

Abstract
More than 20 mutations in the gene encoding A-type lamins (LMNA) cause progeria, a rare premature aging disorder. The major pathognomonic hallmarks of progeria cells are seen as nuclear deformations or blebs that are related to the redistribution of A- and B-type lamins within the nuclear lamina. However, the functional significance of these progeria-associated blebs remains unknown. We have carried out an analysis of the structural and functional consequences of progeria-associated nuclear blebs in dermal fibroblasts from a progeria patient carrying a rare point mutation p.S143F (C428T) in lamin A/C. These blebs form microdomains that are devoid of major structural components of the nuclear envelope (NE)/lamina including B-type lamins and nuclear pore complexes (NPCs) and are enriched in A-type lamins. Using laser capture microdissection and comparative genomic hybridization (CGH) analyses, we show that, while these domains are devoid of centromeric heterochromatin and gene-poor regions of chromosomes, they are enriched in gene-rich chromosomal regions. The active form of RNA polymerase II is also greatly enriched in blebs as well as nascent RNA but the nuclear co-activator SKIP is significantly reduced in blebs compared to other transcription factors. Our results suggest that the p.S143F progeria mutation has a severe impact not only on the structure of the lamina but also on the organization of interphase chromatin domains and transcription. These structural defects are likely to contribute to gene expression changes reported in progeria and other types of laminopathies.
Keywords: blebs, chromatin organization, lamins, progeria, transcription
Abbreviations
- DAPI
4′,6-diamidino-2-phenylindole
- DNA
deoxyribonucleic acid
- dUTP
2′-deoxyuridine 5′ triphosphate
- PBS
phosphate buffered saline
- SSC
saline sodium citrate
Introduction
The nuclear lamina is a fibrous meshwork comprised of nuclear lamins and lamina-associated proteins.1 The lamina, which underlies the inner nuclear membrane, determines nuclear size, shape and stability and anchors chromatin at the nuclear periphery.1 A fraction of lamins is also found throughout the nucleoplasm2 where their functions are less well understood.3
Lamins are type V intermediate filament proteins that are grouped into A- and B-types. While B-type lamins [lamin B1 (LB1), lamin B2 (LB2)] are ubiquitously expressed, A-type lamins [lamin A (LA), lamin C (LC)] are primarily expressed in differentiated cells.4,5 It has been shown however, that detectable levels of LA/C exist in mouse embryonic stem cells.6 More than 400 disease mutations in the LMNA gene have been reported, causing at least 15 distinct human diseases, collectively termed the “laminopathies” (http://www.umd.be/LMNA/).7 These diseases include dilated cardiomyopathy, Emery-Dreifuss muscular dystrophy (EDMD), limb girdle muscular dystrophy 1B (LGMD1B), familial partial lipodystrophy, peripheral neuropathy (Charcot-Marie-Tooth disease type 2, CMT2B), Hutchinson Gilford progeria syndrome (HGPS) and other atypical progerias. Of all the laminopathies, HGPS shows the most severe phenotype. HGPS patients suffer from premature aging of skin, bone and the cardiovascular system. Typically they die at an average age of 13 y from complications of atherosclerosis (e.g. myocardial infarction or stroke). Progeria can result from numerous LMNA mutations, which can lead to slightly different clinical phenotypes. The most common mutation is a silent point mutation (1824 C > T), which activates a cryptic splice site resulting in a 50 amino acid deletion at the C-terminus of LA. This mutated form of LA (LAΔ50 or progerin) is accumulated in the nucleus and further interferes with the organization of chromatin and transcription.8,9
Lamins are involved in gene regulation at different levels. There is evidence that lamins can bind DNA directly, and some of the lamin-DNA interactions are mediated through GAGA rich lamina-associated sequences.10,11 In addition, LA can associate with several transcription factors through lamin-associated proteins including LAP2α.12 Interestingly, progerin interacts with a specific subset of repressed genes that are not bound by wild-type LA, suggesting that mutations of lamins may directly alter gene expression.10
Beyond the transcriptional control described above, lamins are thought to affect gene activity at the epigenetic level by reorganizing chromosomes and subchromosomal regions within the nucleus. The lamina is believed to create a transcriptionally repressive environment, which is supported by the fact that some genes change their positions toward a more interior localization upon transcriptional activation.13,14 It has been reported that certain gene-poor chromosomes are relocated to the nuclear interior in proliferating laminopathy cells including cells with mutations causing HGPS, EDMD, LGMD, Dunnigan-type familial partial lipodystrophy (FPLD), Mandibuloacral dysplasia type A (MADA) and CMT2B.15 Furthermore, it has been shown that the expression of progerin triggers mesenchymal stem cell differentiation and activates Notch signaling by releasing the nuclear co-activator SKIP from the nuclear periphery.16
One of the hallmarks of progeria is the occurrence of nuclear deformations including nuclear herniations, lobulations and protrusions.9 We have previously reported that a rare progeria mutation (E145K) in the central rod domain of LA/C is characterized by multilobulated nuclei and centrally clustered centromeres due to defects in postmitotic nuclear assembly.17 The most common progeria mutation (G608G) also leads to the formation of single or multiple nuclear “blebs,” which have been defined as nuclear microdomains with enlarged A-type lamin meshworks and a loss of B-type lamin meshworks.9 However, a detailed analysis of the genetic content of these blebs of progeria nuclei and their transcriptional activity has not been carried out.
In the present study, we have determined which chromosomal regions are located inside blebs in progeria patient cells and whether the formation of these blebs has an impact on gene expression. For this purpose, we have investigated cells from a progeria patient with the p.S143F mutation. This missense mutation is in close proximity to the E145K mutation but results in a progeroid and myopathy phenotype and the cells typically contain nuclei with one large bleb.18,19 Our results show that gene-rich chromosomal regions are preferentially located in blebs and that transcription is not globally inhibited or reduced in this compartment. However, we have observed a loss of the co-activator SKIP, from the lamina region of nuclear blebs when compared to the remainder of the nucleus in p.S143F fibroblasts. Our results suggest that while the transcription machinery does not seem to be compromised in the LA-rich bleb compartment, the precise regulation of transcription by certain activators and repressors may be altered.
Results
Nuclear blebs in p.S143F progeria cells are stable nuclear microdomains
Immunofluorescence analysis showed that cultured skin fibroblasts from a patient carrying the 428 C > T mutation (p.S143F) in the LMNA gene frequently displayed misshapen nuclei, as previously reported (Fig. 1A).18,19 The number of cells with nuclear blebs increased with the accumulation of passage number and was ∼22% by passage number 15 (p15) and ∼60% by p40. 69% of cells with abnormally shaped nuclei showed a single large nuclear bleb, while 19% showed 2 blebs and 12% 3 or more blebs (n > 200). These LA/C rich blebs had very little or no LB1/2 (Fig. 1A) and were similar to those found in fibroblasts carrying lamin mutations associated with various diseases and some prostate cancer cell lines.9,20
Figure 1.
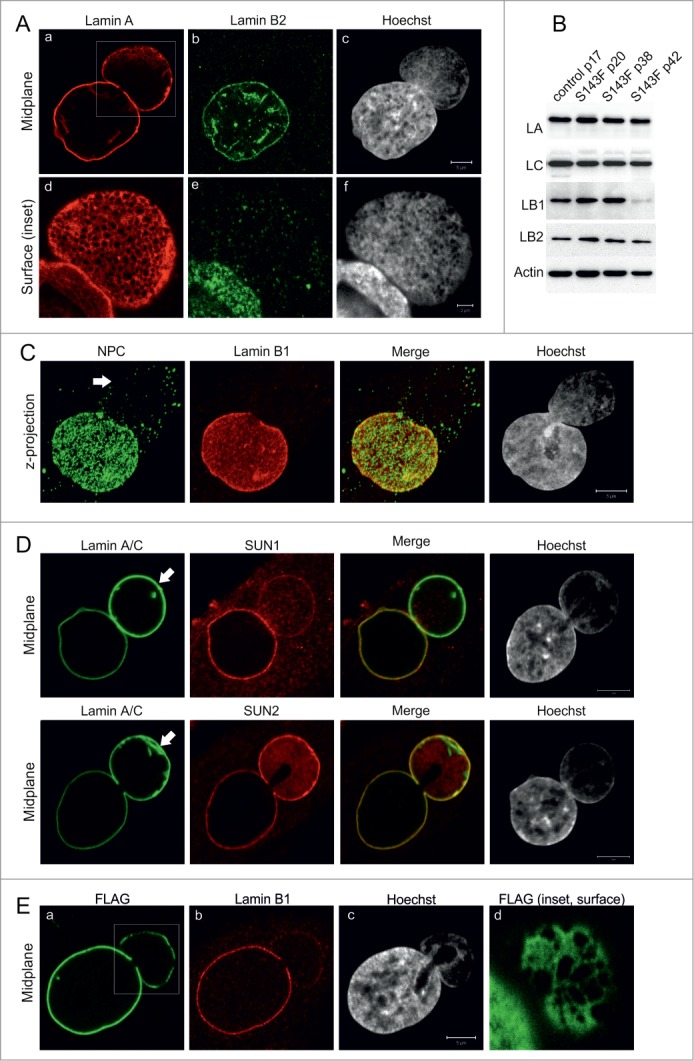
p.S143F mutation in LA induces stable blebs. (A) Primary fibroblasts (p15) from a patient carrying the 428 C > T (p.S143F) mutation in the LMNA gene showed nuclear blebs with enlarged LA/C meshwork and little or no LB2. The cells were stained with antibodies for LA/C and LB2. Hoechst was used to visualize DNA. Single confocal sections from mid-plane (first row) and bleb surface (boxed area, second row) are shown. Note the fine chromatin strands that co-align with the LA/C meshwork in nuclear blebs while DNA staining is generally reduced compared to the main nuclear body. Scale bar 5 μm. (B) The presence of blebs is not due to changes in lamin expression. P.S143F patient fibroblasts at different passages and fibroblasts from a healthy control were harvested and the expression levels of LA/C, LB1 and LB2 were analyzed by western blotting. Actin was used as a loading control. The expression of LB1 was significantly reduced at p42 when the cells stopped proliferating. (C) Blebs are devoid of NPCs. Primary p.S143F fibroblasts (p28) were stained for NPCs and LB1. Hoechst was used to visualize DNA. Z-axis projections of the images in different regions of the whole nucleus are shown. An arrow indicates the bleb. (D) SUN1 and SUN2 localization in blebs. P.S143F fibroblasts were stained for either SUN1 or SUN2, LA/C and Hoechst. Note that SUN1 is markedly reduced while SUN2 is enriched at the NE of blebs. Arrows show blebs. Scale bars 5 μm. (E) The expression of FLAG-tagged p.S143F-LA induces the formation of blebs in cultured HeLa cells. HeLa cells were transiently transfected with a vector encoding FLAG-tagged p.S143F-LA. 72 hours (hrs) after transfection, the cells were stained with anti-FLAG, anti-LB1 and Hoechst. Single mid-plane confocal sections from the whole cell (A-C) and from the surface of the bleb (boxed area, D) are shown. Scale bars 5 μm.
In addition to progeria cells and certain prostate cancer cell lines, we previously described similar blebs induced by silencing LB1 expression in HeLa cells.3 Therefore, the expression levels of LA/C, LB1 and LB2 in p.S143F patient fibroblasts were determined by immunoblotting at different passages. There were no detectable changes in the expression levels of these lamins from p20 to p38 when compared to control fibroblasts, indicating that the presence of blebs is not related to loss of LB1 (Fig. 1B). However, a dramatic reduction of LB1 expression was eventually detected at p42 when the cells stopped proliferating. This finding is consistent with other studies, which showed that the loss of LB1 is a senescence marker in normal diploid cells.21,22
A more detailed analysis of the nuclear envelope (NE) showed several abnormalities in the structure of blebs in p.S143F fibroblasts. Although blebs were occasionally larger in size than the main nuclear body, they were devoid of or had significantly fewer NPCs (Fig. 1C) similar to blebs in HGPS cells with the G608G mutation.9 We also verified that the organization of NPCs was related to the loss of SUN1 in the bleb compartment.23 In contrast, SUN2 was evenly distributed on both the rim of the main nuclear body and the bleb compartment, and it sometimes appeared to be enriched in blebs along with LA (Fig. 1D).
In order to verify that the formation of blebs is due to the p.S143F mutation in the LMNA gene, HeLa cells were transfected with either FLAG-tagged p.S143F (FLAG-p.S143F-LA) or wild type LA (FLAG-WT-LA) as a control. Approximately 17% of cells expressing FLAG-p.S143F-LA showed morphologically similar nuclear blebs with reduced amounts of LB1 and LB2 at 72 h post-transfection, while only ∼0.5% of the cells ectopically expressing FLAG-WT-LA contained structures similar to blebs (Fig. 1E; p = 2.5E-05). These results demonstrate that the p.S143F mutation in LA/C induces large nuclear blebs that have the structural features previously reported for progeria associated nuclear blebs.9 Therefore, p.S143F fibroblasts provide a useful model to study the structure and function of blebs in more detail.
Nuclear blebs are enriched in gene-rich chromosomal regions
We next analyzed the properties of the chromatin structures within blebs. The reduced Hoechst staining intensity in the blebs relative to the remainder of the nucleus suggested that blebs contain primarily euchromatin (Fig. 1, 2). In support of this, we found that the great majority of centromeres were located in the main nuclear body, while few, if any were found in blebs as determined by staining with the CREST antiserum (derived from a patient with scleroderma), which recognizes centromere proteins (Fig. 2A).24 In order to further study the properties of bleb-associated chromatin, the distribution of methylated and acetylated histones was determined by immunofluorescence. Staining for acetylated histone H3 (AcH3), a marker for transcriptionally active euchromatin, showed an increased intensity in ∼75% of blebs (Fig. 2B; n = 100) while staining for histone H3 trimethylated at lysine 9 (H3K9me3), a marker for facultative heterochromatin, was reduced in ∼40% of blebs (Fig. 2C; n = 100). These results indicate that blebs contain mostly transcriptionally active euchromatin.
Figure 2.

Pericentric heterochromatin and epigenetic modifications of chromatin in blebs. (A) p.S143F fibroblasts (p17) were stained for LA/C, LB1 and centromeres. Hoechst was used to visualize DNA. The maximum projection of series of z-sections covering the entire nucleus are shown. A single centromere spot is found in the bleb region (arrow). Scale bar 5 μm. (B) p.S143F fibroblast (p17) stained for LA/C and acetylated histone H3 (AcetH3). A single mid-plane confocal section is shown. The intensity for AcetH3 is increased in the bleb region (arrow). Scale bar 5 μm. (C) p.S143F fibroblast (p17) stained for LA/C and histone H3 trimethylated at lysine 9 (H3K9me3). A single mid-plane confocal section is shown. H3K9me3 is reduced in the bleb region (arrow). Scale bar 5 μm.
To determine which chromosomal regions are preferentially located inside the blebs of the progeria nuclei, we isolated pools of blebs by laser microdissection and analyzed their chromosome content by CGH, using DNA from dissected whole nuclei from the same membrane slide as a reference (Fig. 3A).3 All CGH profiles from 4 separate experiments showed an overrepresentation of a gene-rich region of chromosome 6p (containing the MHC genes) in blebs. The gene-rich chromosomes 17 and 19 were also overrepresented in 4 and 3 out of 4 profiles, respectively. In contrast, the gene-poor chromosome 18 was underrepresented in 3 out of 4 profiles.
Figure 3.

Gene-rich chromosomal regions are preferentially located in blebs.(A) Two examples of CGH profiles from pools of 20–25 blebs each, microdissected from p.S143F patient fibroblasts (p22) and hybridized against a pool of 15–20 whole nuclei from the same sample. The fluorescence intensities are depicted for each chromosome as an intensity ratio profile. Green and red bars indicate overrepresented and underrepresented chromosomal regions within nuclear blebs, respectively. Note the overrepresentation of regions of gene-rich chromosomes 17 and 19 and the underrepresentation of chromosome 18. (B) CGH results were verified by FISH using whole chromosome painting probes for chromosomes 17, and 18. A z-projected nucleus is shown. Note that both chromosomes 18 are located in the main nuclear body, while chromosomes 17 appear in the bleb. Scale bar 5 μm.
The CGH data were verified by fluorescence in situ hybridization (FISH) analysis with whole chromosome painting probes (WCPs). Despite the fact that chromosomes 17 and 18 are similar in size, we found that chromosome 17 was present in 84% of blebs, while only 25% of blebs contained chromosome 18 (n = 100) (Fig. 3B). In addition, the gene-rich short arm of chromosome 6 (6p) was located in 64% of the blebs, while the gene-poorer long arm of chromosome 6 (6q) was detected in 29% of the blebs (n = 100). In summary, the results obtained by CGH and FISH strongly suggest that chromosomal regions are not randomly distributed. Gene-rich chromosomal regions preferentially localize to the bleb compartment.
Transcription is active within blebs
Based on the predominantly gene-rich chromatin structures located within blebs, we speculated that these LA-rich bleb domains may be sites for active gene expression. Staining for RNA polymerase II (Pol II) showed an increased staining intensity in 86% of blebs (n = 50) when compared to the main body of the nucleus (Fig. 4A). In addition, the active form of RNA Pol II phosphorylated at Ser2 (Pol IIo) appeared to be excessively accumulated in 90% of blebs (n = 80), suggesting that the chromosomal regions within these domains are actively transcribed.
Figure 4.
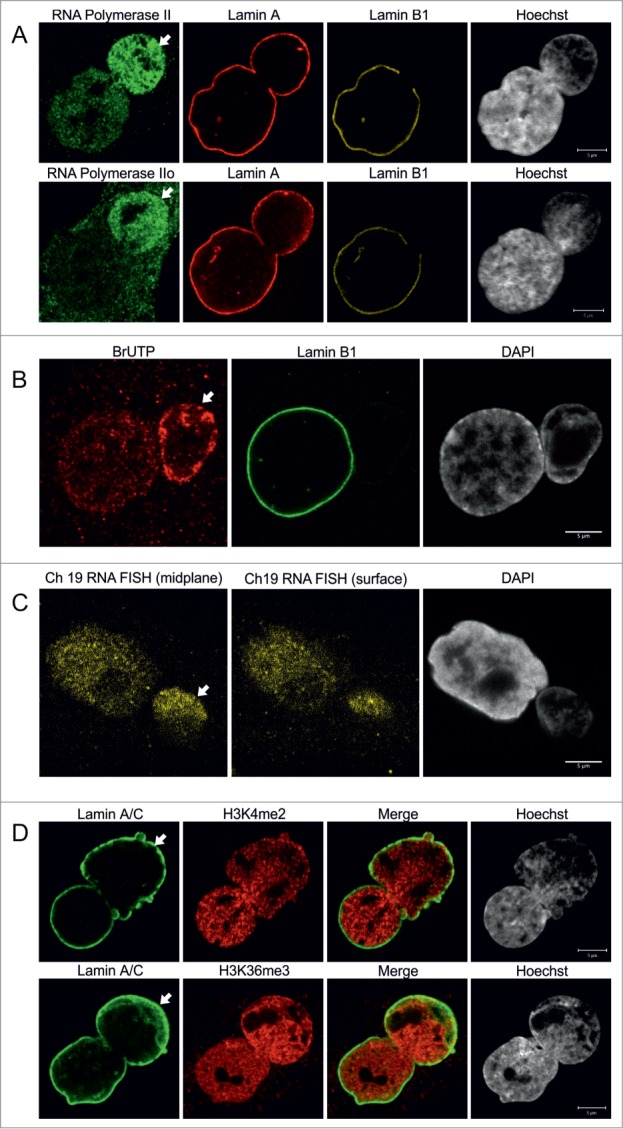
Bleb-associated genetic regions are actively transcribed. (A) Primary p.S143F fibroblasts (p33) were stained for either RNA polymerase II (first row) or active RNA polymerase II (Pol IIo, second row), LA and LB1. Hoechst was used to visualize DNA. Single mid-plane confocal sections from the whole cell are shown. RNA polymerase II and active RNA polymerase II (Pol IIo) are enriched in the blebs (arrows). Scale bars 5 μm. (B) Transcriptional activity in p.S143F fibroblasts (p30) was visualized by BrUTP incorporation into nascent RNA by scratch labeling. DAPI was used to visualize DNA. A single mid-plane confocal section is shown. Note the intense BrUTP staining in the bleb compartment (arrow). Scale bar 5 μm. (C) p.S143F fibroblasts (p36) were subjected to RNA FISH with a probe for a highly transcribed region on chromosome 19. DAPI was used to visualize DNA. Single confocal sections from mid-plane and nuclear surface are shown. FISH signals are detectable in the bleb region (arrow). Scale bar 5 μm. (D) p.S143F fibroblasts (p21) were stained for LA/C and histone H3 trimethylated at lysine 4 or lysine 36. Hoechst was used to visualize DNA. Single mid-plane confocal sections are shown. Both histone marks are detectable in the bleb region (arrows). Scale bars 5 μm.
To investigate whether nascent RNA is located within blebs, we incorporated BrUTP by scratch labeling followed by fixation and staining for BrUTP at 10, 20 and 30 min. Nascent RNA could be detected in 97% of blebs (n = 36) and typically the intensity of the BrUTP staining was equal to or even more intense than in the main nuclear body (Fig. 4B). This is likely related to the relatively high concentration of gene-rich chromosomal regions in blebs.
We further investigated the expression of genetic loci within blebs by RNA FISH. For this purpose, we chose a probe that spans a transcriptionally active region on chromosome 19. RNA FISH signals were detected throughout the nucleoplasm including the bleb region, and larger foci likely representing the actual transcription loci were occasionally detected (Fig. 4C). These observations were further confirmed by staining for other epigenetically modified histones. Histone H3 dimethylated at lys4 (H3K4me2) which marks promotor regions and therefore visualizes transcription initiation, as well as histone H3 trimethylated at Lys36 (H3K36me3), a marker for transcription elongation, were both observed in association with chromatin in blebs (Fig. 4D, n = 100 for both). In summary, these results suggest that transcription initiation and elongation can efficiently take place in the LA-rich domains within blebs.
Finally, we tested by indirect immunofluorescence whether the formation of blebs has an impact on the distribution of transcription factors that have been shown to play a role in progeria. The nuclear co-activator SKIP is known to be lost from the periphery in progerin-expressing mesenchymal stem cells, to activating genes involved in the Notch pathway, which in turn, trigger differentiation.16 We noticed that there was a significant decrease in SKIP signals inside blebs (n = 50), compared to the remainder of the nucleus (Fig. 5). However, this did not appear to be a general feature of transcription factors as other factors including p53 and pRb were equally distributed throughout the nucleus including blebs (Fig. 5 and data not shown).
Figure 5.
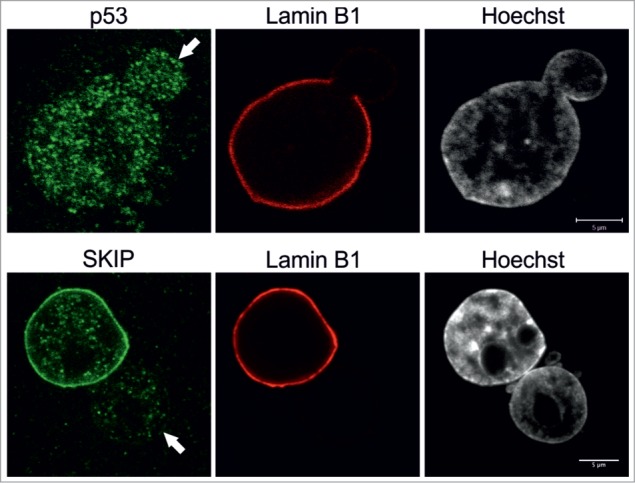
Nuclear co-activator SKIP is reduced in blebs. Primary p.S143F fibroblasts (p36 and p25, respectively) were stained for either SKIP or p53 and LB1. Hoechst was used to visualize DNA. Single mid-plane confocal sections from the whole cell are shown. SKIP is localized at the lamina in the main nuclear body but absent in the blebs (indicated with arrows) while p53 is equally distributed throughout the nucleoplasm. Scale bars 5 μm.
Discussion
Nuclear blebs have been described as one of the hallmarks of HGPS patient cells bearing the mutation G608G, which causes the permanent farnesylation of LA. Blebs occur more frequently when the cells age in culture. This is likely due to the accumulation of progerin, the mutant form of LA.9 Some reports have shown that blocking the farnesylation of prelamin A by farnesyl transferase inhibitors (FTIs) or N6-isopentenyladenosine improves nuclear morphology (e.g., reduced blebbing) of HGPS cells.25-27 Since it has been reported that progeria patients benefit from FTI (lonafarnib) treatment28, this suggests that the nuclear shape abnormalities are correlated with disease progression.
We previously reported that cells from a patient with a very rare progeria mutation (E145K) are characterized by multilobulated nuclei and clustering of centromeres in the middle of the nucleus because of anaphase defects.17 These lobulations, however, have some LB1 and are structurally distinct from the larger blebs found in nuclei of fibroblasts derived from patients bearing the more frequently encountered G608G progeria mutation. Similar large nuclear blebs are also commonly found in cells expressing other laminopathy-associated mutant forms of lamins which do not result in permanent farnesylation, but little is known about their functional significance.29
In this study, we have characterized the structure and functions of nuclear blebs in dermal fibroblasts from a progeria patient carrying a rare C428T mutation (p.S143F) in the LMNA gene. This particular mutation was selected as patient's cells typically contain a single large nuclear bleb even in early passage cultures.19 Nuclear blebs in p.S143F cells were devoid of B-type lamins and similar to blebs seen in progerin expressing cells.9 The large size of these p.S143F-associated LA enriched bleb domains represents an ideal model for studies of their structural and functional properties.
Our studies by immunofluorescence revealed several structural alterations in the p.S143F blebs. They had no or very few NPCs, and SUN1, a component of the LINC complex, was greatly decreased in the bleb compartment compared to other regions of the NE. In contrast, SUN2 was detected at the NE of both the bleb compartment and the main nuclear body, which was similar to the distribution of LA. The latter observation is in line with reports showing that LA is required for the NE localization of SUN2 but not of SUN1.30-32
There is evidence that the localization of SUN1 in the NE might require binding to chromatin in interphase.33 Further support for this comes from the findings that both SUN proteins appear to function in the removal of membranes from chromatin during NE breakdown in mitosis34 and that SUN1 has been shown to interact with telomeres during meiotic prophase I.35 Taken together, our observations and those from other laboratories suggest that the formation of LA/C-rich blebs devoid of gene-poor heterochromatin, interferes in some way with the localization of SUN1 to the NE.
Other alterations in the LINC complex have been linked to the progression of laminopathies.36 For example, it has been reported that nesprin-2 is greatly reduced throughout the nuclear periphery including the blebs and that the re-expression of the nesprin-2 giant isoform restores a normal nuclear phenotype in p.S143F progeria cells.19 For future research, it will be very important to study the impact of the LINC complex on nuclear shape changes and their role in other laminopathies.36
Our results also show that the LA/C-rich blebs in p.S143F nuclei are devoid of centromeric heterochromatin and are enriched in euchromatin. Using the DNA derived from microdissected blebs, CGH- and FISH-analyses show that the LA-rich domains contain mainly gene-rich chromosomal regions, such as chromosomes 17 and 19, while gene-poor chromosomes such as chromosome 18 are more likely to reside in the main nuclear body. In normal interphase cell nuclei, gene-rich chromosome regions are typically found in the nuclear interior, and gene-poor chromosomes are typically detected at the nuclear periphery.37 This interphase arrangement has been reported to be of great importance for gene activation and transcription as changes in intranuclear position can affect gene silencing or activation.13 Interestingly, it has been shown that gene-poor chromosomes such as 13 and 18 are relocated to the nuclear interior in proliferating laminopathy cells (with mutations causing HGPS, EDMD, LGMD, FPLD, MADA and CMT2B), and this type of arrangement is frequently found in quiescent or senescent normal cells.15,38 Furthermore, the chromosomal content of LA/C-rich blebs in p.S143F progeria patient cells is similar to the blebs induced by LB1-silencing in the nuclei of HeLa cells.3
Based upon the available data and our present findings, it appears that gene-poor chromosome regions are mainly associated with B-type lamins, while gene-rich regions are more closely associated with A-type lamins as seen in blebs. This arrangement is likely established in daughter cells as nuclei reassemble. Our results support the finding by Guelen et al, who showed that LB1 is connected with large gene-poor chromatin domains in human fibroblasts.39 Our observations are also in agreement with McCord et al, who found that gene-poor genomic regions showed decreased levels of H3K27me3 and a reduced association with LA/C in HGPS cells.40 Furthermore, it has been proposed that targeting of chromatin to the lamina is mediated by the interactions of LB1 with multiple dispersed sequence elements (termed LASs).11 These LASs are bound by the transcriptional repressor cKrox in a complex with HDAC3 and Lap2β, supposedly to maintain heterochromatin at the lamina.11
In our earlier study on LB1-silenced cells, we observed that the gene-rich chromosomal regions residing in the blebs of LB1-silenced cells could not be transcribed likely due to a stalling effect of Pol II.3 This is a major difference to our results on p.S143F cells, where we have shown that transcription takes place in the blebs. Apparently, the reduction of LB1 in blebs does not necessarily lead to a stalling of Pol II. This difference in transcription between the blebs of progeria cells and LB1-silenced HeLa cells is puzzling, but this may partly be explained by the amount of LB1 in the whole cell nuclei. While HeLa cells with blebs were devoid of detectable LB1, p.S143F cells with blebs still had significant amounts of LB1 in the non-blebbed regions of the nucleus. This amount of LB1 may be sufficient for transcription to take place in the LB-deficient bleb compartment.
Our results also showed a loss of the transcription factor SKIP within the lamina regions of the p.S143F blebs. SKIP is normally associated with the lamina compared to the nuclear interior. However, p53 appears to be distributed throughout the blebbed and non-blebbed regions of p.S143F nuclei. It was reported by others that the expression of progerin triggers mesenchymal stem cell differentiation by releasing SKIP from the nuclear periphery, which in turn activates genes of the Notch pathway.16 Our results imply that tethering of certain transcription factors, such as SKIP, to the lamina region41,42 can be compromised in LA-rich blebs. Future studies are required to determine whether the altered distribution of SKIP within the bleb compartment has an impact on the transcription of SKIP-regulated genes.
In conclusion, the large LA-rich nuclear blebs in the p.S143F form of progeria contain gene-rich regions, which can be transcribed despite the fact that the expression of the B-type lamins was significantly reduced. However, the distribution of transcriptional repressors and activators such as SKIP can be compromised. These findings should ultimately lead to new insights into our understanding of the alterations in gene expression and nuclear organization in both progeria patients and in other types of laminopathies.
Materials and Methods
Cell culture
Dermal fibroblasts from a progeria patient carrying the 428 C > T (p.S143F) mutation in the LMNA gene have been previously described.18,19 Cells from a healthy donor (AG08470, derived from a 10-year-old female) were obtained from the NIA Aging Cell Culture Repository, Coriell Institute for Medical Research (Camden, NJ) and cultured as described before.9 HeLa cells were obtained from the ATCC and cultured in DMEM supplemented with 10% FCS and antibiotics. For transfection, ∼3 × 105 HeLa cells were plated into a 35 mm culture dish one day before transfection. Two μg of DNA plasmids were introduced using the TransIT-HeLa MONSTER transfection kit (Mirus cat.# MIR2900) in accordance with the manufacturer's instructions. The medium was replaced with fresh medium 24 later later and the cells were fixed 72 h post-transfection. Transfection efficiencies were between 25–30% for all the cultures as confirmed with anti-FLAG staining and immunofluorescence in 3 separate experiments.
DNA plasmids
The pFLAG-CMV2-WT-LA vector has been previously described.17 The vector was converted to pFLAG-CMV2-p.S143F-LA with the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene, cat.# 200522) using the primers 5′-gctcagtgcggcctccttggagttcagca-3′ and 5′-tgctgaactccaaggaggccgcactgagc-3′. All the vectors were verified by DNA sequencing.
Indirect immunofluorescence
Cells grown on glass coverslips were fixed in methanol at −20°C for 5 minutes or in 3.7% formaldehyde in PBS for 10 minutes on ice, followed by permeabilization with 0.1% Triton X-100 in PBS for 10 minutes at room temperature. Primary antibodies used were mouse monoclonal anti-LA/C (1:300; JoL2, Chemicon, cat.# MAB3211), rabbit polyclonal anti-LA (1:1000)43, rabbit polyclonal anti-LB1 (1:1000)2, mouse monoclonal anti-LB2 (1:100; LN43, Abcam, cat.# ab8983), rabbit polyclonal anti-H3K9me3 (1:1000, Abcam, cat.# ab8898) and rabbit polyclonal anti-H3K27me3 (1:1000, Abcam, cat.# ab108245), rabbit polyclonal anti-H3K4me2 (1:1000, Abcam, cat.# ab7766), rabbit polyclonal anti-H3K36me3 (1:1000, Abcam, cat.# ab9050, rabbit polyclonal anti-AcH3 (1:1000, Upstate/Merck Millipore, cat.# 06–599), mouse monoclonal anti-Pol II (1:1000, 4H8, Abcam, cat.# ab5408), mouse monoclonal anti-Pol II phosphorylated at Ser2 (H5, 1:100, Abcam, cat.# ab24758), mouse monoclonal anti-FLAG (1:400, clone M2, Sigma, cat.# F1804), rabbit polyclonal anti-SUN1 and anti-SUN2 (1:40 and 1:20; kindly provided by Didier Hodzic)44,45, goat polyclonal anti-LB1 (1:200 Santa Cruz cat.# sc-6217), mouse anti-BrUTP (1:100, Caltag Laboratories, clone Br-3), rabbit polyclonal anti-p53 (1:100, Imgenex, cat.# IMG-533) and rabbit polyclonal anti-SKIP (NCOA62) (1:100, Abcam, cat.# ab153887). Centromeres were visualized with human CREST antiserum (1:100; provided by Dr. Bill Brinkley, Baylor College of Medicine, TX, USA), and NPCs were detected with mouse monoclonal antibody 414 (1:1000; Covance, cat.# MMS-120R). The secondary antibodies used were goat anti-mouse IgG-Alexa Fluor 488 (Molecular Probes, cat.# A11017), goat anti-mouse IgG-Alexa Fluor 568 (Molecular Probes, cat.# A11031), goat anti-rabbit IgG-Alexa Fluor 488 (Molecular Probes, cat.# A11008), goat anti-rabbit IgG-Alexa Fluor 568 (Molecular Probes, cat.# A11036), goat-anti-human IgG-Alexa Fluor 633 (Molecular Probes, cat.# A21091), donkey anti-mouse Alexa Fluor 488 (Molecular Probes, cat# A21202), donkey anti-goat Alexa Fluor 568 (Molecular Probes, cat.# A11057) and donkey anti-rabbit Cy5 (Jackson Immuno-Resarch Laboratories, Inc., cat.# 71–176–152). DNA was stained with Hoechst 33258 (1 μg/ml in PBS, Molecular Probes, Inc. cat.# H3569) or DAPI.
Fixed and stained cells were imaged with a Zeiss LSM 510 META (Carl Zeiss) microscope using oil immersion objective lenses (PlanApochromat, 63×, 1.40 NA; PlanApochromat 100×, 1.40 NA; Carl Zeiss) and a Leica SP5 using an 100× oil immersion objective. For statistical analysis of primary fibroblasts, a minimum of 100–300 randomly selected cells were analyzed for each passage number and study group. In experiments involving transfection, a minimum of 300 cells were analyzed for nuclear shape in each experiment and the mean +/− SD from 3 separate experiments was determined. Statistical analysis was carried out using the students T-test.
Laser microdissection and comparative genomic hybridization (CGH) analysis
The experimental procedure was previously described.3 Progeria cells were grown on PEN membrane slides (Carl Zeiss) and analyzed by immunofluorescence with anti-LA/C and anti-LB2 to identify nuclei containing LB2-deficient nuclear blebs. Pools of 20–30 blebs were collected from the samples by laser microdissection and prepared for CGH as described.3 Whole nuclei on the same membrane were dissected and served as controls for the CGH experiments. The amplified DNA from isolated nuclear blebs was labeled with digoxigenin-11-dUTP. and the amplified DNA from whole nuclei was labeled with biotin 16-dUTP. The labeled DNA was mixed with an excess of human Cot-1 DNA (Roche, cat.# 05480647001) and 5 ul of salmon testis DNA (Invitrogen, cat.# AM9680). This preparation was hybridized onto normal 46XY metaphase spreads. The biotinylated and digoxigenin-labeled DNA probes were detected with avidin-Cy3.5 (Rockland Inc, cat.# A003–12) and mouse anti-digoxigenin-FITC (Roche, cat.# 11207741910). Chromosomes were counterstained with DAPI. Ratios between control and test DNA were evaluated using the Zeiss Isis CGH software (Carl Zeiss) to identify regions that were over- or underrepresented in nuclear blebs. Four pools each containing 20–25 blebs were analyzed by CGH.
Fluorescence in situ hybridization (FISH) with whole chromosome painting probes (WCPs)
Cells were fixed and pretreated for FISH as described before using DNA from flow sorted chromosomes 17, 18, 6p and 6q.46 FISH probes were indirectly labeled by either Nick-Translation or DOP-PCR with digoxigenin-dUTP or biotin-dUTP. Images were taken using a Leica SP5 confocal microscope with a 100× immersion oil objective lens.
Visualization of nascent RNA by BrUTP scratch labeling
Cells were grown on glass coverslips to 90% confluency. After removal of culture medium, 20 μl of 5 mM BrUTP diluted in complete culture medium was added to the coverslips, which were scratched with the tip of a hypodermic needle as described.47 Immediately after scratching, prewarmed growth medium was added to flood the coverslips and the cells were incubated for 10–20 min at room temperature. Cells were fixed in 4% paraformaldehyde in PBS for 10 min at room temperature and permeabilized with 0.5% Triton X-100 in PBS for 5 min at room temperature. Primary antibodies used were goat anti-LB1 (Santa Cruz, cat.# sc-6217) and mouse anti-BrUTP (Caltag Laboratories, clone Br-3), The secondary antibodies used were donkey anti-mouse Alexa 594 (Life technologies, cat.# A-21203) and donkey anti-goat DyLight 488 (Jackson Immuno Research, cat.# 705–486–147).
RNA Fluorescence in situ hybridization (RNA FISH)
For RNA FISH, a BAC FISH probe (RP11–43N16), which spans a GC-rich region on chromosome 19q13.32 and contains at least 6 genes was used, which was earlier shown to be highly expressed in normal female fibroblasts (Thomas Cremer, personal communication). The probe was hybridized onto cells using a protocol modified from (http://www.singerlab.org/protocols) and the protocol described by Edith Heard (personal communication). Briefly, cells were grown on coverslips, washed with PBS, permeabilized for 5 min in 0,5% Triton X-100 in cytoskeletal (CSK) buffer on ice, fixed in 4% paraformaldehyde in PBS on ice, washed with PBS, equilibrated in 2× SSC buffer and the denatured probe was hybridized onto the cells at 37°C over night. After hybridization, cells were washed twice in 50% formamide in 2× SSC (pH 7.2 – 7.4) at 42°C and twice in 2 × SSC (pH 7.0) at 50 C followed by counterstaining with DAPI.
Immunoblotting
Cells were collected and processed for SDS-PAGE as previously described.17 The protein concentration of each sample was determined using a BCA kit (Pierce cat.# 23227), and 20 μg of each sample was loaded on SDS-gels. Proteins were transferred onto nitrocellulose membranes and blocked with 5% fat-free milk for 1 h. Primary antibodies used were rabbit polyclonal anti-LA and anti-LC (both 1:500)43, rabbit polyclonal anti-LB1 (1:500)2, mouse monoclonal anti-LB2 (1:300; LN43, Abcam) and mouse monoclonal anti-α-actin as a loading control (1:600, Sigma, cat.# A4700). The secondary antibodies used were horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (KPL, cat.# 074–1809), goat anti-rabbit IgG (KPL, cat.# 074–1516) and donkey anti-goat IgG (KPL, all 1:15000). After enhanced chemiluminescense reaction, signals were collected with Kodak Imaging Station (Carestream).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
KB was supported by the Human Frontier Science Program. PT was supported by Sigrid Jusélius Foundation, the Finnish Medical Foundation and the Finnish Cultural Foundation. RDG received support from the Progeria Research Foundation, NCI and TS is supported by the NIGMS.
References
- 1. Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, Goldman RD. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev 2008; 22:832-53; PMID:18381888; http://dx.doi.org/ 10.1101/gad.1652708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moir RD, Montag-Lowy M, Goldman RD. Dynamic properties of nuclear lamins: lamin B is associated with sites of DNA replication. J Cell Biol 1994; 125:1201-12; PMID:7911470; http://dx.doi.org/ 10.1083/jcb.125.6.1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shimi T, Pfleghaar K, Kojima S, Pack CG, Solovei I, Goldman AE, Adam SA, Shumaker DK, Kinjo M, Cremer T, et al. The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev 2008; 22:3409-21; PMID:19141474; http://dx.doi.org/ 10.1101/gad.1735208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rober RA, Weber K, Osborn M. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. Development 1989; 105:365-78; PMID:2680424 [DOI] [PubMed] [Google Scholar]
- 5. Stuurman N, Heins S, Aebi U. Nuclear lamins: their structure, assembly, and interactions. J Struct Biol 1998; 122:42-66; PMID:9724605; http://dx.doi.org/ 10.1006/jsbi.1998.3987 [DOI] [PubMed] [Google Scholar]
- 6. Eckersley-Maslin MA, Bergmann JH, Lazar Z, Spector DL. Lamin A/C is expressed in pluripotent mouse embryonic stem cells. Nucleus 2013; 4:53-60; PMID:23324457; http://dx.doi.org/ 10.4161/nucl.23384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schreiber KH, Kennedy BK. When lamins go bad: nuclear structure and disease. Cell 2013; 152:1365-75; PMID:23498943; http://dx.doi.org/ 10.1016/j.cell.2013.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003; 423:293-8; PMID:12714972; http://dx.doi.org/ 10.1038/nature01629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 2004; 101:8963-8; PMID:15184648; http://dx.doi.org/ 10.1073/pnas.0402943101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kubben N, Adriaens M, Meuleman W, Voncken JW, van Steensel B, Misteli T. Mapping of lamin A- and progerin-interacting genome regions. Chromosoma 2012; 121:447-64; PMID:22610065; http://dx.doi.org/ 10.1007/s00412-012-0376-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zullo JM, Demarco IA, Pique-Regi R, Gaffney DJ, Epstein CB, Spooner CJ, Luperchio TR, Bernstein BE, Pritchard JK, Reddy KL, et al. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell 2012; 149:1474-87; PMID:22726435; http://dx.doi.org/ 10.1016/j.cell.2012.04.035 [DOI] [PubMed] [Google Scholar]
- 12. Dorner D, Vlcek S, Foeger N, Gajewski A, Makolm C, Gotzmann J, Hutchison CJ, Foisner R. Lamina-associated polypeptide 2alpha regulates cell cycle progression and differentiation via the retinoblastoma-E2F pathway. J Cell Biol 2006; 173:83-93; PMID:16606692; http://dx.doi.org/ 10.1083/jcb.200511149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kosak ST, Skok JA, Medina KL, Riblet R, Le Beau MM, Fisher AG, Singh H. Subnuclear compartmentalization of immunoglobulin loci during lymphocyte development. Science 2002; 296:158-62; PMID:11935030; http://dx.doi.org/ 10.1126/science.1068768 [DOI] [PubMed] [Google Scholar]
- 14. Zink D, Amaral MD, Englmann A, Lang S, Clarke LA, Rudolph C, Alt F, Luther K, Braz C, Sadoni N, et al. Transcription-dependent spatial arrangements of CFTR and adjacent genes in human cell nuclei. J Cell Biol 2004; 166:815-25; PMID:15364959; http://dx.doi.org/ 10.1083/jcb.200404107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Meaburn KJ, Cabuy E, Bonne G, Levy N, Morris GE, Novelli G, Kill IR, Bridger JM. Primary laminopathy fibroblasts display altered genome organization and apoptosis. Aging Cell 2007; 6:139-53; PMID:17274801; http://dx.doi.org/ 10.1111/j.1474-9726.2007.00270.x [DOI] [PubMed] [Google Scholar]
- 16. Scaffidi P, Misteli T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat Cell Biol 2008; 10:452-9; PMID:18311132; http://dx.doi.org/ 10.1038/ncb1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Taimen P, Pfleghaar K, Shimi T, Moller D, Ben-Harush K, Erdos MR, Adam SA, Herrmann H, Medalia O, Collins FS, et al. A progeria mutation reveals functions for lamin A in nuclear assembly, architecture, and chromosome organization. Proc Natl Acad Sci U S A 2009; 106:20788-93; PMID:19926845; http://dx.doi.org/ 10.1073/pnas.0911895106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kirschner J, Brune T, Wehnert M, Denecke J, Wasner C, Feuer A, Marquardt T, Ketelsen UP, Wieacker P, Bonnemann CG, et al. p.S143F mutation in lamin A/C: a new phenotype combining myopathy and progeria. AnnNeurol 2005; 57:148-51; PMID:15622532; http://dx.doi.org/ 10.1002/ana.20359 [DOI] [PubMed] [Google Scholar]
- 19. Kandert S, Luke Y, Kleinhenz T, Neumann S, Lu W, Jaeger VM, Munck M, Wehnert M, Muller CR, Zhou Z, et al. Nesprin-2 giant safeguards nuclear envelope architecture in LMNA S143F progeria cells. Hum Mol Genet 2007; 16:2944-59; PMID:17881656; http://dx.doi.org/ 10.1093/hmg/ddm255 [DOI] [PubMed] [Google Scholar]
- 20. Helfand BT, Wang Y, Pfleghaar K, Shimi T, Taimen P, Shumaker DK. Chromosomal regions associated with prostate cancer risk localize to lamin B-deficient microdomains and exhibit reduced gene transcription. J Pathol 2012; 226:735-45; PMID:22025297; http://dx.doi.org/ 10.1002/path.3033 [DOI] [PubMed] [Google Scholar]
- 21. Shimi T, Butin-Israeli V, Adam SA, Hamanaka RB, Goldman AE, Lucas CA, Shumaker DK, Kosak ST, Chandel NS, Goldman RD. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev 2011; 25:2579-93; PMID:22155925; http://dx.doi.org/ 10.1101/gad.179515.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Freund A, Laberge RM, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell 2012; 23:2066-75; PMID:22496421; http://dx.doi.org/ 10.1091/mbc.E11-10-0884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu Q, Pante N, Misteli T, Elsagga M, Crisp M, Hodzic D, Burke B, Roux KJ. Functional association of Sun1 with nuclear pore complexes. J Cell Biol 2007; 178:785-98; PMID:17724119; http://dx.doi.org/ 10.1083/jcb.200704108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moroi Y, Peebles C, Fritzler MJ, Steigerwald J, Tan EM. Autoantibody to centromere (kinetochore) in scleroderma sera. Proc Natl Acad Sci U S A 1980; 77:1627-31; PMID:6966403; http://dx.doi.org/ 10.1073/pnas.77.3.1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cao K, Capell BC, Erdos MR, Djabali K, Collins FS. A lamin A protein isoform overexpressed in Hutchinson-Gilford progeria syndrome interferes with mitosis in progeria and normal cells. Proc Natl Acad Sci U S A 2007; 104:4949-54; PMID:17360355; http://dx.doi.org/ 10.1073/pnas.0611640104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Glynn MW, Glover TW. Incomplete processing of mutant lamin A in Hutchinson-Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum Mol Genet 2005; 14:2959-69; PMID:16126733; http://dx.doi.org/ 10.1093/hmg/ddi326 [DOI] [PubMed] [Google Scholar]
- 27. Bifulco M, D'Alessandro A, Paladino S, Malfitano AM, Notarnicola M, Caruso MG, Laezza C. N6-isopentenyladenosine improves nuclear shape in fibroblasts from humans with progeroid syndromes by inhibiting the farnesylation of prelamin A. FEBS J 2013; 280:6223-32; PMID:24112551; http://dx.doi.org/ 10.1111/febs.12544 [DOI] [PubMed] [Google Scholar]
- 28. Gordon LB, Kleinman ME, Miller DT, Neuberg DS, Giobbie-Hurder A, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland R, Snyder BD, et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 2012; 109:16666-71; PMID:23012407; http://dx.doi.org/ 10.1073/pnas.1202529109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Muchir A, Medioni J, Laluc M, Massart C, Arimura T, van der Kooi AJ, Desguerre I, Mayer M, Ferrer X, Briault S, et al. Nuclear envelope alterations in fibroblasts from patients with muscular dystrophy, cardiomyopathy, and partial lipodystrophy carrying lamin A/C gene mutations. Muscle Nerve 2004; 30:444-50; PMID:15372542; http://dx.doi.org/ 10.1002/mus.20122 [DOI] [PubMed] [Google Scholar]
- 30. Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, Burke B, Stahl PD, Hodzic D. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol 2006; 172:41-53; PMID:16380439; http://dx.doi.org/ 10.1083/jcb.200509124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Haque F, Mazzeo D, Patel JT, Smallwood DT, Ellis JA, Shanahan CM, Shackleton S. Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. J Biol Chem 2010; 285:3487-98; PMID:19933576; http://dx.doi.org/ 10.1074/jbc.M109.071910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liang Y, Chiu PH, Yip KY, Chan SY. Subcellular localization of SUN2 is regulated by lamin A and Rab5. PLoS One 2011; 6:e20507; PMID:21655223; http://dx.doi.org/ 10.1371/journal.pone.0020507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haque F, Lloyd DJ, Smallwood DT, Dent CL, Shanahan CM, Fry AM, Trembath RC, Shackleton S. SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol Cell Biol 2006; 26:3738-51; PMID:16648470; http://dx.doi.org/ 10.1128/MCB.26.10.3738-3751.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Turgay Y, Champion L, Balazs C, Held M, Toso A, Gerlich DW, Meraldi P, Kutay U. SUN proteins facilitate the removal of membranes from chromatin during nuclear envelope breakdown. J Cell Biol 2014; 204:1099-109; PMID:24662567; http://dx.doi.org/ 10.1083/jcb.201310116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ding X, Xu R, Yu J, Xu T, Zhuang Y, Han M. SUN1 is required for telomere attachment to nuclear envelope and gametogenesis in mice. Dev Cell 2007; 12:863-72; PMID:17543860; http://dx.doi.org/ 10.1016/j.devcel.2007.03.018 [DOI] [PubMed] [Google Scholar]
- 36. Mejat A, Misteli T. LINC complexes in health and disease. Nucleus 2010; 1:40-52; PMID:21327104; http://dx.doi.org/ 10.4161/nucl.1.1.10530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Croft JA, Bridger JM, Boyle S, Perry P, Teague P, Bickmore WA. Differences in the localization and morphology of chromosomes in the human nucleus. J Cell Biol 1999; 145:1119-31; PMID:10366586; http://dx.doi.org/ 10.1083/jcb.145.6.1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bridger JM, Boyle S, Kill IR, Bickmore WA. Re-modelling of nuclear architecture in quiescent and senescent human fibroblasts. Curr Biol 2000; 10:149-52; PMID:10679329; http://dx.doi.org/ 10.1016/S0960-9822(00)00312-2 [DOI] [PubMed] [Google Scholar]
- 39. Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008; 453:948-51; PMID:18463634; http://dx.doi.org/ 10.1038/nature06947 [DOI] [PubMed] [Google Scholar]
- 40. McCord RP, Nazario-Toole A, Zhang H, Chines PS, Zhan Y, Erdos MR, Collins FS, Dekker J, Cao K. Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Rese 2013; 23:260-9; PMID:23152449; http://dx.doi.org/ 10.1101/gr.138032.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mattout-Drubezki A, Gruenbaum Y. Dynamic interactions of nuclear lamina proteins with chromatin and transcriptional machinery. Cell Mol Life Sci 2003; 60:2053-63; PMID:14618255; http://dx.doi.org/ 10.1007/s00018-003-3038-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Heessen S, Fornerod M. The inner nuclear envelope as a transcription factor resting place. EMBO Rep 2007; 8:914-9; PMID:17906672; http://dx.doi.org/ 10.1038/sj.embor.7401075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dechat T, Shimi T, Adam SA, Rusinol AE, Andres DA, Spielmann HP, Sinensky MS, Goldman RD. Alterations in mitosis and cell cycle progression caused by a mutant lamin A known to accelerate human aging. Proc Natl Acad Sci U S A 2007; 104:4955-60; PMID:17360326; http://dx.doi.org/ 10.1073/pnas.0700854104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hodzic DM, Yeater DB, Bengtsson L, Otto H, Stahl PD. Sun2 is a novel mammalian inner nuclear membrane protein. J Biol Chem 2004; 279:25805-12; PMID:15082709; http://dx.doi.org/ 10.1074/jbc.M313157200 [DOI] [PubMed] [Google Scholar]
- 45. Olins AL, Hoang TV, Zwerger M, Herrmann H, Zentgraf H, Noegel AA, Karakesisoglou I, Hodzic D, Olins DE. The LINC-less granulocyte nucleus. Eur J Cell Biol 2009; 88:203-14; PMID:19019491; http://dx.doi.org/ 10.1016/j.ejcb.2008.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Solovei I, Cavallo A, Schermelleh L, Jaunin F, Scasselati C, Cmarko D, Cremer C, Fakan S, Cremer T. Spatial preservation of nuclear chromatin architecture during three-dimensional fluorescence in situ hybridization (3D-FISH). Exp Cell Res 2002; 276:10-23; PMID:11978004; http://dx.doi.org/ 10.1006/excr.2002.5513 [DOI] [PubMed] [Google Scholar]
- 47. Schermelleh L, Solovei I, Zink D, Cremer T. Two-color fluorescence labeling of early and mid-to-late replicating chromatin in living cells. Chromosome Res 2001; 9:77-80; PMID:11272795; http://dx.doi.org/ 10.1023/A:1026799818566 [DOI] [PubMed] [Google Scholar]