

Abstract
Eight new and 10 known compounds, were isolated from an organic extract of the bulbs of Bellevalia eigii as part of a search for anticancer leads from native plants of Jordan. Of these, the series of 16 homoisoflavonoids (1-16), comprise the seven new analogues 7-O-methyl-3′-hydroxy-3,9-dihydropunctatin (3), 6-hydroxy-7-O-methyl-3,9-dihydropunctatin (6), 7,4′-di-O-methyl-3′-hydroxy-3,9-dihydropunctatin (9), 7-O-methylpunctatin (10), 7-O-methyl-3′-hydroxypunctatin (13), 5-hydroxy-7,8-dimethoxychroman-4-one (14), and 7-O-methyl-8-demethoxy-3-hydroxy-3,9-dihydropunctatin (15), as well as the known ferulic acid-derived acrylamide (17), and the new methylthioacrylate bellegimycin (18). The structures were elucidated using a set of spectroscopic and spectrometric techniques; the absolute configurations of compounds 1–9, 15, and 16 were determined using ECD spectroscopy, while a modified Mosher’s ester method was used for compound 18. Optical rotation data for the known compounds 1, 2, and 8 are reported here for the first time. The cytotoxic activities of all compounds were evaluated using the MDA-MB-435 (melanoma) and HT-29 (colon) cancer cell lines. Compounds 4 and 9 were the most potent on the latter cell line with IC50 values of 1.0 and 1.1 μM, respectively. Compounds 1-18 were assessed for antimicrobial activity using a collection of bacteria and fungi; compounds 4 and 12 showed promising activity against the bacterium Mycobacterium smegmatis with MIC values of 17 and 24 μg/mL, respectively.
Homoisoflavonoids, a rare class of natural compounds, are structurally related to the more widely distributed flavonoids. In comparison with flavonoids, homoisoflavonoids have one additional carbon atom in their skeleton. More than 240 natural homoisoflavonoids have been reported with a wide range of biological activities, including antimicrobial, antimutagenic, anti-oxidant, and anti-inflammatory effects.1 Homoisoflavonoids can be classified into five types: 3-benzyl-4-chromanones, 3-benzyl-3-hydroxy-4-chromanones, 3-benzylidene-4-chromanones (E or Z), 3-benzylchrom-2-en-4-ones, and the scillascillins.2
Being a floral bridge between the continents of Asia, Africa, and Europe, with ecologically diverse natural habitats, Jordan is richer in biodiversity than many realize.3,4 More than 2000 plant species were reported to grow in the wild,3 with only a small percentage explored for bioactive secondary metabolites. As part of a long-term research project to examine the flora of Jordan for anticancer leads,5–8 Bellevalia eigii Feinbrun was investigated. B. eigii is a perennial plant belonging to the family Asparagaceae, subfamily Scilloideae, which may also be treated as a separate family Hyacinthaceae.2,9 Eleven species were reported to grow in the wild in Jordan.3 B. eigii, which is known locally as “the Jordan Valley onion”, is known for its toxicity among local people. Promising cytotoxic activity of the organic extract of the bulbs led to the isolation and characterization of compounds 1-18, including 16 homoisoflavonoids, nine known (1, 2, 4, 5, 7, 8, 11, 12, and 16) and seven new analogues 7-O-methyl-3′-hydroxy-3,9-dihydropunctatin (3), 6-hydroxy-7-O-methyl-3,9-dihydropunctatin (6), 7,4′-di-O-methyl-3′-hydroxy-3,9-dihydropunctatin (9), 7-O-methylpunctatin (10), 7-O-methyl-3′-hydroxypunctatin (13), 5-hydroxy-7,8-dimethoxychroman-4-one (14), and 7-O-methyl-8-demethoxy-3-hydroxy-3,9-dihydropunctatin (15). The known phenolic amide, N-trans-feruloyltyramine (moupinamide) (17) and a new methylthioacrylate derivative, bellegimycin (18) were also isolated. The absolute configurations of compounds 1–9, 15, and 16 were determined using ECD spectroscopy, while for 18 a modified Mosher’s ester method was used. There has been growing interest in homoisoflavonoids because of their broad range of biological activities, including cytotoxic and antimicrobial effects.1 Hence, these compounds were tested for their cytotoxicity in the MDA–MB–435 (melanoma) and HT–29 (colon) cancer cell lines and for antimicrobial activity in an assemblage of bacteria and fungi.
RESULTS AND DISCUSSION
The CH2Cl2 extract of the air-dried bulbs of B. eigii was reconstituted in a mixture of 5:4:1 H2O:CHCl3:MeOH. The organic layer was separated and dried under reduced pressure. To the dried organic extract was added equal volumes of CH3CN–MeOH and hexanes. The CH3CN–MeOH fraction was evaporated to dryness. The brine shrimp test was utilized5,10,11 as an initial screen for cytotoxic activity; a promising LC50 value of 28.1 μg/mL was observed. The organic extract was then fractionated using flash chromatography to produce 10 fractions. Of these, fractions 3–8 showed interesting HPLC profiles, and hence were further purified using HPLC (preparative and semipreprative). Compounds 1–18 were isolated, the purities of which were evaluated by UPLC (Figure S1).
Sixteen homoisoflavonoids 1–16 were isolated and identified using HRMS, 1D/2D NMR, and ECD data. The experimental and reported spectroscopic and spectrometric data were used to define the structures of the known analogues. Compound 1 (68.6 mg), obtained as a vitreous solid, had a molecular formula of C18H18O6 as established by HRESIMS and 13C NMR data. The NMR, HRMS, and ECD data12 (Figures S2 and S3, Supporting Information) identified 1 as the known 3-benzylchroman-4-one-type homoisoflavonoid, 7-O-methyl-3,9-dihydropunctatin, which was described for the first time by Adinolfi and coauthors in 1984 from the bulbs of Muscari comosum.13 The specific rotation of 1 was determined as −92 (c 0.1, MeOH)] for the first time.
In addition to 1, the following known homoisoflavonoids were isolated and identified, 8-O-demethyl-7-O-methyl-3,9-dihydropunctatin12–14 (2) [4.6 mg, −82 (c 0.1, MeOH)]; 5,7-dihydroxy-3-(3′-hydroxy-4′-methoxybenzyl)-4-chromanone (4) (1.9 mg);12,14,15 5,6-dihydroxy-3-(4-hydroxybenzyl)-7-methoxy-4-chromanone (5) (0.9 mg);16 4′,5,7-trihydroxyhomoisoflavanone (7) (1.1 mg);14,16 3′,4′,5,7-tetrahydroxyhomoisoflavanone (8) [1.0 mg, −90 (c 0.1, MeOH)];17 demethyleucomin (11) (0.9 mg);18 punctatin (12) (0.5 mg);19 and isomuscomosin (16) (0.5 mg).20 The specific rotations of 2 and 8 were not reported previously and are hence provided herein.

Compound 3 (10.2 mg) was isolated as a yellowish oil with a molecular formula of C18H18O7 as determined by HRESIMS and NMR data (Tables 1 and 2, Figure S5, Supporting Information). Analysis of the NMR data of 3 indicated a 3-benzylchroman-4-one homoisoflavonoid with structural similarity to 1. However, the aromatic A2B2 system of ring B in 1 was replaced by an ABM spin system in 3 (δH 6.73, s; 6.79, d, J = 7.5; and 6.62, d, J = 7.5, for H-2′, H-5′, and H-6′, respectively), indicating the presence of a 1,3,4-trisubstituted benzene ring. These data along with a deshielded C-3′ resonance in 3 (δC 144.0) relative to 1 (δC 115.7) and a 16 amu difference in the HRMS data indicated oxygenation at C-3′ in 3. Further examination of the 2D NMR data established the structure of 3 (Figure 1) as 7-O-methyl-3′-hyroxy-3,9-dihydropunctatin. The absolute configurations of 3-benzylchroman-4-one-type homoisoflavonoids have been determined by electronic circular dichroism (ECD) spectroscopy,12 where a negative Cotton effect in the 287–295 nm region of the ECD curves is indicative of a 3R configuration.12 As such, a negative Cotton effect at 299 nm in the ECD spectrum of compound 3 (Δε = −17.7) supported an R-configuration at C-3 (Figure 2).
Table 1.
1H NMR Data for Compounds 3, 6, 9, 10, 13, and 14 (500 MHz in CDCl3) and 15 (700 MHz in Methanol-d4)a
| position | 3b | 6b | 9b | 10b | 13c | 14b | 15c |
|---|---|---|---|---|---|---|---|
| 2 | 4.17, dd (11.5, 6.9) | 4.17, dd (11.5, 7.5) | 4.18, dd (11.5, 7.5) | 5.34, d (1.2) | 5.40, s | 4.52, t (6.3) | 3.98, d (11.3) |
| 4.30, dd (11.5, 3.4) | 4.31, dd (11.5, 4.0) | 4.33, dd (11.5, 4.6) | 4.08, d (11.3) | ||||
| 3 | 2.77, m | 2.83, m | 2.83, m | 2.79, t (6.3) | |||
| 6 | 6.09, s | 6.09, s | 6.11, s | 6.19, s | 6.08, s | 6.08, d (2.1) | |
| 8 | 6.06, d (2.1) | ||||||
| 9 | 2.64, dd (13.8, 10.3) | 2.72, dd (13.8, 10.3) | 2.66, dd (13.8, 10.3) | 7.79, t (1.2) | 7.73, s | 3.88, s | 2.89, d (14.1) |
| 3.07, dd (13.8, 4.0) | 3.14, dd (13.8, 4.6) | 3.16, dd (13.8, 4.6) | 2.91, d (14.1) | ||||
| 10 | 3.87, s | 4.09, s | 3.88, s | 3.88, s | 3.88, s | 3.77, s | 3.83, s |
| 11 | 3.76, s | 3.80, s | 3.76, s | 3.76, s | 3.72, s | ||
| 2′ | 6.73, s | 7.10, d (8.6) | 6.80, d (2.3) | 7.20, d (7.5) | 6.85, s | 7.07, d (8.5) | |
| 3′ | 6.79, d (8.6) | 6.91, d (7.5) | 6.71, d (8.5) | ||||
| 5′ | 6.79, d (7.5) | 6.79, d (8.6) | 6.78, d (8.6) | 6.91, d (7.5) | 6.87, d (8.6) | 6.71, d (8.5) | |
| 6′ | 6.62, d (7.5) | 7.10, d (8.6) | 6.69, dd (8.6, 2.3) | 7.20, d (7.5) | 6.81, d (8.6) | 7.07, d (8.5) | |
| 7′ | 3.87, s | ||||||
| 5-OH | 12.02, s | 11.66 | 12.05, s | 12.68, s | 11.99, s | ||
| 6-OH | 5.15, s | ||||||
| 3′-OH | 5.59, s | ||||||
| 4′-OH | 4.75, s | 5.72, br. s |
δ in ppm, mult (J in Hz).
In CDCl3.
In methanol-d4.
Table 2.
13C NMR Data for 10 (100 MHz), 3, 6, 9, 13, 14 (125 MHz) and for 15 (175 MHz) in CDCl3
| position | 3a | 6a | 9a | 10a | 13b | 14a | 15b |
|---|---|---|---|---|---|---|---|
| 2 | 69.4 | 69.6 | 69.5 | 67.8 | 69.1 | 67.1 | 73.1 |
| 3 | 47.0 | 47.6 | 47.0 | 127.6 | 127.9 | 36.8 | 73.7 |
| 4 | 198.5 | 199.6 | 198.3 | 185.8 | 187.1 | 196.2 | 200.5 |
| 4a | 102.4 | 103.7 | 102.5 | 103.2 | 104.1 | 103.3 | 102.3 |
| 5 | 160.5 | 145.2 | 160.4 | 161.1 | 160.4 | 160.3 | 165.7 |
| 6 | 93.2 | 130.9 | 93.1 | 93.4 | 94.1 | 93.1 | 96.2 |
| 7 | 161.6 | 148.9 | 161.5 | 161.5 | 162.8 | 161.5 | 169.9 |
| 8 | 129.3 | 133.2c | 129.6 | 129.5 | 130.6 | 129.6 | 95.0 |
| 8a | 153.8 | 147.7 | 153.9 | 153.0 | 154.5 | 154.0 | 164.4 |
| 9 | 32.4 | 32.0 | 32.3 | 137.6 | 139.4 | 56.4 | 40.8 |
| 10 | 56.5 | 61.5 | 56.4 | 56.4 | 56.9 | 61.6 | 56.5 |
| 11 | 61.6 | 61.9 | 61.6 | 61.6 | 61.7 | ||
| 1′ | 130.6 | 130.0 | 130.9 | 127.1 | 127.2 | 126.9 | |
| 2′ | 116.3 | 130.5 | 115.3 | 132.5 | 118.4 | 132.9 | |
| 3′ | 144.0 | 115.8 | 145.9 | 116.1 | 147.0 | 116.0 | |
| 4′ | 142.7 | 154.5 | 145.6 | 157.3 | 149.8 | 157.7 | |
| 5′ | 115.7 | 115.8 | 110.9 | 116.1 | 116.7 | 116.0 | |
| 6′ | 121.8 | 130.4 | 120.9 | 132.5 | 125.2 | 132.9 | |
| 7′ | 56.2 |
In CDCl3.
methanol-d4.
obtained from HMBC
Figure 1.

Key COSY and HMBC correlations of 3, 6, 9, 10, 13–15, and 18.
Figure 2.

ECD spectra for A) (3) [0.09 mM, MeOH, cell length 2 cm], B) (6) [0.09 mM, MeOH, cell length 2 cm], C) (9) [0.08 mM, MeOH, cell length 2 cm], and D) (15) [0.09 mM, MeOH, cell length 2 cm].
Compound 6 (0.3 mg), which was obtained as a yellow amorphous powder, had a molecular formula of C18H18O7 as determined by HRESIMS and NMR data (Tables 1 and 2, Figure S8, Supporting Information). The NMR data of 6 indicated a 3-benzylchroman-4-one-type homoisoflavonoid with structural similarity to 1. However, the aromatic proton singlet of ring A (δH 6.09 for H-6) in 1 was replaced by an exchangeable proton singlet (δH 5.15) in 6. The deshielded C-6 (δC 130.9) resonance in 6 relative to that in 1 (δC 93.1), along with a 16 amu difference in the HRMS data between 6 and 1, indicated hydroxylation at C-6, which was confirmed by HMBC correlations from the 6-OH proton to C-5 and C-7. The structure of compound 6 was inferred by further analysis of the 2D NMR data (Figure 1). The trivial name 6-hydroxy-7-O-methyl-3,9-dihydropunctatin was assigned to compound 6. A negative Cotton effect at 293 nm in the ECD spectrum of compound 6 (Δε = −6.1), indicated an R-configuration at C-3 (Figure 2).
Compound 9 (1.1 mg) was obtained as a white amorphous powder. HRESIMS and NMR data indicated a molecular formula of C19H20O7 (Tables 1 and 2, Figure S11, Supporting Information). Analysis of the NMR data indicated compound 9 as a 3-benzylchroman-4-one-type of homoisoflavonoid. The compound showed high structural similarity to 3. Compound 9 showed an extra methoxy group, as indicated by 1H and 13C NMR data (δH/δC 3.87/56.2), consistent with the 14 amu difference in the HRMS data of 9 relative to 3. An HMBC correlation from the 7′-OCH3 protons to C-4′ (δC 145.6) confirmed its connectivity. The structure of compound 9 was deduced by further inspection of the 2D NMR, including COSY and HMBC spectra (Figure 1). Compound 9 was given the trivial name 7,4′-di-O-methyl-3′-hydroxy-3,9-dihydropunctatin. The ECD spectrum of 9 showed a negative Cotton effect (Δε = −21.1) at 297 nm, indicating an R-configuration at C-3 (Figure 2).
Compound 10 (6.7 mg), with a molecular formula of C18H16O6, was isolated as a yellow amorphous powder (Tables 1 and 2; Figure S12, Supporting Information). Analyses of the spectroscopic and spectrometric data suggested 10 as a homoisoflavonoid derivative showing structural similarity with 1. Key differences were replacement of the signals for H-3/C-3 and H2-9/C-9 in 1 by a quaternary olefinic carbon (δC 127.6) and an olefinic methine (δH/δC 7.79/137.6, t, J = 1.2 Hz) in 10, respectively. The H2-2 chemical shift (δH 5.34, d, J = 1.2 Hz) in 10 was deshielded relative to that in 1. The carbonyl carbon in 10 (δC 185.8) was shielded relative to 1 (δC 198.5), indicating conjugation with a double bond. These data, along with a 2 amu difference between 1 and 10, indicated the presence of a Δ3,9 double bond in 10. The geometry of the double bond was established as E, deduced from the long-range coupling between H2-2 and H-9 and by the unusual deshielding of the olefinic proton H-9 (δH 7.79).18,21 The structure of 10 was established as a 3-benzylidene-4-chromanone-type homoisoflavonoid by further examination of the 2D NMR data (Figure 1). The trivial name 7-O-methylpunctatin was given to compound 10.
Compound 13 (0.7 mg), with a molecular formula of C18H16O7, was obtained as a yellow amorphous powder (Tables 1 and 2, Figure S15, Supporting Infromation). The NMR data of 13 indicated a 3-benzylidene-4-chromanone-type homoisoflavonoid with structural similarity to 10 including the E-configuration of the Δ3,9 double bond. However, 13 lacked the aromatic A2B2 spin system of ring B, which was replaced by an ABC aromatic proton spin system (δH 6.85, s; 6.87, d, J = 8.6; and 6.81, d, J = 8.6, for H-2′, H-5′, and H-6′, respectively), indicative of a 1,3,4-trisubstituted benzene ring. These data, along with a deshielded C-3′ resonance in 13 (δC 147.0) relative to 10 (δC 116.1) and a 16 amu difference in the HRMS data, indicated oxygenation at C-3′ in 13. The structure of 13 was established by further inspection of the 2D NMR data such as COSY and HMBC experiments (Figure 1), and was named as 7-O-methyl-3′-hydroxypunctatin.
Compound 14 (0.4 mg) was isolated as a white amorphous powder with a molecular formula of C11H12O5 (Tables 1 and 2, Figure S16, Supporting Information). Key structural features of 14 include two mutually coupled methylenes (δH 4.52, t, J = 6.3 Hz and 2.79, t, J = 6.3 Hz, for H2-2 and H2-3, respectively). The deshielded singlet, δH 11.99, was attributed to the hydrogen-bonded 5-hydroxy group in ring A, which also bore the proton responsible for the singlet at δH 6.08 and the two methoxy groups (δH 3.88 and 3.77 for 9- and 10-OCH3, respectively). The structure of 14 was established by further examination of the HMBC NMR data (Figure 1), and was defined as 5-hydroxy-7,8-dimethoxychroman-4-one.
Compound 15 (0.4 mg) was obtained as a white amorphous powder with a molecular formula of C17H16O6 (Tables 1 and 5; Figure S17, Supporting Information), establishing an index of hydrogen deficiency of 10. Analyses of the HRMS and NMR data suggested 15 as a homoisoflavonoid derivative with structural similarity with 7, including 1H NMR signals of an aromatic A2B2 system (δH 7.07 and 6.71, 2H, d, J = 8.5 Hz) and two meta coupled aromatic protons (δH 6.08, d, J = 2.1 Hz and 6.06, d, J = 2.1 Hz, for H-6 and H-8, respectively). A key difference was replacement of the H-3/C-3 (δH/δC 2.81/48.2) functionality in 7 by a tertiary oxygenated carbon (δC 73.7) in 15 indicating hydroxylation at C-3 in 15 relative to 7. Compound 15 showed a methoxy group (δH/δC 3.83/56.5), located at C-7 as indicated by an HMBC correlation from H3-10 to C-7. The structure of 15 was deduced by further examination of the 2D NMR data such as COSY and HMBC spectra (Figure 1), and was named 7-O-methyl-8-demethoxy-3-hydroxy-3,9-dihydropunctatin. The ECD spectrum of 15 showed a negative Cotton effect (Δε = −22.4) at 291 nm, indicating an S-configuration for C-3 (Figure 2).12
Table 5.
Antimicrobial Activities of Compounds 4, 7, 12, and 13
| Minimal inhibitory concentration (μg/mL)a
|
|||||
|---|---|---|---|---|---|
| compound | S. aureus | E. coli | M. smegmatis | C. albicans | A. niger |
| 4 | >138 | >138 | 17 | >138 | >138 |
| 7 | >125 | >125 | 63 | >125 | >125 |
| 12 | >48 | >48 | 24 | >48 | >48 |
| 13 | 93 | >93 | 47 | >93 | >93 |
| Vancomycinb | 0.25 | --- | --- | --- | --- |
| Ampicillinb | --- | 8 | --- | --- | --- |
| Ciprofloxacinb | --- | --- | 2 | --- | --- |
| Amphotericin Bb | --- | --- | --- | 16 | 31 |
Compounds 1–3, 5–6, 8–11, 14–18 were inactive
Positive controls
In addition to compounds 1–16, one known phenolic amide derivative N-trans-feruloyltyramine (17) was isolated and its structure was identified by comparison of its recorded and reported22 NMR and HRMS data.
Compound 18 (1.2 mg) was isolated as a colorless oil with a molecular formula of C12H14O3S as revealed by HRESIMS, 1H, 13C and edited-HSQC NMR data (Figure S20, Supporting Information), indicating an index of hydrogen deficiency of six. Inspection of the NMR data showed signals characteristic of six aromatic carbons and five aromatic protons constituting an AA′BB′C spin system suggestive of a monosubstituted benzene ring (Table 3, Figure S20, Supporting Information). COSY data identified two spin systems H-2′/H-3′/H-4′/H-5′ and 1-OH/H-1/H2-2 with C-1 and C-2 being oxygenated and C-1 attached to the benzene ring. HMBC correlation from 1-OH to C-1′ confirmed C-1 as the attachment point of the aliphatic side chain with the aromatic ring. 13C NMR data indicated an ester functionality (δC 165.6), which was conjugated with the Δ5,6 double bond. The two olefinic protons showed a coupling constant that supported an E configuration (JH-5/H-6 = 15.1 Hz). Further analysis of the NMR data indicated a methyl group (δH/δC 2.33/14.6) that was attached to a sulfur atom. An HMBC correlation from 8-CH3 to C-6 confirmed the sulfur atom as the attachment point of the S-CH3 group to C-6. Also, an HMBC correlation from H2-2 to C-4 confirmed the connectivity of C-2 to the ester functionality. Further examination of the NMR data yielded the planar structure of 18, which was ascribed the trivial name, bellegimycin. The absolute configuration of 18 was assigned via a modified Mosher’s ester method,23 establishing the (1R) configuration (Figure 3). This is the first report of a suphur-containing compound in the Hyacinthaceae. Although the compound may be biosyntheized in B. eigii, there is a possibility it could arise as an artifact. However, we could not prove this one way or the other. The compound was detected in the crude organic extract by HRESIMS, which excludes the possibility of contamination during the fractionation/purification processes.
Table 3.
NMR Data for 18 (500 MHz for 1H, 100 MHz for 13C) in CDCl3
| position | δC | δH mult (J in Hz) |
|---|---|---|
| 1 | 72.9 | 4.98, ddd (8.7, 3.2, 3.2) |
| 2 | 69.5 | 4.21, dd (11.9, 8.7) |
| 4.33, dd (11.9, 3.2) | ||
| 4 | 165.6 | |
| 5 | 112.5 | 5.69, d (15.1) |
| 6 | 148.5 | 7.79, d (15.1) |
| 8 | 14.6 | 2.33, s |
| 1′ | 139.9 | |
| 2′, 6′ | 126.4 | 7.38, dd (7.5, 1.7) |
| 3′, 5′ | 128.8 | 7.36, dd (7.5, 6.9) |
| 4′ | 128.4 | 7.31, tt (6.9, 1.7) |
| 1-OH | 2.57, d (3.2) |
Figure 3.
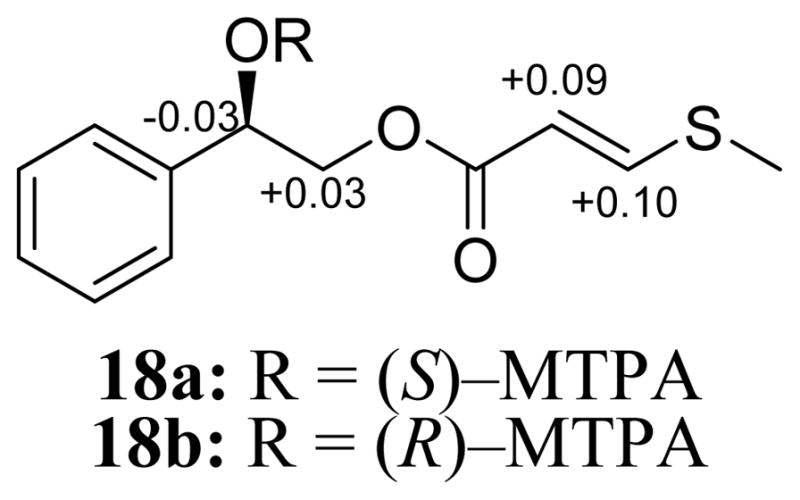
ΔδH values [Δδ (in ppm) = δS – δR] obtained for (S)– and (R)–MTPA esters 18a and 18b, respectively of bellegimycin (18) in pyridine–d5.
The cytotoxicities of compounds 1–18 were measured against the MDA–MB–435 (melanoma) and HT–29 (colon) cancer cell lines. Compounds 4 and 9 were the most potent on the MDA–MB–435 cell line with IC50 values of 1.0 and 1.1 μM, respectively (Table 4). Compound 4 was reported previously to be active against colon cancer (HT-29 cell line ED50 = 2.78 μM) and breast cancer (MDA-MB-435 cell line ED50 = 1.33 μM).24 The cytotoxicity data of the compounds enabled preliminary structure-activity relationships considerations. Introducing an exo-double bond increased the activity significantly. Although compounds 1 and 3 were inactive, compounds 10 and 13 with exo-double bonds showed IC50 values of 13.2 and 4.6 μM, respectively, against the MDA-MB-435 cell line. Introducing a 3′-OH group to compound 10 (13.2 μM) increased the cytotoxic activity (IC50 = 4.6 μM) for compound 13 three fold. Reducing the polarity of highly oxygenated compounds increased the activity as well. For example, while the highly oxygenated compound 8 was inactive, compound 4, with a 4′-methoxy substituent was active (IC50 = 1.0 μM) against the MDA-MB-435 cell line. The same trend was observed for compounds 3 and 9, where the latter with a 4′-methoxy substituent was more active (IC50 = 1.1 μM) against the MDA-MB-435 cell line. In general, less polar substituents contributed to the activity more than the exo-double bond, as seen in compounds 11 and 12, both having exo-double bonds, but they were inactive.
Table 4.
Cytotoxic Activity Evaluation of Active Compounds 4, 9, 10, and 13 Against Colon and Breast Cancer Cell Lines.
| compounda | IC50 values in μMb
|
|
|---|---|---|
| MDA–MB–435 | HT–29 | |
| 4 | 1.0±0.1 | >20 |
| 9 | 1.1±0.1 | 17.3±3.1 |
| 10 | 13.2±0.4 | >20 |
| 13 | 4.6±0.1 | >20 |
| Vinblastinec | 0.0005 | 0.0045 |
Compounds 1–3, 5–8, 11, 12, 14–18 were inactive, IC50 values >20 μM.
IC50 is the concentration to inhibit 50% of growth with a 72 h incubation.
Positive control.
Compounds 1–18 were evaluated for antimicrobial activity using a group of bacteria and fungi (Table 5). Compounds 4 and 12 showed promising activity against the bacterium, Mycobacterium smegmatis, with MIC values of 17 and 24 μg/mL, respectively. As M. smegmatis is used as a surrogate for the pathogenic Mycobacterium species (e.g., M. tuberculosis and M. avium) whose susceptibility to antimicrobials can differ from that of M. smegmatis,25 the susceptibility of pathogenic Mycobacterium species to these novel compounds will be measured in the future studies.
EXPERIMENTAL SECTION
General Experimental Procedures
Description of the NMR, HRESIMS, UV, ECD, IR, UPLC, and HPLC instrumentation were as reported previously.25,26
Plant Material
Bulbs of B. eigii were collected in March/May 2012 from the campus of the Jordan University of Science and Technology (JUST), Irbid, Jordan. The plant material was identified by Mohammad Al-Gharaibeh, Plant Taxonomist, Faculty of Agriculture, JUST. A voucher specimen (PHS-118) was deposited in the herbarium of the Faculty of Pharmacy, JUST. The bulbs were cleaned of mud, sliced into small pieces, and air dried at rt in a well-ventilated area.
Extraction and Isolation
Air-dried bulbs of B. eigii were ground to a powder using a Retsch Mühle mill (RETSCH GmbH, Haan, Germany). About 0.5 kg of powdered bulbs were extracted exhaustively with CH2Cl2 using a Soxhlet apparatus. The solvent was evaporated under reduced pressure to yield about 3 g of CH2Cl2 extract, which was reconstituted in a 2.5 L mixture of 5:4:1 H2O:CHCl3:MeOH. The mixture was stirred for 30 min and left to separate in a separatory funnel. The organic layer was collected and evaporated to dryness under reduced pressure. To the dried organic extract was added 200 mL of 1:1 MeOH/CH3CN and 200 mL of hexanes and shaken vigorously in a separatory funnel. The dried MeOH/CH3CN layer (1.1 g) was dissolved in CHCl3 and mixed with Celite 545. Normal-phase flash chromatography was run using a gradient solvent system of hexanes-CHCl3-MeOH, the flow rate used was 40 mL/min, and column volumes were 53.3 over a total run time of 63.9 min to yield 10 fractions. Fraction 4 (96.0 mg) was purified using preparative HPLC linked with a YMC column using a gradient system of 70:30 to 90:10 of MeOH-H2O (0.1% formic acid) over 15 min at a flow rate of 12 mL/min to yield compounds 1 (65.7 mg) and 10 (5.8 mg). Fraction 5 (54.3 mg) was subjected to preparative HPLC over a Gemini column using a gradient system of 50:50 to 60:40 of MeOH-H2O (0.1% formic acid) over 30 min at a flow rate of 21.24 mL/min to yield four sub-fractions. Further semipreparative HPLC purifications of the sub-fractions yielded compounds 2 (2.7 mg), 4 (1.9 mg), 5 (0.9 mg), 6 (0.3 mg), 15 (0.4 mg), and 16 (0.5 mg). Preparative HPLC purification of fraction 6 (53.4 mg) over a Gemini column, at a flow rate of 21.24 mL/min and a gradient system of 50:50 to 60:40 of MeOH-H2O (0.1% formic acid) over 30 min yielded six subfractions. Further semipreparative HPLC purifications of the subfractions yielded compounds 2 (1.9 mg), 3 (10.2 mg), 12 (0.5 mg), and 13 (0.74 mg). Preparative HPLC purification of fraction 3 (99.1 mg) over a Gemini column using a gradient system of 50:50 to 60:40 of MeOH-H2O (0.1% formic acid) over 30 min at a flow rate of 21.24 mL/min yielded five subfractions. Further semipreparative purifications of the subfractions yielded compounds 1 (2.9 mg), 9 (1.1 mg), 14 (0.9 mg), and 18 (1.2 mg). Fraction 7 (84.6 mg) was purified using preparative HPLC that was connected with a Gemini column. The mobile phase used was a gradient system of 50:50 to 60:40 over 30 min to 100:00 over 10 min of MeOH-H2O (0.1% formic acid) at a flow rate of 21.24 mL/min. The three subfractions were purified to afford compounds 7 (1.1 mg) and 11 (0.9 mg). Fraction 8 (118.1 mg) was purified using preparative HPLC over a Gemini column and a gradient system of 50:50 to 100:00 over 15 min of MeOH-H2O (0.1% formic acid) at a flow rate of 21.24 mL/min. The four subfractions were further purified to yield compounds 8 (1.0 mg) and 17 (0.6 mg).
7-O-Methyl-3,9-dihydropunctatin (1): vitreous solid; −92 (c 0.1, MeOH)]; UV (MeOH) λmax (log ε) 343 (3.26), 289 (3.79), 224 (3.72) nm; ECD (0.9 × 10−4 M, MeOH), see Figure S2, Supporting Information; HRESIMS m/z 331.1169 [M + H]+ (calcd for C18H18O6, 331.1176).
8-O-Demethyl-7-O-methyl-3,9-dihydropunctatin (2): light yellow amorphous powder; −82 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 366 (3.29), 293 (3.72), 245 (3.62), 223 (3.63) nm; ECD (0.9 × 10−4 M, MeOH), see Figure S2, Supporting Information; HRESIMS m/z 317.1012 [M + H]+ (calcd for C17H16O6, 317.1020).
7-O-Methyl-3′-hydroxy-3,9-dihydropunctatin (3): yellowish oil; = −86 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 343 (3.31), 289 (3.76), 224 (3.67) nm; ECD (c 0.9 × 10−4 M, MeOH) λ (Δε) 238 (+5.2) nm, 272 (+6.8) nm, 299 (−17.8) nm (Figure 2); IR (diamond) vmax 3325, 2934, 1627, 1518, 1496, 1445, 1374, 1255, 1200, 1104, 1010, 954, 749 cm−1; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Tables 1 and 2; HRESIMS m/z 347.1118 [M + H]+ (calcd for C18H19O7, 347.1125).
6-Hydroxy-7-O-methyl-3,9-dihydropunctatin (6): yellowish amorphous powder; = −14 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 376 (2.79), 288 (3.42), 223 (3.48) nm; ECD (c 0.9 × 10−4 M, MeOH)λ (Δε) 261 (+2.9) nm, 293 (−6.1) nm (Figure 2); 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Tables 1 and 2; HRESIMS m/z 347.1116 [M + H]+ (calcd for C18H19O7, 347.1125).
3′,4′,5,7-Tetrahydroxyhomoisoflavanone (8): yellow amorphous powder; −89 (c 0.1, MeOH)]; UV (MeOH) λmax (log ε) 290 (3.72), 230 (3.66) nm; ECD (1.0 × 10−4 M, MeOH), see Figure S2, Supporting Information; HRESIMS m/z 303.0859 [M + H]+ (calcd for C16H14O6, 303.0863).
7,4′-di-O-Methyl-3′-hydroxy-3,9-dihydropunctatin (9): white amorphous powder; = −66 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 342 (3.21), 289 (3.71), 225 (3.60) nm; ECD (c 0.8 × 10−4 M, MeOH)λ (Δε) 236 (+7.5) nm, 272 (+4.8) nm, 297 (−21.1) nm (Figure 2); 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Tables 1 and 2; HRESIMS m/z 361.1276 [M + H]+ (calcd for C19H21O7, 361.1282).
7-O-Methylpunctatin (10): yellow amorphous powder; UV (MeOH) λmax (log ε) 391 (3.49), 378 (3.51), 237 (3.42) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 100 MHz), see Tables 1 and 2; HRESIMS m/z 329.1017 [M + H]+ (calcd for C18H17O6, 329.1020).
7-O-Methyl-3′-hydroxypunctatin (13): dark yellow amorphous powder; UV (MeOH) λmax (log ε) 378 (3.75), 264 (3.47), 222 (3.63) nm; 1H NMR (methanol-d4, 500 MHz) and 13C NMR (methanol-d4, 125 MHz), see Tables 1 and 2; HRESIMS m/z 345.0963 [M + H]+ (calcd for C18H17O7, 345.0969).
5-Hydroxy-7,8-dimethoxychroman-4-one (14): white amorphous powder; UV (MeOH) λmax (log ε) 339 (2.44), 288 (3.01), 237 (2.83), 215 (2.93) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Tables 1 and 2; HRESIMS m/z 225.0758 [M + H]+ (calcd for C11H13O5, 225.0758).
7-O-Methyl-8-demethoxy-3-hydroxy-3,9-dihydropunctatin (15): white amorphous powder; = −63 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 289 (3.66), 226 (3.65) nm; ECD (c 0.9 × 10−4 M, MeOH)λ (Δε) 261 (+12.8) nm, 291 (−22.4) nm, 310 (+3.7) nm, 333 (−6.5) nm (Figure 2); 1H NMR (methanol-d4, 700 MHz) and 13C NMR (methanol-d4, 175 MHz), see Tables 1 and 2; HRESIMS m/z 317.1011 [M + H]+ (calcd for C17H17O6, 317.1020).
Bellegimycin [(R)-2-hydroxy-2-phenylethyl-(E)-3-(methylthio)acrylate (18)]: colorless oil; = −8 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 274 (3.25), 212 (2.73) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 100 MHz), see Tables 1 and 2; HRESIMS m/z 239.0732 [M + H]+ (calcd for C12H15O3S, 239.0736).
Preparation of the (R)- and (S)-MTPA Ester Derivatives of Bellegimycin (18)
To 0.13 mg of compound 18 were added 400 μL of pyridine-d5, and the solution was transferred into an NMR tube. To initiate the reaction, 20 μL of S-(+)-α-methoxy-α-(trifluoromethyl)phenylacetyl (MTPA) chloride were added with careful shaking and then monitored immediately by 1H NMR at the following time points: 0, 5, 10, 15, and 30 min. The reaction was found to be complete in 30 min, yielding the mono (R)-MTPA ester derivative (18b) of 18. 1H NMR data of 18b (500 MHz, pyridine-d5): δH 6.67 (1H, dd, J = 8.6, 3.4, H-1), 4.64 (2H, m, H2-2), 5.81 (1H, d, 14.9, H-5), 7.98 (1H, d, 14.9, H-6). In an analogous manner, 0.13 mg of compound 18 dissolved in 400 μL pyridine-d5 was reacted in a second NMR tube with 20 μL (R)(−)-α-MTPA chloride for 30 min, to afford the mono (S)-MTPA ester (18a). 1H NMR data of 18a (500 MHz, pyridine-d5): δH 6.64 (1H, dd, J = 8.6, 4.0, H-1), 4.67 (2H, m, H2-2), 5.90 (1H, d, 14.9, H-5), 8.08 (1H, d, 14.9, H-6).
Cytotoxicity Assay
Compounds (1–18) were tested for cytotoxicity against the MDA-MB-43527 human melanoma (HTB-129, ATCC) and the HT-29 human colon cancer (HTB-38,ATCC) cell lines as described previously.26,28 Briefly, the cell lines were propagated at 37 °C in 5% CO2 in RPMI 1640 medium, which was supplemented with 10% fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin. The cells were harvested by trypsinization while in their log phase and washed twice to remove all traces of enzyme. In every well of a 96-well plate (Microtest 96, Falcon), about 5,000 cells were seeded and incubated overnight in 5% CO2 at 37 °C. Tested compounds dissolved in DMSO were incubated with the cells for 72 h at 37 °C and evaluated for viability with a commercial absorbance assay (CellTiter 96 AQueous One Solution Cell Proliferation Assay, Promega Corp, Madison, WI). IC50 values are expressed in μM relative to the solvent (DMSO) control. Vinblastine was used as a positive control.
Antimicrobial Assay
Minimal inhibitory concentrations (MICs) of compounds (1–15) were measured against a collection of bacteria and fungi by broth microdilution in 96-well microtitre plates as described previously.26 Briefly, in 96-well microtitre plates, a 2-fold dilution series of the tested compounds was prepared in a 50 μL volume of 1/10-strength BHIB+S. The resulted dilution series was inoculated with 50 μL of each cell suspension. After incubation for 4 days, the growth, as noted by turbidity, was scored visually. MIC, which is defined as the lowest concentration of drug resulting in absence of turbidity compared with negative control, was measured for each compound in triplicate. All measurements were made in duplicate.
Supplementary Material
Acknowledgments
This research was supported in part by the Deanship of Research, Jordan University of Science and Technology, Irbid, Jordan and via program project grant P01 CA125066 from the National Cancer Institute/National Institutes of Health, Bethesda, MD, USA.
Footnotes
Notes
The authors declare no competing financial interest.
UPLC chromatograms of compounds 1–18, ECD spectra of known compounds, and 1H and 13C NMR spectra for compounds 1–18. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lin LG, Liu QY, Ye Y. Planta Med. 2014;80:1053–1066. doi: 10.1055/s-0034-1383026. [DOI] [PubMed] [Google Scholar]
- 2.Mulholland DA, Schwikkard SL, Crouch NR. Nat Prod Rep. 2013;30:1165–1210. doi: 10.1039/c3np70008a. [DOI] [PubMed] [Google Scholar]
- 3.Al-Eisawi DM. Field Guide to Wild Flowers of Jordan and Neighbouring Countries. Jordan Press Foundation Al Rai; Amman: 1998. [Google Scholar]
- 4.Feinbrun-Dothan N. Flora Palestina. The Israel Academy of Sciences and Humanities; Jeruselum: 1986. [Google Scholar]
- 5.Alali FQ, El-Elimat T, Li C, Qandil A, Alkofahi A, Tawaha K, Burgess JP, Nakanishi Y, Kroll DJ, Navarro HA, Falkinham JO, Wani MC, Oberlies NH. J Nat Prod. 2005;68:173–178. doi: 10.1021/np0496587. [DOI] [PubMed] [Google Scholar]
- 6.Alali FQ, Amrine CSM, El-Elimat T, Alkofahi A, Tawaha K, Gharaibah M, Swanson SM, Falkinham JO, III, Cabeza M, Sánchez A, Figueroa M, Oberlies NH. Phytochem Lett. 2014;9:96–101. [Google Scholar]
- 7.Alali FQ, Gharaibeh AA, Ghawanmeh A, Tawaha K, Qandil A, Burgess JP, Sy A, Nakanishi Y, Kroll DJ, Oberlies NH. Nat Prod Res. 2010;24:152–9. doi: 10.1080/14786410902941097. [DOI] [PubMed] [Google Scholar]
- 8.Alali FQ, Tahboub YR, Ibrahim ES, Qandil AM, Tawaha K, Burgess JP, Sy A, Nakanishi Y, Kroll DJ, Oberlies NH. Phytochemistry. 2008;69:2341–6. doi: 10.1016/j.phytochem.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 9.Al-Eisawi DM. Jordan Press, Foundation Al-Rai. 1998. pp. 1–2. [Google Scholar]
- 10.McLaughlin JL, Rogers LL. Drug Inf J. 1998;32:513–524. [Google Scholar]
- 11.Meyer BN, Ferrigni NR, Putnam JE, Jacobsen LB, Nichols DE, McLaughlin JL. Planta Med. 1982;45:31–34. [PubMed] [Google Scholar]
- 12.Adinolfi M, Barone G, Corsaro MM, Mangoni L, Lanzetta R, Parrilli M. Tetrahedron. 1988;44:4981–4988. [Google Scholar]
- 13.Adinolfi M, Barone G, Belardini M, Lanzetta R, Laonigro G, Parrilli M. Phytochemistry. 1984;23:2091–2093. [Google Scholar]
- 14.Adinolfi M, Lanzetta R, Laonigro G, Parrilli M, Breitmaier E. Magn Reson Chem. 1986;24:663–666. [Google Scholar]
- 15.Adinolfi M, Barone G, Lanzetta R, Laonigro G, Mangoni L, Parrilli M. Phytochemistry. 1985;24:624–626. [Google Scholar]
- 16.Koorbanally C, Mulholland DA, Crouch NR. Biochem Syst Ecol. 2006;34:588–592. doi: 10.1016/s0305-1978(00)00073-9. [DOI] [PubMed] [Google Scholar]
- 17.Hawksworth DL. Mycol Res. 1991;95:641–655. [Google Scholar]
- 18.Silayo A, Ngadjui BT, Abegaz BM. Phytochemistry. 1999;52:947–955. [Google Scholar]
- 19.Corsaro MM, Lanzetta R, Mancino A, Parrilli M. Phytochemistry. 1992;31:1395–1397. [Google Scholar]
- 20.Adinolfi M, Barone G, Giordano F, Lanzetta Michelangelo Parrilli R. Tetrahedron. 1990;46:6565–6574. [Google Scholar]
- 21.Heller W, Tamm C. In: Progress in the Chemistry of Organic Natural Products. Herz W, Grisebach H, Kirby GW, editors. Vol. 40. Springer; Vienna: 1981. pp. 105–152. [Google Scholar]
- 22.Yoshihara T, Yamaguchi K, Takamatsu S, Sakamura S. Agric Biol Chem. 1981;45:2593–2598. [Google Scholar]
- 23.Hoye TR, Jeffrey CS, Shao F. Nat Protoc. 2007;2:2451–2458. doi: 10.1038/nprot.2007.354. [DOI] [PubMed] [Google Scholar]
- 24.Abegaz B. Phytochem Rev. 2002;1:299–310. [Google Scholar]
- 25.El-Elimat T, Figueroa M, Raja HA, Graf TN, Swanson SM, Falkinham JO, III, Wani MC, Pearce CJ, Oberlies NH. Eur J Org Chem. 2014;2015:109–121. doi: 10.1002/ejoc.201402984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.El-Elimat T, Figueroa M, Raja HA, Swanson SM, Falkinham JO, Lucas DM, Grever MR, Wani MC, Pearce CJ, Oberlies NH. J Antibiot. 2015;68:191–196. doi: 10.1038/ja.2014.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rae J, Creighton C, Meck J, Haddad B, Johnson M. Breast Cancer Res Treat. 2007;104:13–19. doi: 10.1007/s10549-006-9392-8. [DOI] [PubMed] [Google Scholar]
- 28.El-Elimat T, Raja HA, Falkinham JO, Day CS, Oberlies NH. J Nat Prod. 2014;77:2088–2098. doi: 10.1021/np500497r. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.