imaging. In vivo, this allows the locations of molecules to be determined relative to the higher-order structures of cells, while in vitro it allows one to observe a large ensemble of individual immobilized molecules, obtaining statistically reliable datasets in a minimum amount of time. The most common types of detectors used for single-molecule fluorescence imaging are charge coupled device (CCD) cameras, particularly intensified CCDs (iCCDs) and electron-multiplied CCDs (emCCDs). While emCCDs generally offer higher quantum efficiency than iCCD photocathodes, iCCDs (in which each pixel operates similarly to a photomultiplier tube) offer amplification prior to the introduction of noise sources such as image readout. Depending on signal-to-noise ratio and excitation power, setups utilizing these cameras can readily achieve time resolutions on the order of tens of milliseconds. If one is concerned with achieving the highest-possible time resolution, typically down to tens of microseconds (König et al. 2013; Phelps et al. 2013), one may sacrifice the advantages of a large field of view and instead image one molecule at a time onto a single-photon counting avalanche photodiode (APD). This is accomplished by placing a pinhole in the emission path to allow only the signal from one molecule to reach the detector.
To maximize the detection of true signals and rejection of spurious signals, the optical path from excitation to detection must be optimized. For optimal stability and rejection of background, this region of the optical setup should be thoroughly shielded from dust, mechanical disturbance and outside light. High-quality optical filters are required to transmit the emission of the fluorophore in use while rejecting scattered laser light. To take advantage of multiplexed excitation and detection, it is necessary to separate signals from different fluorescent species (the donor and acceptor in smFRET, for example). By utilizing dichroics that transmit the emission of one of the fluorescent species while reflecting the emission of the other, their fluorescence signals can be separated and projected onto different cameras (Fig. 1b) or different parts of a single camera’s active area (Fig. 1a). An especially compact implementation is the Sagnac interferometer, which requires only a single dichroic and two mirrors, with the same dichroic separating the two signals and re-collimating them onto parallel paths to be directed at adjacent regions of a single camera (Lee et al. 2013).
Single-molecule fluorescence microscopy is commonly performed on glass or quartz slides using immobilized fluorophore-labeled biomolecules, but can also be performed in living bacteria or eukaryotic cells. The in vitro approach (Fig. 1c) permits the sample conditions to be strictly controlled by the use of purified components, allowing for low background noise and slow photobleaching. For in vivo experiments (referring here mostly to those in single live cells), the cell largely determines the sample conditions, creating new challenges and opportunities (Fig. 1d). Recent advancements in direct labeling strategies and genetic engineering of fluorescent protein fusions, along with improved single molecule detection using superresolution fluorescence imaging methods, such as photoactivatable localization microscopy (PALM) and stochastic optical reconstruction microscopy (STORM), have facilitated the transition to direct visualization of fluorescently-labeled biomolecules in living or fixed cells (Li and Xie 2011; Patterson et al. 2010; Pitchiaya et al. 2014). In vivo single-molecule fluorescence microscopy is revolutionizing how biomolecules are detected in their natural environment, wherein almost any biological phenomenon can be investigated, including microRNA-protein assembly (Pitchiaya et al. 2012), mRNA transport (Grunwald et al. 2011; Ma et al. 2013; Park et al. 2014), splicing (Vargas et al. 2011; Waks et al. 2011), gene expression (Martin et al. 2013; Raj et al. 2006) and protein assembly (Xu et al. 2013). In addition to biological applications, single-molecule fluorescence microscopy techniques have contributed to great progress in several other research areas. We will exemplify the versatility of single-molecule fluorescence techniques by discussing their application to nucleic acids in particular.
The specifics of the ideal optical setup, probe(s) and sample(s) will vary among different applications of single-molecule fluorescence detection. In the remainder of this review, we discuss the application of single-molecule fluorescence to enzymology and mechanistic studies, systems biology, and nanotechnology. We highlight the technical considerations relevant to each of these applications, and present recent work that highlights developments in these fields.
Enzymology and structural biology through single-molecule fluorescence
Enzyme function derives from the unique combination of structure and dynamics, and single-molecule fluorescence techniques are uniquely suited for the study of enzymology and structural biology for a number of reasons. The catalysis of a chemical reaction is necessarily a dynamic process, involving at a minimum the steps of substrate binding, catalysis and product release, often involving conformational rearrangements (understood here as a domain of structural biology). The study of these processes is therefore greatly enhanced by techniques like single-molecule fluorescence that report on structure and function in real time, essentially allowing one to ‘watch’ an enzymatic process as it occurs without the need for synchronization of molecules to reach a detectable ensemble-averaged signal. In addition, complex catalysts like proteins and macromolecular machines are often heterogeneous, and single-molecule techniques enable the differentiation of different subpopulations, as exemplified in studies of functionally different folded species of the hairpin ribozyme (Ditzler et al. 2008; Zhuang et al. 2002) as well as other RNA (Marek et al. 2011), DNA (Hyeon et al. 2012) and protein molecules (Liu et al. 2013). Finally, single-molecule fluorescence offers large flexibility in experimental observables, such as through the choice and placement of probes, allowing experiments to be fine-tuned to address the scientific question of interest.
One of the most common approaches to enzymology is single-molecule fluorescence resonance energy transfer (smFRET, Fig. 2a) (Roy et al. 2008). In this approach, a pair of donor and acceptor fluorophores is placed strategically so that specific states of the system of interest place them in close proximity to each other, allowing efficient energy transfer from the excited donor to the acceptor (a ‘high FRET’ state), while other states place them far apart (a ‘low FRET’ state). For typical donor-acceptor pairs, FRET is sensitive to donor-acceptor distance changes over the range of ~20–80 Ångstroms, making it well suited for the study of local conformational changes, or global conformational changes in small systems. Examples of the use of smFRET in enzymology include the sampling of active conformations by small ribozymes (Bokinsky et al. 2003; Ditzler et al. 2008; Liu et al. 2007; McDowell et al. 2010; Pereira et al. 2008; Rueda et al. 2004; Silva and Walter 2009; Zhuang et al. 2002), and conformational changes in the pre-mRNA within the spliceosome (Abelson et al. 2010; Krishnan et al. 2013). In addition, the conformations sampled by riboswitches in ligand-free and ligand-bound states were investigated (Suddala et al. 2013). For large complexes, colocalization single-molecule spectroscopy (CoSMoS, Fig. 2b) (Hoskins et al. 2011) allows for the determination of binding and dissociation times and kinetics of multiple components, labeled with different fluorescent dyes to enable separate detection. This technique has, for example, been applied to the spliceosome to study the binding and dissociation of the U1, U2 and U5 snRNPs (Hoskins et al. 2011). Colocalization and superresolution fluorescence microscopy techniques (discussed in more detail in the systems biology and nanotechnology sections) are also well suited to report on higher-order structure.
Fig. 2.

Single-molecule approaches used in enzymology and structural biology. a Single-molecule FRET, with the PreQ1 riboswitch as an example. b Colocalization single-molecule spectroscopy, applied to the assembly of the spliceosome. c Tracking DNA polymerase activity using fluorescently-labeled NTPs. d Visualizing enzymatic activity using fluorogenic substrates (Eid et al. 2009; Gorris et al. 2007; Hoskins et al. 2011; Suddala et al. 2013)
While smFRET and CoSMoS report on the conformations and assembly states of enzymes or macromolecular complexes, other approaches are available that report directly on substrate binding and the catalysis of chemical reactions. Fluorescent NTP analogues have been used to directly visualize the binding and dissociation of NTPs (Fig. 2c), as was done to study DNA replication (Eid et al. 2009). These experiments often require additional considerations to prevent the high concentrations of NTPs required (~µM) from generating an excessive fluorescence background signal. To address this challenge, techniques have been developed that utilize zero mode waveguides to confine the detection area to a small volume around a molecule of interest (Eid et al. 2009; Elting et al. 2013). Arguably the most direct observation of enzyme catalysis is permitted by fluorogenic substrates or cofactors (Fig. 2d), which were used in the context of a flavoenzyme in some of the first reported single-molecule enzymology studies (Lu et al. 1998; Lu and Liu 2006). This approach is confined to the increasing number of enzymes for which such substrates exist, with some of the most popular being resorufin-based compounds such as resorufin-β-D-galactopyranoside (RGP), a fluorogenic substrate of β-galactosidase (English et al. 2006). Finally, the toolbox of spectroscopic observables that can be measured under physiological conditions at the single-molecule level continues to expand, with recent reports of single-molecule absorption (Chong et al. 2010; Gaiduk et al. 2010; Kukura et al. 2010) and fluorescence detected linear dichroism (smFLD) (Phelps et al. 2013), which has been applied to the study of bacteriophage T4 helicase and primase. In addition, single-molecule studies using UV excitation have enabled observation of local nucleic acid structure through the use of fluorescent base analogues (Alemán et al. 2014). The use of an infrared laser to induce temperature jumps, combined with single-molecule FRET (termed laser-assisted single-molecule refolding, LASR), yields new insight into the folding and unfolding of macromolecules (Zhao et al. 2010).
The wide variety of recent studies in single-molecule enzymology and structural biology extend from individual proteins and ribozymes to megaDalton macromolecular machines. The next section will highlight some specific studies that provide examples of the expanding scope and depth of single-molecule fluorescence studies, applied to a variety of RNA and protein systems.
The hammerhead ribozyme
Small catalytic RNAs, RNA enzymes or ribozymes have provided a fertile platform for single-molecule fluorescence studies due to their favorable size and ease of labeling with organic fluorophores. In the hammerhead ribozyme, which is found in satellite RNAs associated with certain plant viruses where it has a critical enzymatic function during satellite RNA replication (Walter and Perumal 2009), the conformational dynamics leading to catalysis were investigated (McDowell et al. 2010). This work has both structural and catalytic components, as they investigated the relationship between loop-loop tertiary interactions and the sampling of catalytically active conformations. By placing a FRET donor and acceptor on the ends of Stem I and Stem II (Fig. 3a), changes in the proximity of loops within each of these stems could be monitored through changes in FRET. The extended hammerhead ribozyme used in this work exhibited at least four distinguishable FRET states, in contrast to fewer observed in the minimal hammerhead ribozyme that lacks these loops. A state with a FRET efficiency of ~0.7 became significantly populated only at high magnesium ion concentration, and only in the catalytically active wild-type ribozyme (Fig. 3b), suggesting that this state corresponded to the catalytically active conformation. This state was absent or less populated in mutant ribozymes in which the tertiary loop-loop interactions were diminished. Nevertheless, this state was visited only transiently even under optimum conditions for catalysis. Molecular dynamics simulations were performed on ribozymes with intact, partially disrupted, and fully disrupted loop-loop interactions. It was found that both of these disruptions significantly reduced the ribozyme’s sampling of favorable, near-180° in-line attack angles, consistent with the lower chemical reactivity of ribozymes with disrupted loop-loop interactions (McDowell et al. 2010).
Fig. 3.
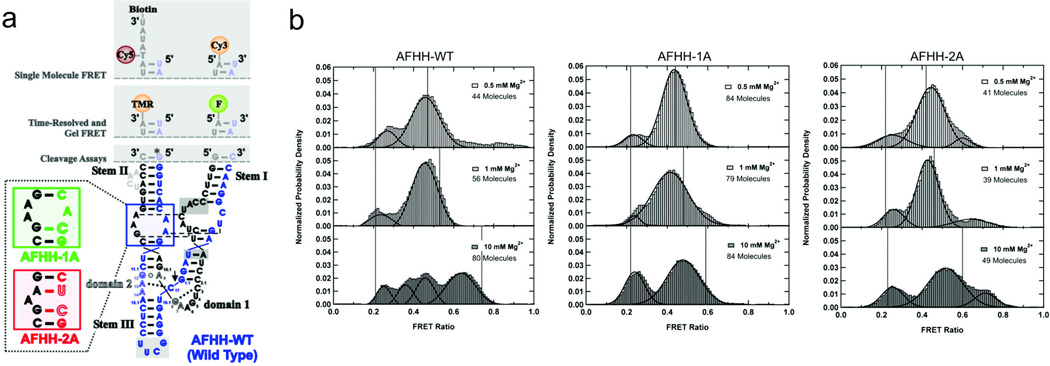
Single-molecule FRET investigation of the hammerhead ribozyme. a Full-length hammerhead ribozyme used in the studies discussed. The labeling sites for single-mol ecule FRET are indicated at the top, and the mutations used to disrupt loop-loop interactions are shown in the bottom-left. b Single-molecule FRET histograms showing the effect of magnesium concentration and loop mutations on the conformations adopted by wild-type (left panel) and two mutant (center and right panels) ribozymes. The active, high-FRET conformation is sampled only by the wild-type ribozyme, and only under high magnesium conditions (McDowell et al. 2010)
Pre-mRNA dynamics in splicing
While smFRET is now regularly applied to individual proteins and RNAs, its application to large macromolecular complexes has been slower due to challenges associated with complex purification and data analysis. The application of smFRET to the large RNA-protein complexes of the spliceosome (Fig. 4) (Abelson et al. 2010; Krishnan et al. 2013) therefore illustrates how the envelope can be pushed in the field. The spliceosome carries out the process of splicing by excising non-protein coding introns from precursor messenger RNAs (pre-mRNAs) and ligating together protein-coding exons. Splicing was studied using a pre-mRNA harboring the yeast Ubc4 intron, which is short enough to permit chemical synthesis while being efficiently spliced in vitro. In initial work, a donor fluorophore was placed adjacent to the 5' splice site and an acceptor fluorophore was placed near the 3' splice site, allowing changes in the proximity of these two sites to be monitored throughout the process of splicing (Abelson et al. 2010). Blocks were introduced into the splicing cycle to allow for the pre-mRNA conformation and dynamics to be monitored at well-controlled points in the cycle. Pre-mRNA was found to dynamically fold and unfold in buffer, in the absence of any spliceosomal components. Addition of yeast extract (to provide all of the necessary spliceosomal protein and RNA components) that had been depleted of ATP led to an enrichment of low-FRET states. Upon addition of +ATP extract, the pre-mRNA favored higher-FRET conformations, indicating that the 3' and 5' splice sites were brought into closer proximity. Meanwhile, a substrate with a mutation in the branch point sequence showed little change with time or ATP. This substrate would be expected to stall prior to any major steps in splicing, suggested that the changes with time and ATP observed for the wild-type substrate were indeed the result of spliceosome assembly. A major observation in this work was that for any given pair of FRET states, both forward and reverse transitions occurred, often with approximately the same rates, indicating that the spliceosome operates near equilibrium (Abelson et al. 2010). This suggests a re-evaluation of the role of ATP in splicing, implying that the numerous ATPases involved function by unlocking intrinsic fluctuations in the complex, rather than driving rearrangements forward.
Fig. 4.

smFRET studies of splicing. a The spliceosome is immobilized on a slide through an affinity tag on the NTC. b Cy3 (green) and Cy5 (red) intensity traces of spliceosomes that had been stalled by a mutation in Prp2, then chased through step 1 of chemistry by the addition of active Prp2, Spp2, ATP and Cwc25. Under these conditions, the pre-mRNA explored mostly mid- and high-FRET states. The light blue trace in the lower plot is a hidden Markov model (HMM) fit to the FRET trace (black). c Transition occupancy density plot made from the HMM fits to 156 molecules under the conditions described in b. L1, L2, M and H indicate, respectively, low-FRET states 1 and 2, a mid-FRET state, and a high-FRET state. This plot shows that under these conditions, there is a population that dynamically samples states L2, M and H, and another population that exists stably in H. d Kinetic map showing the rate constants extracted for transitions between different FRET states under conditions that stall the spliceosome before step 1 of chemistry (“B* condition”) or after step 1 (“C condition”) (Krishnan et al. 2013)
More recent work (Krishnan et al. 2013) focused specifically on the first step of chemistry in splicing, in which the branchpoint adenosine attacks the 5' splice site to generate a lariat exon intermediate. This work used a different approach to stalling and immobilizing spliceosomal complexes. Yeast extract was prepared with spliceosomes stalled before step 1 by a heat-sensitive mutation in the ATPase Prp2, and these complexes were immobilized on a slide through an affinity tag on the nineteen complex (NTC) (Fig. 4a). It was found that the stalled complexes adopted a relatively static low-FRET state. Addition of Prp2, Spp2 and ATP unlocked transitions between this low-FRET state and mid- and high-FRET states, and allowed first step chemistry to occur, albeit inefficiently. Further addition of Cwc25 greatly increases the occupation of the high-FRET state (Fig. 4b,c), and increases the efficiency of first-step splicing. These observations are consistent with the spliceosome acting as a biased Brownian ratchet as envisioned by Feynman for molecular machines operating at the nanoscale (Feynman 1963) - Prp2 unlocks thermal fluctuations between low- and high-FRET states, and Cwc25 acts as a “pawl” that captures the high-FRET state, facilitating first step chemistry (Krishnan et al. 2013).
Other groups have also applied single-molecule fluorescence spectroscopy to the study of large macromolecular machines. In Hoskins et al. (Hoskins et al. 2011), spliceosomal components such as the U1 snRNP and the NTC were labeled with fluorophores, and colocolization single-molecule spectroscopy (CoSMoS) was used to determine the order and kinetics of their assembly on the pre-mRNA substrate. Single-molecule fluorescence has also been extended to the ribosome. A variety of different labeling schemes and experimental configurations have been used to probe relative motion between the 30S and 50S subunits in initiation complexes, the roles of initiation factors in assembly of elongation-competent ribosomes, the dynamics of tRNAs and critical regions of the ribosome during elongation, and the conformational changes that result in termination (Aitken et al. 2010; Chen et al. 2012). For example, Uemura et al. (Uemura et al. 2010) used fluorescently-labeled tRNAs to track the tRNA occupation of different binding sites on the ribosome. This required the use of zero mode waveguides to permit physiological concentrations of tRNAs. In addition, work by Chen et al. (Chen et al. 2011) measured FRET between different tRNAs as well as between tRNAs and ribosomal proteins in order to track ribosome translocation.
The preQ1 riboswitch as a paradigm for mechanistic single-molecule studies
Riboswitches are structured RNA motifs that regulate the expression of up to 4% of all bacterial genes (Breaker 2011). While they themselves are not enzymes, they share many properties with enzymes such as conformational dynamics and ligand (“substrate”) binding, and different types of riboswitches regulate the activities of enzymes such as RNA polymerase and the ribosome. A 2013 study of the preQ1 riboswitch (Suddala et al. 2013) provided insight into the “activation” of partially folded RNA structures by magnesium ions and ligands, yielding results that are generally applicable to enzymes as well. The ligand preQ1, which the riboswitch responds to, is an intermediate on the biosynthetic pathway of queuosine, a derivative of guanine that is incorporated into certain tRNAs. Despite very similar sequences in the ligand binding domain, the preQ1 riboswitches in the species Bacillus subtilis (Bsu) and Thermanaerobacter tengcongensis (Tte) regulate gene expression by different mechanisms. The Bsu riboswitch operates by inducing transcription termination in the presence of preQ1, while the Tte riboswitch inhibits translation by sequestering part of the Shine-Dalgarno sequence in the presence of preQ1.
Both the Bsu and the Tte riboswitches contain a stem-loop structure (with the stem denoted P1 and the loop denoted L1), as well as an AU-rich 3' tail that under conditions of high ligand and/or Mg2+ concentration can interact with L1 (forming a helix denoted P2) (Fig. 5a). In a recent study (Suddala et al. 2013), a donor fluorophore was placed on the end of the 3' tail and an acceptor fluorophore was placed in L1, so that when the P2 helix formed, a high-FRET state would result (Fig. 2a). The central question of this work, which is relevant for enzymes as well, was where the riboswitches operate on the continuum of conformational selection to induced fit mechanisms. In a conformational selection mechanism, the riboswitch samples multiple conformations in the absence of ligand, and the ligand binds to the one that most closely resembles the ligand-bound state. In an induced fit mechanism, the ligand instead binds to a conformation that is different from that of the bound state, and induces a structural change into the bound conformation. Both riboswitches were found to dynamically sample both high- and mid-FRET states in the absence of ligand, and increasing concentrations of ligand increased the percentage of molecules in the high-FRET state. For the Tte, but not the Bsu riboswitch, this change was accompanied by an increase in the mean FRET value of each state (Fig. 5b). This observation provided the first clue that Bsu operates by conformational selection, with ligand selecting the high-FRET state for binding but not modifying the structures of the two states (Fig. 5c). Conversely, it suggested that Tte operates by induced fit, with ligand modifying the underlying states upon binding (Suddala et al. 2013).
Fig. 5.

Study of Bsu and Tte PreQ1 riboswitches using smFRET. a Crystal structures of the Bsu (colored) and Tte (grey) riboswitches, showing their close structural similarity. b Both riboswitches exhibited dynamically interconverting high- and low-FRET states, with the population of the high-FRET state increasing with increasing concentration of PreQ1 (upper pair of plots). In only the Tte riboswitch, however, the mean FRET values of the two states also changed with PreQ1 concentration (lower pair of plots). c These results, along with other experiments and modeling, suggested that the Tte riboswitch follows an induced fit mechanism of ligand-induced folding, while the Bsu riboswitch follows a conformational selection mechanism (Suddala et al. 2013)
Coarse-grained TOPRNA simulations, which represent each nucleotide using three pseudo-atoms, were used to identify the structures of the mid- and high-FRET states (Suddala et al. 2013). Structures with the tail partially docked led to inter-fluorophore distances of 35–45 Å, consistent with the mean FRET value and broad distribution of the mid-FRET state. Meanwhile, the fully folded conformation produced a narrow distance distribution centered at 25 Å, consistent with the high-FRET state. Further investigation utilized Gō-model simulations to compare the folding pathways of the two riboswitches. These simulations found that in the Bsu riboswitch, ligand binding does not occur until the P1 helix is almost fully formed, while in the Tte riboswitch, ligand binding occurs concomitantly with P1 folding. This further supported the assignment of the conformational selection mechanism to the Bsu and the induced fit mechanism to the Tte riboswitch (Fig. 5c). These same mechanisms are found in both ribozymes and protein enzymes, and can be highly relevant to the design of drugs targeting them.
Future directions in single-molecule enzymology and mechanistic studies
Single-molecule techniques are being applied to an ever-expanding set of targets, beyond basic RNA, DNA and protein systems. Recently, Zhou et al. used fluorogenic substrates to image the activity of individual gold nanoparticle catalysts (Zhou et al. 2013). In addition, Jimenez et al. determined the sequences of enzymes from ancestral species and used single-molecule force spectroscopy to study their reaction mechanisms and folding stability (Perez-Jimenez et al. 2011). Single-molecule techniques are also increasingly applied to systems that are directly relevant to human health. These include the intrinsically disordered viral oncoprotein E1A (Ferreon et al. 2013), and the effects on protein folding of mutations related to Parkinson’s disease (Ferreon et al. 2010). Clearly, the variety of systems to which such single-molecule techniques can be applied is limited only by our imagination.
Future directions in this field include translating in vitro observations into biologically relevant discoveries by studying macromolecular structure and function under increasingly in vivo-like conditions. Such conditions include experiments in the presence of chemical crowding agents such as polyethylene glycol (Dupuis et al. 2014; Strulson et al. 2012), in cell extract (Abelson et al. 2010; Krishnan et al. 2013) and ultimately, in live cells (Martin et al. 2013). In addition, the study of natural enzymes inspires the design of novel enzymes and enzyme systems. The construction of such systems has been facilitated by the development of approaches like DNA nanotechnology, which allows multiple enzymes to be positioned in space in layouts that benefit catalysis (Fu et al. 2014). Finally, single-molecule ‘crystal structures’ have become a real possibility thanks to the development of x-ray free electron lasers (XFELs). With the structures photosystem I nanocrystals (Chapman et al. 2011) and single molecules of 2,5-diiodobenzonitrile starting to be solved by X-ray diffraction from XFELs (Küpper et al. 2014), it may only be a matter of time before we can solve the full ‘crystal structure’ of a single protein molecule.
Single-molecule systems biology
In the preceding section, we focused on recent scientific advancements using in vitro single-molecule fluorescence microscopy to study surface-constrained biomolecules. The goal of in vitro studies is to characterize biomolecules in a well-controlled environment, but these studies inevitably fall short of recapitulating the complex networks and pathways found in living cells. Recent advances in single-molecule fluorescent microscopy now allow functional biomolecules to be introduced into cells using relatively noninvasive techniques, followed by visualizing their location and movement at high spatial and temporal precision. Systems biology is an emerging multidisciplinary approach that aims to understand the system-wide properties of a biological system, such as a cell, by combining experimental measurements of its components with computational modeling. Biology is fundamentally stochastic in nature, leading to diverse, spatiotemporally inhomogeneous distributions of molecules within as well as across individual cells, even of a clonal cell line or a (tumor) tissue (Li and Xie 2011; Pitchiaya et al. 2014; Xie et al. 2006). The resulting inhomogeneities – involving short-lived and/or rare pathway and reaction intermediates, dispersed cellular localization and time evolution, and a multitude of parallel mechanisms of action and non-linear responses from complex multi-hub networks that lead to emergent behaviors – together form the very foundation of biomolecular function. A case can thus be made that a single-molecule based interrogation in service of systems biology is poised to contribute functionally critical insights into spatiotemporal inhomogeneities and stochastic behaviors of biomolecules, including DNAs, RNAs, proteins and metabolites; features that are inaccessible using ensemble-averaging techniques. To this end, the behaviors of fluorescently-tagged cellular components need to be interrogated in vivo in response to environmental perturbations, including the administration of drugs, potentially aiding the development of therapeutics. In this section, we briefly describe in vivo fluorescent probes, provide an overview of in vivo single-molecule fluorescence microscopy techniques and summarize some recent reports using in vivo single-molecule fluorescence microscopy to study living cells that eventually will lead to a true single-molecule systems biology as previously envisioned (Walter et al. 2008).
Fluorescent probes and microscopy techniques suitable for in vivo studies
To visualize single biomolecules, they must first be genetically modified with a fluorescent protein tag or chemically modified with an organic fluorescent probe (Pitchiaya et al. 2014). Fluorescent protein tags have been most broadly applied for use in intracellular fluorescence microscopy. The prototype fluorescent protein tag, green fluorescent protein (GFP), was first purified from jellyfish Aequorea victoria in the 1960’s (Shimomura et al. 1962) and subsequently cloned for use as a genetic tool in the 1990’s (Prasher et al. 1992). Currently, fluorescent proteins are offered in a variety of colors spanning the visible spectrum, including blue, cyan, green, yellow, and red, and have found use in fluorescence microscopy partly due to the ease with which these sequences can be manipulated in protein expression plasmids to tag a protein of interest. For single-molecule studies, this labeling strategy is particularly useful for imaging localized proteins, protein aggregates or high affinity RNA-protein complexes, such as phage MS2 coat protein-MS2 stem-loop and lambda N peptide-boxB labeling systems (Keryer-Bibens et al. 2008). However, fluorescent proteins have limitations in their use for single-molecule fluorescence microscopy. Adding a fluorescent protein tag may perturb normal protein function and therefore, prior to any in vivo studies, the function of the tagged protein must be tested. As an alternative to fluorescent protein tags, it may be advantageous to chemically modify the biomolecule of interest with a small organic fluorophore, i.e., rhodamine, cyanine, oxazine, BODIPY or perylene derivatives (Harada et al. 2014; Juette et al. 2014; Yuan et al. 2013). Small fluorophores have the advantage of emitting more fluorescent photons and blinking less than fluorescence proteins, thus making them quite suitable for single-molecule studies. These molecules are typically introduced into cells by microinjection or electroporation.
Transfection (either stable or transient) and overexpression of fluorescently-tagged proteins often results in an inhomogeneous cellular distribution and high cellular fluorescence density, thereby obscuring the ability to detect single particles. To address this challenge, super-resolution fluorescence microscopy methods were developed that rely on stochastic switching of fluorophores between bright and dark states — so called ON and OFF states (Johnson-Buck et al. 2013; Walter et al. 2008). These techniques include photoactivated localization microscopy (PALM), point accumulation for imaging in nanoscale topography (PAINT), stochastic optical reconstruction microscopy (STORM), stimulated emission depletion (STED), and fluorescence imaging with one nanometer accuracy (FIONA) (Jungmann et al. 2014; Jungmann et al. 2012; Nienhaus and Nienhaus 2014; Sengupta et al. 2012; Stracy et al. 2014; Walter et al. 2008). Using these techniques, the reconstruction of an object can be achieved at high precision by localizing the centers of the mountain-shaped, diffraction-limited point spread functions (PSFs) of images obtained from single fluorescent molecules associated with the object through a software-based fitting routine. The working principle of how these techniques beat the classical diffraction limit of ~200 nm by a factor of ~10–100-fold has been outlined in a previous review (Walter et al. 2008). PALM serially photoactivates and deactivates many sparse subsets of photoactivatable fluorophores to produce a time sequence of images whose PSF analysis is combined into a super-resolution composite. PAINT uses continuous specific or nonspecific binding and dissociation or photobleaching of diffusing fluorescent probes to an object for high-resolution imaging. A microscope capable of total internal reflection fluorescence (TIRF) or highly inclined and laminated optical sheet microscopy (HILO) is required to reject the background signal from freely diffusing fluorescent probes. Similarly, STORM uses photo-switchable fluorophores to image a stochastically different sparse subset in each switching cycle and then all images are combined into a super-resolution composite. By contrast, STED uses overlapping light beams to cause stimulated emission in the surrounding emitters, reducing the effective focal detection spot in size. 3D imaging using these techniques is made possible by introducing a cylindrical lens between the objective and imaging lens (Huang et al. 2008). FIONA laid the foundation for STORM, PALM and PAINT in that only a single fluorescent molecule is ever deployed within a diffraction-limited area; a time series of images therefore allows for spatiotemporal tracking of this molecule by finding the center of its PSF in each sequential image. A two-color version of FIONA called single-molecule high-resolution colocalization (SHREC) was also demonstrated (Churchman et al. 2005). For specifics on experimental configurations and on choosing the proper tool for a given task, the reader is referred to our previous review (Walter et al. 2008).
Due to the complex milieu of the cell, single-molecule analyses in living cells require special considerations that are reviewed in detail elsewhere (Gahlmann and Moerner 2014; Nienhaus and Nienhaus 2014; Pitchiaya et al. 2014). As mentioned in the introductory section, TIRF microscopy is the most common method for the excitation and detection of single molecules, as it effectively removes spurious signal from background autofluorescence and out-of-focus fluorophores. For in vivo single-molecule studies, however, near-TIRF or HILO microscopy is preferred, wherein a thin sheet of illumination penetrates above the basal membrane of the cell and into the cytoplasm beyond the typical ~100 nm achieved using TIRF (Fig. 1b, d). In addition to customized wide-field objective-type TIRF microscopes, narrow-field reduced illumination microscopes, as used for single-point edge-excitation sub-diffraction (SPEED) microscopy (Ma et al. 2013; Ma and Yang 2010), are capable of detecting single fluorophore-labeled molecules with high spatiotemporal resolution (Figs. 6 and 7). An optimized single-molecule setup permits both tracking of single particles and counting of immobile particles. Following, we provide a general overview of recent advancements in single-molecule applications to study intracellular structure-function relationships of DNAs, RNAs, proteins and metabolites. We place special emphasis on examples involving in vivo DNA, RNA and RNA-protein complex analyses.
Fig. 6.

Tracking and counting single miRNAs using iSHiRLoC. a Psuedocolored, live U2OS cell microinjected with miR-let-7a-1-Cy5 and imaged 4 h post-injection; miRNP particles exhibit very slow (i), corralled (ii), fast (iii), and biased (iv) Brownian diffusion. b Distribution of diffusion coefficient for let-7a-1-Cy5 at various time points. c Pseudocolored, fixed U2OS cell microinjected with miR-let-7a-1-Cy5 and imaged 4 h post-injection (top) and photobleaching steps detected by iSHiRLoC (bottom). d Particle counting time course reveals assembly of multimeric let-7a-1-Cy5 particles (top), but not cxcr4-Cy5 particles (middle). Co-injection of cxcr4-Cy5 with an mRNA target promotes assembly of multimeric cxcr4-Cy5 particles (bottom). e Model established using iSHiRLoC data of miRNA-mediated translational repression and degradation (Pitchiaya et al. 2012)
Fig. 7.

Tracking single mRNPs through the nuclear pore complex in HeLa cells using SPEED microscopy. a Wide-field epifluorescence illumination of four mCherry-tagged mRNPs (red) and GFP-nuclear envelope (green). b A successful mRNP export event through the NPC captured using SPEED microscopy. c An abortive mRNP export event. d, e Single particle tracks (black) and the centroid of the NPC (red) of successful and abortive mRNP export events through the NPC, as shown in b and c, respectively. f, g Export time distributions and single-exponential fit of successful (f) and abortive (g) mRNP export events through the NPC as shown in b and c, respectively (Ma et al. 2013)
Applications to single-celled organisms
In vivo single-molecule studies have been used to characterize DNA replication and repair, nucleoid organization and chromosome segregation and partitioning, predominantly in bacterial model systems, including Caulobacter crescentus, Escherichia coli, and more recently Bacillus subtilis, as reviewed in detail elsewhere (Gahlmann and Moerner 2014; Robinson and van Oijen 2013; Stracy et al. 2014). The introduction of single-molecule techniques has revealed many insights regarding bacterial DNA function under normal conditions and in response to DNA damage. In most cases, these studies rely on photoactivation, photoswitching and photoinduced blinking of fluorescent-tagged proteins and post-acquisition data analyses using PALM and STORM reconstruction to obtain spatial resolutions of 20–40 nm, well below the ~200 nm diffraction limit of light. A recent report by Crawford et al. successfully applied in vivo single-molecule FRET in living E. coli cells upon electroporation of DNA and proteins (up to 100 kDa) labeled with organic FRET pairs (Crawford et al. 2013). This study provided proof-of-principle for the use of organic fluorophores to study DNA and proteins structural rearrangements and demonstrated the versatility of the technique by applying it to yeast Saccharomyces cerevisiae. Ultimately, further insights will be gained by examining the dynamic nature of nucleic acid structural rearrangements and DNA-protein interactions using in vivo single-molecule FRET in both bacteria and eukaryotes.
iSHiRLoC: tracking and counting intracellular microRNAs
Recent genome-wide RNA profiling has revealed an unexpectedly diverse landscape of non-coding RNAs in eukaryotic cells, yielding an emergent field of investigation into non-coding RNA structure-function relationships (Djebali et al. 2012). One particularly important class of such non-coding RNAs are microRNAs (miRNAs). These endogenously expressed ~22-nucleotide-short, double-stranded RNAs assemble into microRNA-protein (miRNP) complexes, upon which their guide strand can bind ~6–8-nucleotide short, complementary, so-called seed sequences in the 3’ untranslated regions (UTRs) of messenger (m)RNAs to repress protein expression via RNA silencing. To date, almost 1,500 unique mammalian miRNAs have been identified that, collectively, represent over 2% of the human genome and are predicted to regulate over 60% of all protein coding genes in a complex network of interactions (Krol et al. 2010; van Kouwenhove et al. 2011). We recently reported on the temporal assembly of miRNP complexes using a technique we termed intracellular single-molecule, high resolution localization and counting, or iSHiRLoC (Fig. 1d) (Pitchiaya et al. 2012; Pitchiaya et al. 2013). By combining live and fixed cell imaging of microinjected, cyanine dye-labeled miRNAs in HeLa cells, we were able to track miRNP assembly and count the number of miRNAs within single particles (Fig. 6). Temporal changes in miRNA diffusion patterns were observed, wherein miRNAs imaged less than 1 hour after microinjection were blurred out when using a 100-ms camera integration time, but later started to diffuse slowly enough to be tracked, indicating their incorporation into large miRNP complexes. Four different types of diffusive motion were observed: virtually immobile, corralled, Brownian and directional (Fig. 6a). Plotting these diffusion constants half-logarithmically revealed at least two Gaussian distributions that we could interpret as fast diffusing miRNP-mRNA complexes (with a diffusion constant of 0.26 µm2/s) and slowly diffusing miRNP-mRNA complexes localized on/in degrading P-bodies (0.034 µm2/s) (Fig. 6b). In fixed cells, photobleaching steps were counted within a particle to distinguish monomeric from multimeric miRNPs (Figs. 6c,d). By comparing miRNAs let-7-a1 (which has many predicted mRNA targets) to artificial cxcr4 (with few predicted mRNA targets), we determined that only let-7-a1, and not cxcr4, displayed a characteristic temporal increase-then-decrease pattern in miRNA multimerization, suggesting that miRNPs were aggregating in P-bodies followed by their dispersal due to the degradation of their mRNA targets (Pitchiaya et al. 2012). Only when cxcr4 was co-microinjected with an mRNA target did it display the same pattern, strongly supporting the notion that these changes are target-dependent. Taken together, these observations established a unifying model of dynamic miRNP-mRNA assembly into P-bodies, wherein mRNAs are translationally repressed, then targeted for degradation (Fig. 6e).
mRNA transport in the cell
Several single-molecule reports have investigated mRNA transport in the cell (Grunwald and Singer 2010; Kalo et al. 2013; Mor et al. 2010; Siebrasse et al. 2012). In a recent report (Ma et al. 2013), the process of mRNA export through the nuclear pore complex (NPC) in eukaryotic cells was examined at an unprecedented spatiotemporal resolution of 8 nm and 2 ms (500 frames per second) using the recently developed single-molecule fluorescence imaging approach of SPEED microscopy (Ma and Yang 2010) (Fig. 7). Using narrow-field illumination, SPEED microscopy illuminates a sample volume area of < 1 mm2, thereby improving time resolution to ~2 ms. This stands in contrast with the more commonly used wide-field illumination schemes of TIRF and HILO, which excite a sample area of > 100 mm2 and can track more particles simultaneously, but only reach time resolutions of 20–100 ms. To increase the field of view during SPEED microscopy, the authors used oblique angle illumination and added a 400 mm pinhole in a standard wide-field microscope. Compared to previous wide-field micropscopy measurements, these modifications improved temporal and spatial resolution by 10- and 3-fold, respectively (Ma et al. 2013).
Experiments were conducted in HeLa or murine embryonic fibroblast (MEF) cell lines expressing spectrally distinct fluorescent proteins used to label the NPC and mRNPs, wherein the NPC contained a fluorescently-tagged protein and the β-actin or luciferase mRNA was labeled using a MS2 labeling system (Fig. 7a) (Ma et al. 2013). Using SPEED microscopy, a single NPC and individual mRNPs were illuminated and detected; single mRNPs interacted with the NPC, and finally either moved from the nuclear to the cytoplasmic side (Fig. 7b) or aborted export (Fig. 7c). The technological advancements of SPEED microscopy enabled several observations that were most likely undetected in prior studies due to their lower spatiotemporal resolution. First, a fast-slow-fast mRNP diffusion pattern was observed when exiting the NPC (Fig. 7d,e), which contrasts the slow-fast-slow pattern previously reported based on measurements with lower spatiotemporal resolution (Grunwald and Singer 2010). Second, mRNPs interacted with the NPC for only ~12 ms (Fig. 7f,g), a 10-fold shorter time than previously reported (Grunwald and Singer 2010). Third, 3D transport routes were derived that revealed that mRNPs interact with the periphery of the NPC, while passively diffusing small molecules follow a central path through the NPC, thus suggesting a multilane traffic model through the NPC. Together, these findings highlight how technological advancements lead to refined insights into biological phenomena such as NPC function as the critical selectivity filter that controls nuclear mRNP export.
Other applications and future directions of intracellular single-molecule fluorescence
Single-molecule fluorescence microscopy can be used to examine RNA localization and expression levels, as well as rates of transcription and splicing (Pitchiaya et al. 2014). Determining the localization and function of long non-coding RNAs (lncRNAs) has become of particular interest in recent years, as little is known about these transcripts. What is known from the few annotated lncRNAs are their functions related to important cellular processes such as X-chromosome inactivation, imprinting, pluripotency, cell lineage commitment, and apoptosis (Batista and Chang 2013; Lee and Bartolomei 2013; Rinn and Chang 2012). Single-molecule fluorescence in-situ hybridization (smFISH) is a technique in which many short, fluorophore-labeled DNA oligonucleotides are directed toward an RNA target sequence. It has been applied to detect significant changes in lncRNA transcript levels during red blood cell maturation (Alvarez-Dominguez et al. 2014) and in somatic tissue differentiation (Kretz et al. 2013). Given the ease with which smFISH can be applied to detect long RNAs, it is likely to be a significant player in future studies of lncRNAs.
Transcription and splicing have been examined using smFISH and MS2 labeling systems (Raj et al. 2006; Shav-Tal et al. 2004; Waks et al. 2011). For example, Tyagi and co-workers directly observed the synthesis of nascent RNA transcripts using smFISH, wherein they found nascent transcripts were generated as intrinsically random bursts dependent on single gene activation and deactivation (Raj et al. 2006). Using an RNA-protein labeling system, the kinetics of co-transcriptional splicing has revealed that RNA Pol II transcribes at a rate of 3–6 kb min−1 (Martin et al. 2013), consistent with previous observations (Darzacq et al. 2007; Singh and Padgett 2009). Co-transcriptional and alternative splicing processes also have been monitored using smFISH, wherein it was found that extensive intronic secondary structure inhibits co-transcriptional splicing (Vargas et al. 2011) and cell-to-cell variability in alternatively spliced isoforms is more apparent in cancerous cell lines due to reduced control of alternative splicing factors (Waks et al. 2011). Together, these single-molecule studies offer insights into transcription and splicing that had gone undetected using ensemble methods.
Metabolites are essential biomolecules that regulate many cellular processes of interest in toxicology; as such it is advantageous to detect intracellular levels of metabolites under normal and stressed conditions. Recently, Paige et al. described an ensemble-based fluorescence technique capable of detecting cell-to-cell variations of intracellular levels of adenosine 5-diphosphate (ADP) and S-adenosylmethionine (SAM) in E. coli (Paige et al. 2012). RNA-based sensors were developed, comprised of a ligand-binding RNA aptamer and Spinach, an RNA mimic of GFP, and function by binding a metabolite to switch on the fluorescence of the small molecule fluorophore. Similar fluorescence-based RNA biosensors have been since used to detect intracellular levels of cyclic di-GMP and cyclic AMP-GMP (Kellenberger et al. 2013), RNAs (Strack et al. 2013), as well as proteins (Song et al. 2013). While these sensors do not yet reach single-molecule sensitivity, future improvement of their binding affinities, fluorescence intensities and metabolite specificities may pave the way to this level.
Together, these recent reports highlight the versatility of single-molecule fluorescence microscopy to study biomolecules in the context of living cells, with their complex milieu of constituents that interact in system-wide networks leading to emergent behaviors. Further technological advances are likely to bring many processes to light that then can be assembled and modeled through single-molecule systems biology.
Nanotechnology: building with DNA blocks
The traditional biological role of DNA has been to carry and pass on genetic information, but DNA has begun to find a new role in the field of materials science (Seeman 2003). Many biotechnology applications of DNA have been developed in the past 30 years, including DNA vaccines, anti-sense DNA and RNA, and anti-gene technology, to name just a few (Ito and Fukusaki 2004). The many functions and nanoscale dimensions of DNA have made it an important asset in the field of nanotechnology. To build a complex DNA structure, all that is needed are the simple thermodynamic base pairing rules that govern formation of well-defined double helical structures between two complementary DNA strands. DNA is the preferred biopolymer in nanotechnology, as it is less susceptible to hydrolysis than RNA and more structurally stable and predictable than proteins. These properties of DNA have enabled the construction of a diverse set of nanostructures and nanodevices through the programmed hybridization of complementary strands (Lu and Liu 2006; Michelotti et al. 2012). In the last three decades, three- and four-way junctions displaying sticky ends have been exploited as two- and three-dimensional DNA building blocks to connect into larger structures. By incorporating protruding strands at specific locations within these nanostructures, proteins, nanoparticles or other DNA structures can be captured to form well-defined patterns. Nucleic acid structures (e.g., DNA aptamers), proteins or nanoparticles can be incorporated into these nanostructures during the self-assembly process, and DNA molecules can be easily modified with a wide range of fluorophores and functional groups to conjugate with proteins and nanoparticles. In addition to biological applications, several efforts have been made in the past few years to generate DNA based ‘smart materials’ with specific chemical and biological functions. The discovery that DNA can also act as a catalyst, or ‘DNAzyme’, has further integrated the power of DNA nanotechnology and molecular biology. Through the development of these powerful approaches, the field of DNA nanotechnology has been significantly broadened and the resulting materials have found use in many other fields and practical applications relevant to toxicology, such as small-molecule sensing, environmental monitoring, medical diagnostics, drug screening, therapeutics, nanoelectronics, and nanophotonics (Lu and Liu 2006; Michelotti et al. 2012). Before nanomaterials can be utilized in real-life applications, however, it is necessary to develop a detailed mechanistic understanding of their behavior using nanoscopic tools, most commonly single-molecule fluorescence microscopy or atomic force microscopy (AFM) (Michelotti et al. 2010; Rueda and Walter 2005; Walter et al. 2008). Studies using these techniques have revealed that DNA nanostructures exhibit heterogeneous behavior at the molecular level. Single-molecule techniques, in particular, are powerful in that they can detect heterogeneities in structure and conformational dynamics, therefore revealing how efficiently a unique conformer performs a specific function. Recent advances in super-resolution fluorescence microscopy (Walter et al. 2008), have revolutionized how single-molecule data are collected and processed. These versatile techniques are compatible with diverse experimental conditions and suitable for complex nanotechnological systems, both in vitro and in vivo.
AFM imaging has long been the main characterization method for DNA-based structures. However, in recent years a variety of other techniques have been adopted, namely high-speed AFM, electron microscopy (EM), and super-resolution fluorescence microscopy. The advantages and limitations of these techniques have recently been reviewed (Jungmann et al. 2012). AFM requires deposition of the sample directly onto a mica surface, which could introduce surface perturbation to the sample. In addition, it is not very sensitive to three-dimensional (3D) features. EM has been recently applied to the characterization of compact 3D DNA nanostructures, enabling the reconstruction of their 3D-architecture from single-particle analysis. However, EM requires expensive and sophisticated instrumentation and laborious data analysis. In addition, imaging is performed under conditions far from the native environment, as a sample has to be either stained (positive stain) or surrounded by stain (negative stain) in transmission electron microscopy (TEM), and the sample must be frozen in cryo-EM experiments.
Fluorescence microscopy offers advantages over these approaches since it is a relatively noninvasive, extremely sensitive, high throughput, and cost effective method that offers fast data acquisition (Jungmann et al. 2012; Michelotti et al. 2010; Rueda and Walter 2005). In addition, fluorescence microscopy allows one to perform experiments in living systems. Due to their small size, the application of fluorescence microscopy to DNA nanostructures was precluded until the recent development of super-resolution methods (introduced in the systems biology section). Unlike mechanical interaction-based imaging such as AFM and optical and magnetic tweezers, super-resolution microscopy is achieved through the remote imaging of fluorescent molecules (Walter et al. 2008). As with any single-molecule technique, fine-tuning of experimental configurations such as microscope design, light sources, optical detectors, probes, and sample environment, as well as an optimal data analysis pipeline, are critical for successful experiments. Given the fact that various techniques are available for super-resolution fluorescence imaging and each of these techniques are specialized to a unique experimental environment, choosing a suitable technique is critical (Walter et al. 2008). Here, we highlight some of the recent advances and applications of super-resolution microscopy in the field of DNA nanotechnology.
Tracking a moving cargo on DNA origami
DNA origami is a two- or three-dimensional structure made by programmed hybridization of a long scaffold DNA and hundreds of short (typically ~32-nucleotide) oligonucleotides called staple strands (Rothemund 2006). These static DNA nanostructures have been widely used as a platform to study biomolecular reactions (Michelotti et al. 2012). In addition to DNA self-assembly into static nanostructures, DNA nanotechnology has recently been exploited to engineer systems with interesting dynamic properties. For example, recent advances in designing and constructing DNA-based devices have yielded molecular ‘walkers’ capable of autonomously changing their physical location over time (Bath and Turberfield 2007; Lund et al. 2010; Zhang and Seelig 2011). The development of walkers was inspired by motor proteins such as myosin and kinesin that move in a step-by-step fashion. Such molecular walkers can be used to pick up, transport and drop off ‘cargo’ in a controlled manner. Some recent examples include directional transportation of streptavidin, gold nanoparticles, and small molecules that are directly attached to the walking oligonucleotides (Cha et al. 2014; He and Liu 2010; Lund et al. 2010). Walkers so far have been designed to use one of two modes of locomotion, either toehold-mediated strand displacement or chemical/catalytic cleavage of substrate oligonucleotides that destabilize the association and pass the walker on to the next attachment point (He and Liu 2010; Lund et al. 2010), both of which allow kinetic control of reaction pathways. There are two main motivations for the use of DNA walkers. First, as demonstrated by He et al. (He and Liu 2010), DNA-templated chemical synthesis can take place upon arrival of the walker at each station. This strategy of chemical synthesis could improve the ease, speed, specificity and efficiency of multistep reaction sequences. Second, cargo can be transported from station to station.
While the motion of molecular walkers had previously been established only indirectly, Lund et al. directly visualized DNA walkers on a DNA origami-based track using super-resolution imaging (Fig. 8) (Lund et al. 2010). In this work, we used biotinylated DNA enzyme (DNAzyme) oligonucleotides to create a molecular ‘spider’ with a streptavidin ‘cargo’ at its core. This spider can walk along a predefined trajectory when its catalytic DNAzyme legs cleave at a single RNA base substrate strands protruding from the origami (Fig. 8a). Cleavage destabilizes the association of the corresponding leg with the origami, but the multiple legs of the spider prevent it from completely dissociating from its track. The cleaved legs explore neighboring substrate sites, allowing the spider to bind and move directionally along the liner track as more and more substrates are cleaved. The walker was guided by patterning the substrate onto a two-dimensional origami landscape; the movement of the cargo was then monitored by super-resolution fluorescence microscopy (Fig. 8b) and AFM imaging. We found that the molecular spider was able to carry out actions such as ‘start’, ‘follow’, ‘turn’ and ‘stop’ on specifically designed origami tracks (Lund et al. 2010). The speed of the walker was determined by position-time trajectories (Fig. 8b, right panel). This work provided proof-of-principle that high-resolution fluorescence microscopy is a powerful tool for monitoring in real-time the nanometer-scale steps of molecular walkers. In the future, by incorporating additional control mechanisms, more complex robotic behavior may be implemented on DNA nanostructures such as interactions between multiple molecular robots, leading to collective systems behavior.
Fig. 8.

Tracking a molecular spider that walks along an origami track. a Schematic of a DNAzyme-based molecular walker on a two-dimensional DNA track. b DNA origami landscape with positions A—E (left). The substrate track (brown), start position (green), stop and control positions (red) and imaging marker (blue) are highlighted. The middle panel shows the movement of the spider tracked by high-resolution fluorescence microscopy. The right panel shows the displacement of the spider trajectory from its initial position as a function of time (Lund et al. 2010)
One of the current challenges in autonomous transport by DNA walkers is their slow speed. Recently developed synthetic molecular transporters move at ~0.5–6 nm min−1 (Cha et al. 2014; Omabegho et al. 2009; Wickham et al. 2011). The speed of toehold-mediated transporters is limited by toehold dissociation and branch migration, and can potentially be improved by optimizing the track spacing, toe length, and scaffold thickness. In the case of DNAzyme-based walkers, reaction efficiency of the DNAzyme, leg and cargo dissociation, and backtracking all limit the overall speed and distance over which the cargo can move (Lund et al. 2010). Another important factor is the inconvenience associated with refilling the substrate for repeated, directed movement. In the future, these limitations may be addressed in part by constructing two- or multi-track DNA nanostructures to parallelize the transporters. Recyclable substrates such as DNA legs with photoresponsive azobenzene moieties (Suzuki et al. 2014) may facilitate substrate refilling.
Chemical imaging on DNA origami by DNA-PAINT
DNA nanostructures can be modified site-specifically with nanoparticles, proteins and nucleic acids, usually through hybridization of complementary DNA sequences. These modified DNA nanostructures have been used in recent years for various research purposes ranging from therapeutics to in vivo applications (Lu and Liu 2006; Michelotti et al. 2012). Successful design of modified nanostructures relies on the understanding of factors that influence hybridization reactions and kinetics of hybridization on the DNA nanostructures (Johnson-Buck and Walter 2014). The hybridization of DNA nanostructures can be probed using several ensemble and single-molecule methods such as AFM, native gel electrophoresis and the TIRF microscopy techniques smFRET and DNA-PAINT. DNA-PAINT, in particular, has become a popular imaging method, as it works through reversible binding of single fluorescently labeled “emitter” oligonucleotides to individual probe sites on the DNA nanostructure (Fig 9). Transient but stochastic binding of the emitter strand allows one to probe a strand-binding position multiple times (Johnson-Buck et al. 2013; Johnson-Buck and Walter 2014). High spatial resolution is achieved by combining 2D Gaussian fits to many individual signals (Jungmann et al. 2012). DNA-PAINT requires no special equipment beyond a fluorescence microscope and is compatible with virtually any experimental conditions, as reviewed elsewhere (Johnson-Buck and Walter 2014; Jungmann et al. 2012; Walter et al. 2008). The technique is simple and easy to implement, usable with virtually all fluorophores, has multi-color capability, and offers minimal photobleaching due to rapid dissociation of the of the emitter strands from the probes. Unlike AFM experiments, the method allows for biological imaging in a hydrated environment, with the molecule of interest tethered to a passivated surface. Therefore, DNA-PAINT can provide “true” 3D images of single molecules by minimizing distortion of the structure of interest by the surface. Here, we will highlight one of the recent applications of DNA-PAINT for studying the hybridization kinetics of single-stranded DNA (ssDNA) emitters to probes on a 2D DNA nanostructure.
Fig. 9.

Imaging of nanostructures using DNA-PAINT. a Schematic of single-molecule set up to monitor chemical changes by two-color DNA-PAINT. Abbreviations: Cy5-labeled ssDNA target ‘T1’, Cy3-labeled ssDNA target ‘T2’, streptavidin ‘STV’, and substrate ‘S’. b Effective volume overlap of neighboring strands on origami predicted by CanDo modeling. c Heterogeneous probe binding pattern revealed by two-color reconstructions of P strands on rectangular DNA origami (Johnson-Buck et al. 2013)
Recently, Johnson-Buck et al. examined the kinetics of oligonucleotide binding to patterned probes on a single rectangular DNA origami, also known as an origami tile. The reconstructed data were generated from the binding patterns of T1 (Cy5 labeled ssDNA emitter strand) and T2 (Cy3 labeled ssDNA emitter strand) over 100–200 binding cycles per probe site (P) (Fig. 9a) (Johnson-Buck et al. 2013). While simultaneously imaging T1 and T2 binding, the substrate sequence (single stranded region of the probe) was cleaved by a DNAzyme in the presence of Zn2+ so that only T2 could continue to bind (Fig. 9a). Furthermore, the relative frequency of T1 and T2 binding to different sequences within the intact probe on the origami tile revealed heterogeneous binding patterns independent of the identity of the fluorophore labeling the emitter strand. Combining results from the computational modeling software Computer-aided engineering for DNA origami (“CanDo”) (Fig. 9b) and our DNA-PAINT experiments, we concluded that the heterogeneous binding of T1 is due to a combination of intrinsic curvature of the origami tile and weak probe–probe interactions within the origami tile that compete for T1 binding. This study provided direct evidence that emitter-probe hybridization on 2D origami tiles is governed by the tile’s geometry and local density of the probe strands.
Like most developing single-molecule techniques, there are further opportunities for optimization of the spatial and temporal resolution of DNA-PAINT. Due to imperfect correction of instrument or DNA nanostructure drift, it remains challenging to resolve inter-probe distances of <20 nm (Johnson-Buck and Walter 2014). Drift is introduced by uncontrollable, thermally induced movements of the sample stage, irregular air currents, vibration of the instrument table, etc. Some of these can be minimized through vibrational isolation of the instrument table. Methods that utilizes fiduciary markers such as quantum dots, floor beads, gold nanoparticles, and gold nanorods are already in use (Jungmann et al. 2014; Lund et al. 2010). To achieve high spatial resolution, depending on the scientific question at hand and desired signal-to-noise ratio (SNR) the bound and unbound time of emitter strand has to be optimized. In general, a longer dwell time for the unbound state is preferred (Jungmann et al. 2012), which can be achieved by reducing the lengths of the probe and/or emitter oligonucleotides. However, faster dissociation of the emitter will result in a low number of photons, generating a weak signal. The dwell time for the bound state depends on the dissociation rate and is independent of concentration, whereas the dwell time for the unbound state depends on both association rate and the concentration of the emitter stand. Therefore, a balance between all of these parameters is necessary in order to optimize the fluorescence ON and OFF times.
DNA nanostructures are also promising platforms to incorporate target specific DNA or RNA sequence as ‘barcodes’ (Lin et al. 2012). The development of barcode-associated nanostructures in combination with DNA-PAINT would allow clinical applications such as multiplexed detection of cancerous miRNAs in biological samples. Similarly, aptamer domains can be assembled onto the nanostructures to detect proteins, nucleic acids and small molecules (Lu and Liu 2006). Such a platform may be exploited in the field of toxicology to detect toxins. The multiplexed detection of individual toxins on such a nanostructure would require highly sensitive super-resolution imaging. Although spatial resolution of DNA-PAINT is currently limited to ~10 nm (Johnson-Buck and Walter 2014; Jungmann et al. 2014), with further development we project that the technique will be a standard tool to study nanotechnological systems of increasing complexity for both in vitro and in vivo applications.
Conclusions
In this review, we have covered the basic optical requirements for single-molecule fluorescence detection. We have also discussed those technical requirements that are specific to the study of enzymology, structural biology, systems biology and nanotechnology, and have presented some noteworthy recent work in which single-molecule fluorescence was applied to these fields of study. In the future, we expect single-molecule fluorescence tools to shed more and more light on these fields, with an emergent impact on toxicology as well as other fields.
ACKNOWLEDGMENTS
This work was supported in part by National Institutes of Health grants R01 GM062357, R01 GM098023 and R21 AI109791, and by the Department of Defense MURI Award W911NF-12-1-0420 to N.G.W.
Footnotes
ETHICAL STANDARDS
The manuscript does not contain clinical studies or patient data.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Contributor Information
Julia R. Widom, Single Molecule Analysis Group, Department of Chemistry, 930 N. University Ave., University of Michigan, Ann Arbor, MI 48109-1055, USA.
Soma Dhakal, Single Molecule Analysis Group, Department of Chemistry, 930 N. University Ave., University of Michigan, Ann Arbor, MI 48109-1055, USA..
Laurie A. Heinicke, Single Molecule Analysis Group, Department of Chemistry, 930 N. University Ave., University of Michigan, Ann Arbor, MI 48109-1055, USA.
Nils G. Walter, Single Molecule Analysis Group, Department of Chemistry, 930 N. University Ave., University of Michigan, Ann Arbor, MI 48109-1055, USA. Single Molecule Analysis in Real-Time (SMART) Center, 930 N. University Ave., University of Michigan, Ann Arbor, MI 48109-1055, USA.
References
- Abelson J, et al. Conformation dynamics of single pre-mRNA molecules in spliceosome assembly. Nat Struct Mol Biol. 2010;17:504–512. doi: 10.1038/nsmb.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken CE, Petrov A, Puglisi JD. Single ribosome dynamics and the mechanism of translation. Annu Rev Biophys. 2010;39:491–513. doi: 10.1146/annurev.biophys.093008.131427. [DOI] [PubMed] [Google Scholar]
- Alemán EA, Silva Cd, Patrich EM, Musier-Forsyth K, Rueda D. Single-molecule fluorescence using nucleotide analogues: a proof-of-principle. J Phys Chem Lett. 2014;5:777–781. doi: 10.1021/jz4025832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Dominguez JR, et al. Global discovery of erythroid long noncoding RNAs reveals novel regulators of red cell maturation. Blood. 2014;123:570–581. doi: 10.1182/blood-2013-10-530683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod D, Burghardt TP, Thompson NL. Total internal reflection fluorescence. Annu Rev Biophys Bioeng. 1984;13:247–268. doi: 10.1146/annurev.bb.13.060184.001335. [DOI] [PubMed] [Google Scholar]
- Banerjee PR, Deniz AA. Shedding light on protein folding landscapes by single molecule fluorescence. Chem Soc Rev. 2014;43:1172–1188. doi: 10.1039/c3cs60311c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bath J, Turberfield AJ. DNA nanomachines. Nat Nano. 2007;2:275–284. doi: 10.1038/nnano.2007.104. [DOI] [PubMed] [Google Scholar]
- Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298–1307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokinsky G, et al. Single-molecule transition-state analysis of RNA folding. Proc Natl Acad Sci USA. 2003;100:9302–9307. doi: 10.1073/pnas.1133280100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breaker Ronald R. Prospects for Riboswitch Discovery and Analysis. Mol Cell. 2011;43:867–879. doi: 10.1016/j.molcel.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha T-G, Pan J, Chen H, Salgado J, Li X, Mao C, Choi JH. A synthetic DNA motor that transports nanoparticles along carbon nanotubes. Nat Nanotechnol. 2014;9:39–43. doi: 10.1038/nnano.2013.257. [DOI] [PubMed] [Google Scholar]
- Chapman HN, Fromme P, Barty A, White TA, Kirian RA, Aquila A. Femtosecond X-ray protein nanocrystallography. Nature. 2011;470:73–77. doi: 10.1038/nature09750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, et al. Single-molecule fluorescence measurements of ribosomal translocation dynamics. Mol Cell. 2011;42:367–377. doi: 10.1016/j.molcel.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Tsai A, O’Leary SE, Petrov A, Puglisi JD. Unraveling the dynamics of ribosome translocation. Curr Opin Struc Biol. 2012;22:804–814. doi: 10.1016/j.sbi.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong S, Min W, Xie XS. Ground-state depletion microscopy: detection sensitivity of single-molecule optical absorption at room temperature. J Phys Chem Lett. 2010;1:3316–3322. [Google Scholar]
- Churchman LS, Ökten Z, Rock RS, Dawson JF, Spudich JA. Single molecule high-resolution colocalization of Cy3 and Cy5 attached to macromolecules measures intramolecular distances through time. Proc Natl Acad Sci USA. 2005;102:1419–1423. doi: 10.1073/pnas.0409487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford R, Torella JP, Aigrain L, Plochowietz A, Gryte K, Uphoff S, Kapanidis AN. Long-Lived Intracellular Single-Molecule Fluorescence Using Electroporated Molecules. Biophys J. 2013;105:2439–2450. doi: 10.1016/j.bpj.2013.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzacq X, Shav-Tal Y, de Turris V, Brody Y, Shenoy SM, Phair RD, Singer RH. In vivo dynamics of RNA polymerase II transcription. Nat Struct Mol Biol. 2007;14:796–806. doi: 10.1038/nsmb1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditzler MA, Rueda D, Mo J, Hakansson K, Walter NG. A rugged free energy landscap separates multiple functional RNA folds throughout denaturation. Nucleic Acids Res. 2008;36:7088–7099. doi: 10.1093/nar/gkn871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis NF, Holmstrom ED, Nesbitt DJ. Molecular-crowding effects on single-molecule RNA folding/unfolding thermodynamics and kinetics. Proc Natl Acad Sci USA. 2014;111:8464–8469. doi: 10.1073/pnas.1316039111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- Elting MW, et al. Single-molecule fluorescence imaging of processive myosin with enhanced background suppression using linear zero-mode waveguides (ZMWs) and convex lens induced confinement (CLIC) Opt Express. 2013;21:1189–1202. doi: 10.1364/OE.21.001189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English BP, et al. Ever-fluctuating single enzyme molecules: Michaelis-Menten equation revisited. Nat Chem Biol. 2006;2:87–94. doi: 10.1038/nchembio759. [DOI] [PubMed] [Google Scholar]
- Ferreon ACM, Ferreon JC, Wright PE, Deniz AA. Modulation of allostery by protein intrinsic disorder. Nature. 2013;498:390–394. doi: 10.1038/nature12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreon ACM, Moran CR, Ferreon JC, Deniz AA. Alteration of the α-synuclein folding landscape by a mutation related to Parkinson's disease. Angew Chem Int Edit. 2010;49:3469–3472. doi: 10.1002/anie.201000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feynman RP. The Feynman lectures on physics. Vol. 1. Addison-Wesley: Massachusetts, USA; 1963. Rachet and pawl; pp. 1–7. [Google Scholar]
- Feynmann RP. Miniaturization. New York: Reinhold Publishing Corporation; 1961. [Google Scholar]
- Fu J, et al. Multi-enzyme complexes on DNA scaffolds capable of substrate channeling with an artificial swinging arm. Nat Nanotechnol. 2014;9:531–536. doi: 10.1038/nnano.2014.100. [DOI] [PubMed] [Google Scholar]
- Gahlmann A, Moerner WE. Exploring bacterial cell biology with single-molecule tracking and super-resolution imaging. Nat Rev Microbiol. 2014;12:9–22. doi: 10.1038/nrmicro3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiduk A, Yorulmaz M, Ruijgrok PV, Orrit M. Room-temperature detection of a single molecule's absorption by photothermal contrast. Science. 2010;330:353–356. doi: 10.1126/science.1195475. [DOI] [PubMed] [Google Scholar]
- Gorris HH, Rissin DM, Walt DR. Stochastic inhibitor release and binding from single-enzyme molecules. Proc Natl Acad Sci USA. 2007;104:17680–17685. doi: 10.1073/pnas.0705411104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunwald D, Singer RH. In vivo imaging of labelled endogenous beta-actin mRNA during nucleocytoplasmic transport. Nature. 2010;467:604–607. doi: 10.1038/nature09438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunwald D, Singer RH, Rout M. Nuclear export dynamics of RNA-protein complexes. Nature. 2011;475:333–341. doi: 10.1038/nature10318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha T, Tinnefeld P. Photophysics of fluorescent probes for single-molecule biophysics and super-resolution imaging. Annu Rev Phys Chem. 2012;63:595–617. doi: 10.1146/annurev-physchem-032210-103340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada T, et al. Activatable organic near-infrared fluorescent probes based on a bacteriochlorin platform: synthesis and multicolor in vivo imaging with a single excitation. Bioconjug Chem. 2014;25:362–369. doi: 10.1021/bc4005238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Liu DR. Autonomous multistep organic synthesis in a single isothermal solution mediated by a DNA walker. Nat Nanotechnol. 2010;5:778–782. doi: 10.1038/nnano.2010.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins AA, et al. Ordered and dynamic assembly of single spliceosomes. Science. 2011;331:1289–1295. doi: 10.1126/science.1198830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Wang W, Bates M, Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008;319:810–813. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyeon C, Lee J, Yoon J, Hohng S, Thirumalai D. Hidden complexity in the isomerization dynamics of Holliday junctions. Nat Chem. 2012;4:907–914. doi: 10.1038/nchem.1463. [DOI] [PubMed] [Google Scholar]
- Ito Y, Fukusaki E. DNA as a ‘Nanomaterial’. J Mol Catal B-Enzym. 2004;28:155–166. [Google Scholar]
- Johnson-Buck A, Nangreave J, Kim D, Bather M, Yan H, Walter NG. Super-resolution fingerprinting detects chemical reactions and idiosyncrasies of single DNA pegboards. Nano Lett. 2013;13:728–733. doi: 10.1021/nl304415b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Buck A, Walter NG. Discovering anomalous hybridization kinetics on DNA nanostructures using single-molecule fluorescence microscopy. Methods. 2014;67:177–184. doi: 10.1016/j.ymeth.2014.02.032. [DOI] [PubMed] [Google Scholar]
- Joo C, Balci H, Ishitsuka Y, Buranachai C, Ha T. Advances in Single-Molecule Fluorescence Methods for Molecular Biology. Annu Rev Biochem. 2008;77:51–76. doi: 10.1146/annurev.biochem.77.070606.101543. [DOI] [PubMed] [Google Scholar]
- Juette MF, Terry DS, Wasserman MR, Zhou Z, Altman RB, Zheng Q, Blanchard SC. The bright future of single-molecule fluorescence imaging. Curr Opin Chem Biol. 2014;20C:103–111. doi: 10.1016/j.cbpa.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungmann R, Avendano MS, Woehrstein JB, Dai M, Shih WM, Yin P. Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat Methods. 2014;11:313–318. doi: 10.1038/nmeth.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungmann R, Scheible M, Simmel FC. Nanoscale imaging in DNA nanotechnology. WIREs Nanomed Nanobiotechnol. 2012;4:66–81. doi: 10.1002/wnan.173. [DOI] [PubMed] [Google Scholar]
- Kalo A, Kafri P, Shav-Tal Y. Single mRNP tracking in living mammalian cells. Methods Mol Biol. 2013;1042:87–99. doi: 10.1007/978-1-62703-526-2_7. [DOI] [PubMed] [Google Scholar]
- Kellenberger CA, Wilson SC, Sales-Lee J, Hammond MC. RNA-Based Fluorescent Biosensors for Live Cell Imaging of Second Messengers Cyclic di-GMP and Cyclic AMP-GMP. J Am Chem Soc. 2013;135:4906–4909. doi: 10.1021/ja311960g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keryer-Bibens C, Barreau C, Osborne HB. Tethering of proteins to RNAs by bacteriophage proteins. Biol Cell. 2008;100:125–138. doi: 10.1042/BC20070067. [DOI] [PubMed] [Google Scholar]
- König SLB, Liyanage PS, Sigel RKO, Rueda D. Helicase-mediated changes in RNA structure at the single-molecule level. RNA Biol. 2013;10:133–148. doi: 10.4161/rna.23507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretz M, et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature. 2013;493:231–235. doi: 10.1038/nature11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan R, Blanco MR, Kahlscheuer ML, Abelson J, Guthrie C, Walter NG. Biased Brownian racheting leads to pre-mRNA remodeling and capture prior to first-step splicing. Nat Struct Mol Biol. 2013;20:1450–1457. doi: 10.1038/nsmb.2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- Kukura P, Celebrano M, Renn A, Sandoghdar V. Single-molecule sensitivity in optical absorption at room temperature. J Phys Chem Lett. 2010;1:3323–3327. [Google Scholar]
- Küpper J, Stern S, Holmegaard L, Filsinger F, Rouzée A, Rudenko A. X-ray diffraction from isolated and strongly aligned gas-phase molecules with a free-electron laser. Phys Rev Lett. 2014;112:083002. [Google Scholar]
- Lackowickz JR. Principles of Fluorescence Spectroscopy. 3 edn. New York City: Springer; 2007. [Google Scholar]
- Lee JT, Bartolomei MS. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell. 2013;152:1308–1323. doi: 10.1016/j.cell.2013.02.016. [DOI] [PubMed] [Google Scholar]
- Lee W, Jose D, Phelps C, Marcus AH, Hippel PHv. A single-molecule view of the assembly pathway, subunit stoichiometry, and unwinding activity of the bacteriophage T4 primosome (helicase-primase) complex. Biochemistry-US. 2013;52:3157–3170. doi: 10.1021/bi400231s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GW, Xie XS. Central dogma at the single-molecule level in living cells. Nature. 2011;475:308–315. doi: 10.1038/nature10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, et al. Submicrometre geometrically encoded fluorescent barcodes self-assembled from DNA. Nat Chem. 2012;4:832–839. doi: 10.1038/nchem.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Baskin RJ, Kowalczykowski SC. DNA unwinding heterogeneity by RecBCD results from static molecules unable to equilibrate. Nature. 2013;500:482–485. doi: 10.1038/nature12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Bokinsky G, Walter NG, Zhuang X. Dissecting the multi-step reaction pathway of an RNA enzyme by single-molecule kinetic "fingerprinting". Proc Natl Acad Sci USA. 2007;104:12634–12639. doi: 10.1073/pnas.0610597104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HP, Xun L, Xie XS. Single-molecule enzyme dynamics. Science. 1998;282:1877–1882. doi: 10.1126/science.282.5395.1877. [DOI] [PubMed] [Google Scholar]
- Lu Y, Liu J. Functional DNA nanotechnology: emerging applications of DNAzymes and aptamers. Curr Opin Biotech. 2006;17:580–588. doi: 10.1016/j.copbio.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Lund K, et al. Molecular robots guided by prescriptive landscapes. Nature. 2010;465:206–210. doi: 10.1038/nature09012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, et al. High-resolution three-dimensional mapping of mRNA export through the nuclear pore. Nat Commun. 2013;4:2414. doi: 10.1038/ncomms3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Yang W. Three-dimensional distribution of transient interactions in the nuclear pore complex obtained from single-molecule snapshots. Proc Natl Acad Sci USA. 2010;107:7305–7310. doi: 10.1073/pnas.0908269107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek MS, Johnson-Buck A, Walter NG. The shape-shifting quasispecies of RNA: one sequence, many functional folds. Phys Chem Chem Phys. 2011;13:11524–11537. doi: 10.1039/c1cp20576e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RM, Rino J, Carvalho C, Kirchhausen T, Carmo-Fonseca M. Live-cell visualization of pre-mRNA splicing with single-molecule sensitivity. Cell Rep. 2013;4:1144–1155. doi: 10.1016/j.celrep.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell SE, Jun JM, Walter NG. Long-range tertiary interactions in single hammerhead ribozymes bias motional sampling toward catalytically active conformations. RNA. 2010;16:2414–2426. doi: 10.1261/rna.1829110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelotti N, de Silva C, Johnson-Buck AE, Manzo AJ, Walter NG. Chapter Six - A Bird's Eye View: Tracking Slow Nanometer-Scale Movements of Single Molecular Nano-assemblies. In: Walter NG, editor. Methods in Enzymology. Vol. 475. Academic Press; 2010. pp. 121–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelotti N, Johnson-Buck A, Manzo AJ, Walter NG. Beyond DNA origami: the unfolding prospects of nucleic acid nanotechnology. WIREs Nanomed Nanobiotechnol. 2012;4:139–152. doi: 10.1002/wnan.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mor A, Ben-Yishay R, Shav-Tal Y. On the right track: following the nucleo-cytoplasmic path of an mRNA. Nucleus. 2010;1:492–498. doi: 10.4161/nucl.1.6.13515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nienhaus K, Nienhaus GU. Fluorescent proteins for live-cell imaging with super-resolution. Chem Soc Rev. 2014;43:1088–1106. doi: 10.1039/c3cs60171d. [DOI] [PubMed] [Google Scholar]
- Omabegho T, Sha R, Seeman NC. A bipedal DNA Brownian motor with coordinated legs. Science. 2009;324:67–71. doi: 10.1126/science.1170336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige JS, Nguyen-Duc T, Song W, Jaffrey SR. Fluorescence imaging of cellular metabolites with RNA. Science. 2012;335:1194. doi: 10.1126/science.1218298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HY, et al. Visualization of dynamics of single endogenous mRNA labeled in live mouse. Science. 2014;343:422–424. doi: 10.1126/science.1239200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson G, Davidson M, Manley S, Lippincott-Schwartz J. Superresolution imaging using single-molecule localization. Annu Rev Phys Chem. 2010;61:345–367. doi: 10.1146/annurev.physchem.012809.103444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira MJB, Nikolova EN, Hiley SL, Jaikaran D, Collins RA, Walter NG. Single VS ribozyme molecules reveal dynamic and hierarchical folding toward catalysis. J Mol Biol. 2008;382:496–509. doi: 10.1016/j.jmb.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Jimenez R, et al. Single-molecule paleoenzymology probes the chemistry of resurrected enzymes. Nat Struct Mol Biol. 2011;18:592–596. doi: 10.1038/nsmb.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps C, Lee W, Jose D, Hippel PHv, Marcus AH. Single-molecule FRET and linear dichroism studies of DNA breathing and helicase binding at replication fork junctions. Proc Natl Acad Sci USA. 2013;110:17320–17325. doi: 10.1073/pnas.1314862110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitchiaya S, Androsavich JR, Walter NG. Intracellular single molecule microscopy reveals two kinetically distinct pathways for microRNA assembly. EMBO Rep. 2012;13:709–715. doi: 10.1038/embor.2012.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitchiaya S, Heinicke LA, Custer TC, Walter NG. Single-molecule fluorescence approaches shed light on intracellular RNAs. Chem Rev. 2014;114:3224. doi: 10.1021/cr400496q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitchiaya S, Krishnan V, Custer TC, Walter NG. Dissecting non-coding RNA mechanisms in cellulo by Single-molecule High-Resolution Localization and Counting. Methods. 2013;63:188–199. doi: 10.1016/j.ymeth.2013.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasher DC, Eckenrode VK, Ward WW, Prendergast FG, Cormier MJ. Primary structure of the Aequorea victoria green-fluorescent protein. Gene. 1992;111:229–233. doi: 10.1016/0378-1119(92)90691-h. [DOI] [PubMed] [Google Scholar]
- Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006;4:e309. doi: 10.1371/journal.pbio.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson A, van Oijen AM. Bacterial replication, transcription and translation: mechanistic insights from single-molecule biochemical studies. Nat Rev Microbiol. 2013;11:303–315. doi: 10.1038/nrmicro2994. [DOI] [PubMed] [Google Scholar]
- Rothemund PWK. Folding DNA to create nanoscale shapes and patterns. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- Roy R, Hohng S, Ha T. A practical guide to single-molecule FRET. Nat Methods. 2008;5:507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda D, Bokinsky G, Rhodes MM, Rust MJ, Zhuang X, Walter NG. Single-molecule enzymology of RNA: Essential functional groups impact catalysis from a distance. Proc Natl Acad Sci USA. 2004;101:10066. doi: 10.1073/pnas.0403575101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda D, Walter NG. Single molecule fluorescence control for nanotechnology. J Nanosci Nanotechno. 2005;5:1–11. doi: 10.1166/jnn.2005.505. [DOI] [PubMed] [Google Scholar]
- Seeman NC. DNA in a material world. Nature. 2003;421:427–431. doi: 10.1038/nature01406. [DOI] [PubMed] [Google Scholar]
- Sengupta P, Van Engelenburg S, Lippincott-Schwartz J. Visualizing cell structure and function with point-localization superresolution imaging. Dev Cell. 2012;23:1092–1102. doi: 10.1016/j.devcel.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shav-Tal Y, Darzacq X, Shenoy SM, Fusco D, Janicki SM, Spector DL, Singer RH. Dynamics of single mRNPs in nuclei of living cells. Science. 2004;304:1797–1800. doi: 10.1126/science.1099754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura O, Johnson FH, Saiga Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J Cell Comp Physiol. 1962;59:223–239. doi: 10.1002/jcp.1030590302. [DOI] [PubMed] [Google Scholar]
- Siebrasse JP, Kaminski T, Kubitscheck U. Nuclear export of single native mRNA molecules observed by light sheet fluorescence microscopy. Proc Natl Acad Sci USA. 2012;109:9426–9431. doi: 10.1073/pnas.1201781109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva Cd, Walter NG. Leakage and slow allostery limit performance of single drug-sensing aptazyme molecules based on the hammerhead ribozyme. RNA. 2009;15:76–84. doi: 10.1261/rna.1346609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Padgett RA. Rates of in situ transcription and splicing in large human genes. Nat Struct Mol Biol. 2009;16:1128–1133. doi: 10.1038/nsmb.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WJ, Strack RL, Jaffrey SR. Imaging bacterial protein expression using genetically encoded RNARNARNA sensors. Nat Methods. 2013;10:873-+. doi: 10.1038/nmeth.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack RL, Disney MD, Jaffrey SR. A superfolding Spinach2 reveals the dynamic nature of trinucleotide repeat-containing RNA. Nat Methods. 2013;10:1219-+. doi: 10.1038/nmeth.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracy M, Uphoff S, Garza de Leon F, Kapanidis AN. In vivo single-molecule imaging of bacterial DNA replication, transcription, and repair. FEBS Lett. 2014 doi: 10.1016/j.febslet.2014.05.026. [DOI] [PubMed] [Google Scholar]
- Strulson CA, Molden RC, Keating CD, Bevilacqua PC. RNA catalysis through compartmentalization. Nat Chem. 2012;4:941–946. doi: 10.1038/nchem.1466. [DOI] [PubMed] [Google Scholar]
- Suddala KC, et al. Single transcriptional and translational preQ1 riboswitches adopt similar pre-folded ensembles that follow distinct folding pathways into the same ligand-bound structure. Nucleic Acids Res. 2013;41:10462–10475. doi: 10.1093/nar/gkt798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Endo M, Yang Y, Sugiyama H. Dynamic assembly/disassembly processes of photoresponsive DNA origami nanostructures directly visualized on a lipid membrane surface. J Am Chem Soc. 2014;136:1714–1717. doi: 10.1021/ja4109819. [DOI] [PubMed] [Google Scholar]
- Uemura S, Aitken CE, Korlach J, Flusberg BA, Turner SW, Puglisi JD. Real-time tRNA transit on single translating ribosomes at condon resolution. Nature. 2010;464:1012–1017. doi: 10.1038/nature08925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kouwenhove M, Kedde M, Agami R. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat Rev Cancer. 2011;11:644–656. doi: 10.1038/nrc3107. [DOI] [PubMed] [Google Scholar]
- Vargas DY, et al. Single-molecule imaging of transcriptionally coupled and uncoupled splicing. Cell. 2011;147:1054–1065. doi: 10.1016/j.cell.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waks Z, Klein AM, Silver PA. Cell-to-cell variability of alternative RNA splicing. Mol Syst Biol. 2011;7:506. doi: 10.1038/msb.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter NG, Huang C, Manzo AJ, Sobhy MA. Do-it-yourself guide: how to use the modern single-molecule toolkit. Nat Methods. 2008;5:475–489. doi: 10.1038/nmeth.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter NG, Perumal S. The small ribozymes: Common and diverse features observed through the FRET lens. In: Walter NG, Woodson SA, Batey RT, editors. Non-protein coding RNAs. Heidelberg, Germany: Springer-Verlag; 2009. pp. 103–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Moffitt JR, Dempsey GT, Xie XS, Zhuang X. Characterization and development of photoactivatable fluorescent proteins for single-molecule-based superresolution imaging. Proc Natl Acad Sci USA. 2014;111:8452–8457. doi: 10.1073/pnas.1406593111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham SFJ, Endo M, Katsuda Y, Hidaka K, Bath J, Sugiyama H, Turberfield AJ. Direct observation of stepwise movement of a synthetic molecular transporter. Nat Nanotechnol. 2011;6:166–169. doi: 10.1038/nnano.2010.284. [DOI] [PubMed] [Google Scholar]
- Wildenberg SMvd, Prevo B, Peterman EJ. A brief introduction to single-molecule fluorescence methods. Methods Mol Biol. 2011;783:81–99. doi: 10.1007/978-1-61779-282-3_5. [DOI] [PubMed] [Google Scholar]
- Xia T, Li N, Fang X. Single-molecule fluorescence imaging in living cells. Annu Rev Phys Chem. 2013;64:459–480. doi: 10.1146/annurev-physchem-040412-110127. [DOI] [PubMed] [Google Scholar]
- Xie XS, Yu J, Yang WY. Living Cells as Test Tubes. Science. 2006;312:228–230. doi: 10.1126/science.1127566. [DOI] [PubMed] [Google Scholar]
- Xu K, Zhong G, Zhuang X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science. 2013;339:452–456. doi: 10.1126/science.1232251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Lin W, Zheng K, He L, Huang W. Far-red to near infrared analyte-responsive fluorescent probes based on organic fluorophore platforms for fluorescence imaging. Chem Soc Rev. 2013;42:622–661. doi: 10.1039/c2cs35313j. [DOI] [PubMed] [Google Scholar]
- Zhang DY, Seelig G. Dynamic DNA nanotechnology using strand-displacement reactions. Nat Chem. 2011;3:103–113. doi: 10.1038/nchem.957. [DOI] [PubMed] [Google Scholar]
- Zhao R, Marshall M, Alemán EA, Lamichhane R, Feig A, Rueda D. Laser-assisted single-molecule refolding (LASR) Biophys J. 2010;99:1925–1931. doi: 10.1016/j.bpj.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Choudhary E, Andoy NM, Zou N, Chen P. Scalable parallel screening of catalyst activity at the single-particle level and subdiffraction resolution. ACS Catal. 2013;3:1448–1453. [Google Scholar]
- Zhuang X, Kim H, Pereira MJB, Babcock HP, Walter NG, Chu S. Correlating structural dynamics and function in single ribozyme molecules. Science. 2002;296:1473–1476. doi: 10.1126/science.1069013. [DOI] [PubMed] [Google Scholar]