

Abstract
Amyotrophic lateral sclerosis (ALS) is a severely debilitating neurodegenerative disease linked to mutations in various genes implicated in cytoplasmic RNA metabolism. Recent studies from genetic models have also helped reveal connections between various ALS-linked factors and RNA-DNA hybrid (R-loop) regulation. Here, we examine how such hybrid-regulatory processes are pointing to a key role for the nucleus in ALS. We also present a potential molecular mechanism in which hybrids may represent at least one of the long sought after missing links between different ALS genes. Our opinion is that RNA-DNA hybrids will play a key role in deciphering ALS and other human diseases.
Keywords: Amyotrophic lateral sclerosis (ALS), RNA-DNA hybrid, R-loop, genome instability, stress granules, C9ORF72, SOD1, Senataxin, Ataxin2, TDP43, FUS
Beyond cytoplasmic RNA Dysregulation in ALS
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the loss of upper motor neurons in the cortex as well as lower motor neurons in the brainstem and spinal cord.1 ALS cases are presented as 10% familial and approximately 90% sporadic in nature.1 This relentlessly progressive disease is strongly linked to cytoplasmic proteinaceaous aggregates associated with mutations in a number of genes including SOD1 (superoxide dismutase), TDP43 (TAR DNA-binding protein 43; a.k.a. TARDP), FUS (fused in sarcoma), SETX (senataxin), and ATXN2 (ataxin-2), as well as repeat expansions at C9ORF72 (chromosome 9 open reading frame 72) (Table 1).2-8 Studies have pointed to the convergence of these seemingly disparate mutations onto RNA metabolism.3-9 Specifically, a hallmark of ALS pathology is the accumulation of cytoplasmic aggregates containing translationally inert RNA and associated proteins, called stress granules.9 Interestingly, the RNA-binding proteins TDP43, FUS, and ATXN2 strongly associate with these stress granules and are critical for their formation.10,11 Moreover, mutations in these proteins as well as SOD1 and C9ORF72 are thought to induce the aberrant accumulation of stress granules and related RNA processing foci.12,13 Thus, disruption of cytoplasmic RNA metabolism is closely associated with ALS.
Table 1.
ALS gene products proposed to play a role in an overarching pathobiological mechanism implicating nuclear RNA-DNA hybrids and cytoplasmic stress granules.*
| ALS-linked gene | Suspected function/impact within ALS | Estimated % of FALS and SALS | Refs |
|---|---|---|---|
| C9ORF72 (Chromosome 9 open reading frame 72) | Codes for protein of unknown function. Intronic HRE generates G4DNA leading to shortened nucleolar-disrupting transcripts. | 40% and 7% | 7, 16, 41, 43 |
| SOD1 (Superoxide dismutase 1) | Converts free radicals to hydrogen peroxide. Mutations may cause various cellular defects including genome and RNA metabolism-destabilizing free radical build-up. | 20% and 3% | 2 |
| ATXN2 (Ataxin-2; PBP1 in yeast) | RNA binding protein that interacts with polyA-binding factors. Important for stress granule formation in yeast and human. Yeast protein key to RNA-DNA hybrid and R-loop suppression. Mutations may increase ALS risk by promoting hybrid accumulation at various genomic locations, which may also lead to aberrant transcript accumulations and RNA metabolism stress. | <1% and 5% | 6, 15 |
| TDP43 (TAR DNA-binding protein 43) | RNA binding protein involved in multiple mRNA processing activities. Mutations deplete protein from the nucleus and engage it in cytoplasmic ribonucleoprotein aggregates such as stress granules. | 5% and 1% | 3, 64, 68 |
| FUS (Fused in sarcoma) | RNA binding protein with proposed roles in RNA metabolism. Mutations mimic and lead to TDP43 pathological features. | 4% and <1% | 5, 63 |
| SETX (Senataxin; Sen1 in yeast) | Resolves RNA-DNA hybrids in both yeast and human. Mutations may increase ALS risk by promoting hybrid accumulation at various genomic locations, which may also lead to aberrant transcript accumulations and RNA metabolism stress. | Unknown | 47 |
For a full list of ALS genes, please see reference 37.
However, it remains unclear if aberrant cytoplasmic RNA regulation is a driver or rather a bystander in ALS pathobiology. Could the dysregulation of cytoplasmic RNA metabolism be preventing RNA-binding proteins such as TDP-43, FUS, and/or ATXN2 from performing or promoting disease-suppressing nuclear functions? Could such nuclear roles help maintain overall genome structure and function? Recently uncovered connections between some ALS gene products and genome-destabilizing nucleic acid structures containing RNA-DNA hybrids (or R-loops) may help provide answers to these questions.6,14-17 Here we explore how crosstalk between RNA-DNA hybrids and ribonucleoprotein bodies linked to RNA metabolism may lead to an overarching mechanism of ALS pathobiology. We also discuss how this mechanism could point to new connections between different genes commonly mutated in ALS patients. In addition, we highlight important future directions for research that promises to reveal even more intriguing links between RNA-DNA hybrid regulatory processes, neurodegenerative diseases in particular, and human health in general. We note that our overall goal here is to present a broad and thought-provoking hypothesis that may connect seemingly unrelated processes without conducting a detailed review of the field.
RNA-DNA hybrids, R-loops, and Their Regulatory Processes
The ability of newly generated transcripts to ultimately leave their site of transcription is critical to overall cell function. However, it is becoming increasingly clear that newly synthesized RNA can invade the DNA duplex behind an advancing RNA polymerase.12,13, 17 This invasion generally occurs before the emerging transcript can be assembled into competing RNA-protein complexes.18,19 Importantly, cells do have an arsenal of molecules that help prevent or eliminate such events.
Invasion of a DNA duplex by an emerging transcript creates a so-called R-loop structure, which harbours an RNA-DNA hybrid opposite a single-stranded DNA region (Fig. 1A). The ability of RNA to reinvade the DNA duplex is favored by the presence of guanine clusters (G-clusters) especially near the 5′ end of the emerging transcript.20 In addition, such R-loop structures may be further stabilized if the displaced single-stranded DNA is prone to form a type of looped 3-dimensional structure known as G-quadruplex-DNA (G4DNA).21 G4DNA sequences are abundant across the eukaryotic genome and are especially enriched within rDNA repeats and telomeres.22,23 Furthermore, G4DNA-containing regions can generate G4RNA-harbouring transcripts, which could further compromise downstream RNA-regulatory processes.24
Figure 1.
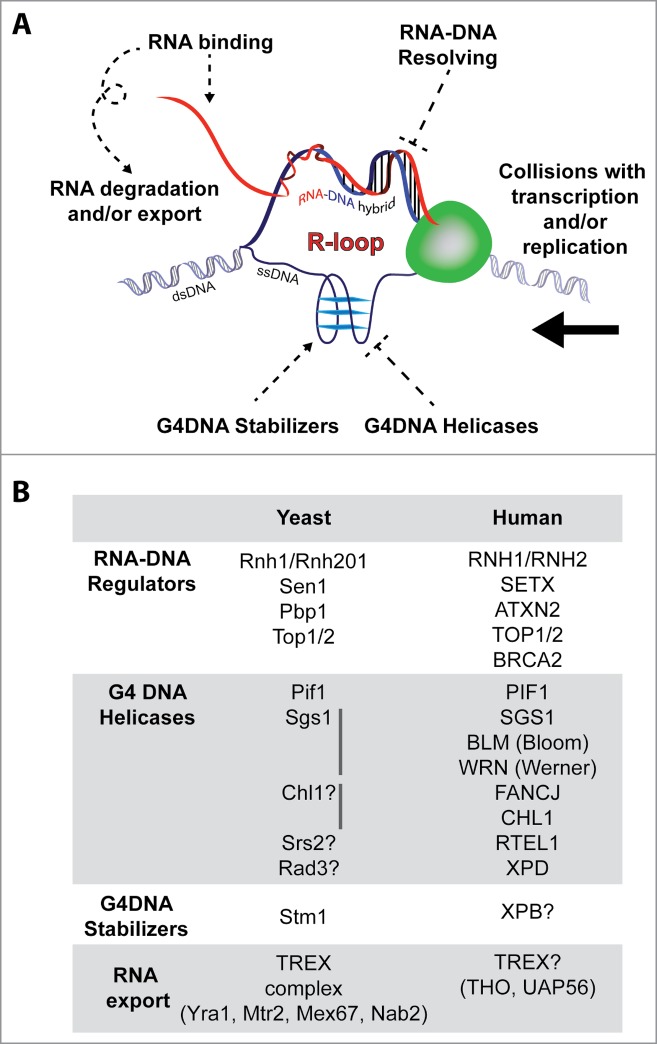
R-loop regulatory factors. (A) RNA modulatory processes involving RNA binding, degradation, and export cooperate with RNA-DNA hybrid resolving/suppressing factors and G4DNA helicases to suppress R-loop accumulation. On the other hand, G4DNA stabilizing factors can promote R-loop accumulation. R-loop accumulation can lead to genome-destabilizing collisions with transcription and/or replication machinery. (B) Examples of yeast and human R-loop regulators that may or may not be linked to ALS pathobiology.
Although R-loops can provide an additional level of regulation of gene expression and possibly other chromosomal processes, aberrant R-loop accumulation is a major threat to genome stability.17,25, 26 Therefore, cells have evolved mechanisms to control R-loop levels across the genome (Fig. 1).17 First, the suppression of transcription, rapid degradation, and/or export of hybrid-prone transcripts greatly limit excessive R-loop accumulation.12,14, 18,27, 28 Second, conserved RNaseH enzymes can degrade the RNA component of formed hybrids.29 Third, conserved enzymes including the Pif1 helicase can limit R-loops by unwinding G4DNA and/or resolving RNA-DNA hybrids.30-32
Factors linked to various diseases including premature aging and cancer have also been linked to R-loop regulation.26,33-36 Intriguingly, a string of recent studies is revealing unexpected links between RNA-DNA hybrids and a number of factors that are typically mutated in ALS patients. Together, these studies raise the possibility that RNA-DNA hybrids could play a key role in ALS pathobiology.
RNA-DNA hybrids May Link C9ORF72, SETX, and ATXN2
Deciphering ALS has proven difficult as the disease is linked to mutations in an unexpectedly large number of genes (Table 1) (please also see ref. 37 for a full list of ALS genes). However, statistical data support the idea that a relatively small number of these genes such as C9ORF72 are causal with the majority of other genes being disease modifiers (Table 1).38-40 For example, ALS modifier gene mutations may decrease the age of onset, hasten disease progression, or increase the severity of ALS pathobiology observed in patients carrying a causal mutation.38,40 It remains difficult to completely eliminate the possibility that particularly severe mutations/disruptions of genes currently viewed as modifiers may actually be sufficient to trigger ALS.38 Importantly, the fact that ALS is linked to many seemingly different genes has often hindered research aiming to decipher the pathobiological processes underlying the disease. However, recent studies are pointing to molecular connections between the different ALS genes. This may ultimately help decode the genetics of this devastating disease.
R-loops within C9ORF72 - Hexanucleotide (GGGGCC) repeat expansions (HRE) within the first introns of the C9ORF72 locus constitute the most common genetic mutation in ALS patients, representing approximately 40% and 7% of familial and sporadic cases, respectively (Table 1).7,8, 16,41 Of note, these patients typically present with an overlapping disease termed frontotemporal dementia (FTD).42 Although events leading to C9ORF72 HRE remain unclear, C9ORF72 HRE DNA sequences constitute G4DNA-containing R-loops that interfere with RNA Pol II transcriptional elongation and lead to the generation of truncated G4RNA-containing C9ORF72 transcripts.16,43 These truncated transcripts bind ribonucleoproteins including the nucleolar protein nucleolin, which in turn leads to the aggregation of aborted transcripts and associated factors within the nucleus and cytoplasm.16,43 Consistent with this finding, C9ORF72 HRE ALS patient cortex tissues show evidence of nucleolar stress.16 In addition, motor neurons derived from induced pluripotent stem cells of C9ORF72 HRE patients exhibit nucleolin mislocalization.16 Furthermore, the expression of (GGGGCC)21 abortive transcripts mimics nucleolin pathological signatures observed in C9ORF72 HRE ALS patient cells.16 Considering the prevalence of C9ORF72 HRE in ALS cases, these findings point to aberrantly formed/stabilized R-loops within C9ORF72 HRE, as well as subsequent downstream deleterious effects of truncated G4RNA transcripts including nucleolar disruption, as a possible key player in ALS pathobiology.
SETX resolves RNA-DNA hybrids - Additional links between R-loop formation and ALS originated from the study of the yeast protein Sen1. Through its RNA-DNA resolving ability, Sen1 suppresses R-loop accumulation and related transcription-induced recombination events.18 Of note, the ability of Sen1 to resolve RNA-DNA hybrids may also underlie the ability of this protein to mediate transcriptional termination by eliminating hybrids within the 3′UTR-coding region of genes.44,45 SETX is the human ortholog of the yeast SEN1 gene.46 SETX mutations are linked to juvenile ALS (Table 1).47 Of note, SETX mutations are also linked to another neurodegenerative disease called AOA2 (ataxia with occulomotor apraxia type 2), providing additional support for this gene having a critical role in neural function.46 Importantly, similar to yeast Sen1, the human SETX protein unwinds RNA-DNA hybrids and prevents collisions between the transcription and replication machinery at RNA Pol II-transcribed genes across the genome.14,48 Although it remains to be determined if the R-loop accumulation associated with SETX mutations directly leads to neurodegeneration, it is clear that SETX-dependent suppression of RNA-DNA hybrids is important for the maintenance of genome stability.14,48 Therefore, while C9ORF72 HRE links R-loops and possible subsequent nuclear defects to ALS, SETX may point to a broader role for R-loop accumulation across the genome in ALS pathobiology.
Toward a function for the conserved ATXN2 in hybrid suppression - Further support for links between ALS and the nucleus in general, as well as ALS and R-loops in particular, has recently been provided by a study in which we focused on the yeast RNA-binding protein Pbp1 (Pab1-binding protein 1). Eukaryotic rRNA genes (rRNA genes or rDNA) are arranged as tandem repeats on one or more chromosomes and provide the foundation for the nucleolar compartment.49 The high copy number helps sustain ribosomal biogenesis and general protein synthesis. Unequal DNA recombination between rDNA units within the repeats allows cells to increase or decrease the number of rDNA units in response to stress conditions.49 However, deregulated recombination within rDNA repeats can lead to chromosome instability and shorten cellular lifespan.50-52 Importantly, non-coding transcripts are generated from bidirectional promoters within intergenic spacers located between rDNA units.53-55 The disruption of ncRNA-suppressing processes such as silent chromatin assembly, early transcriptional termination, or exosome-mediated degradation leads to the accumulation of these ncRNAs but to varying degrees of rDNA instability.53-55 This suggested that additional mechanisms might be preventing accumulating ncRNAs from destabilizing rDNA repeats. Indeed, we found that Pbp1, which harbours a Like SM (LSM) RNA-binding domain, binds to these intergenic ncRNAs and prevents them from engaging in non-coding RNA-DNA hybrids (ncRNA-DNA).15 Importantly, substituting full length Pbp1 with a Pbp1 mutant that is lacking the Pab1-binding domain but not the LSM RNA-binding domain did not compromise ncRNA-DNA hybrid suppression.15 Moreover, we found that a G4DNA-stabilizing protein called Stm1 further stabilizes R-loops formed in Pbp1-deficient cells. Intriguingly, caloric restriction, a dietary regimen in which the amount of glucose available to cells is decreased, essentially abolished hybrid accumulation in Pbp1-deficient cells.15 This ability of caloric restriction is somehow dependent on the yeast RNaseH enzymes Rnh1 and Rnh201 as well as the G4DNA helicase Pif1, which may also have RNA-DNA hybrid resolving activity.15,32 Of note, Pbp1 also suppresses hybrids at non-rDNA G4DNA sites near telomeres as well as within a couple of tested open reading frames pointing to a potentially broader role in R-loop suppression across the genome.15 Furthermore, we observed that the R-loop suppressing ability of Pbp1 maintains cellular lifespan.
Extreme expansion of a polyglutamine (polyQ) site within human ATXN2, the ortholog of yeast Pbp1, was initially identified in spinal cerebellar ataxia type 2.56-58 More recently, intermediate ATXN2 polyQ expansions were identified as a major risk factor for ALS.6 An intriguing possibility is that ATXN2 polyQ expansions may lead to R-loop accumulation within key coding or non-coding DNA sequences within loci with a central role in neural function. That various functions of Pbp1 and ATXN2 are likely conserved is supported by the fact that both proteins have LSM domains and play a role in the establishment of stress granules under stress, a point that we will further explore below.6,59, 60
How R-loops may link C9ORF72, SETX, and ATXN2 - The disruption of an RNA-DNA hybrid-suppressing role for ATXN2 and/or SETX could lead to R-loop accumulation at the G4DNA-containing C9ORF72 HRE. This may further promote HRE expansion and its downstream effects on truncated G4RNA-containing transcripts as well as the sequestration of nucleolin. Consistent with this rationale, a recent study identified intermediate length polyQ expansion of ATXN2 as a critical disease modifier in C9ORF72 HRE patients.40 Alternatively, C9ORF72 HRE may be operating upstream of wild-type ATXN2. In this case, it is possible that accumulating truncated G4RNA-containing C9ORF72 HRE transcripts may sequester the RNA-binding ATXN2 along with nucleolin within ribonucleoprotein aggregates.16 This may in turn further promote C9ORF72 HRE and help establish a vicious cycle. Through these connections, it is possible that ALS-linked genetic defects in C9ORF72, SETX, and ATXN2 all lead to broader hybrid accumulations throughout the genome leading to chromosomal features that would be particularly destabilizing to the transcriptionally active neurons.
Deleterious Feedback Loop Between RNA-DNA Hybrids and Stress Granules May Connect The Dots Between ALS Genes
While C9ORF72, SETX, and ATXN2 may control the formation and stability of RNA-DNA hybrids, cytoplasmic aggregation/retention of RNA-binding proteins, especially TDP43 and FUS, are a key hallmark of ALS pathology. One possibility is that nuclear R-loops and cytoplasmic ribonucleoprotein aggregates are part of a self-reinforcing loop that gains strength with disease progression (Fig. 2). Alternatively, it is possible that proteins such as TDP43 and FUS play unforeseen direct roles in RNA-DNA hybrid suppression.
Figure 2.
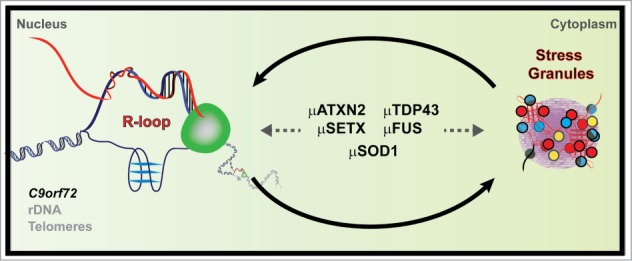
Putative feedback loop between R-loops and stress granules may connect various ALS genes at the molecular level. Mutations (μ) in ALS-linked factors can abrogate their ability to suppress R-loop formation and/or lead to their aberrant sequestration in RNA-processing foci such as cytoplasmic stress granules. Increased R-loop accumulation in turn leads to genomic instability and aberrant transcript accumulations, which in turn further promotes stress granule formation and the cycle continues. Motor neurons are predicted to be particularly sensitive to such a deleterious and vicious cycle.
Both TDP43 and FUS can bind DNA and RNA.61-63 In addition, both proteins have been linked to various nuclear and cytoplasmic steps of mRNA processing and transport.64-66 In contrast, ALS-linked mutant TDP43 is depleted from the nucleus, aggregates within the cytoplasm of affected neurons, and associates with stress granules.67,68 Additionally, FUS accumulates at stress granules within the degenerating neurons of ALS patients.5,11 Interestingly, ATXN2 associates with both TDP43 and FUS and is thought to alter TDP43 toxicity and FUS-related pathology.6,11 Furthermore, ATXN2 is a component of stress granules in yeast and human cells.59,69 Together, these findings point to a number of urgent questions. Are RNA-DNA hybrids accumulating excessively when TDP43 or FUS are aberrantly localized to stress granules? If R-loops are indeed forming, can this be attributed to an ability of mutant TDP43 or FUS to sequester ATXN2 into cytoplasmic stress granules? Alternatively, can nuclear TDP43 and FUS play a role in R-loop suppression? And if so, would that affect C9ORF72 or intersect with the hybrid-suppressing ability of SETX and possibly ATXN2?
Concluding Remarks and Broad Questions
Overall, we have explored how excessive R-loop accumulation may connect the dots between different ALS genes and move us a step closer toward a putative unifying molecular model of ALS pathogenesis. We expect that future work by others and us will put this model to the test. It will also be critical to determine if excessive RNA-DNA hybrid levels are the underlying cause of motor neuron degeneration in ALS. Are hybrids accumulating in ALS patient-derived neurons? Can interventions aiming to alleviate hybrid accumulation result in reduced neurodegeneration? Do increased R-loop levels play a role somewhere along the pathway to degeneration? Does the loss of R-loop regulation act as a trigger that leads to toxic nuclear and cytoplasmic ribonucleoprotein aggregations? Importantly, R-loops are emerging as key players in a number of diseases including cancer and premature aging.30,31, 34 Thus, major efforts are needed to fully investigate connections between R-loops, ALS, and human health.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Karan Joshua Abraham, Kirk Szafranski, Samuel Killackey, and other members of the Mekhail lab for fruitful discussions and comments.
Funding
This work was supported by a Vanier Doctoral Award and an Open Operating Grant from the Canadian Institutes of Health Research (CIHR) respectively to J.S. and K.M., who also holds the Canada Research (CRC) in Spatial Genome Organization.
References
- 1. Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med 2001; 344:1688-700; PMID:11386269; http://dx.doi.org/ 10.1056/NEJM200105313442207 [DOI] [PubMed] [Google Scholar]
- 2. Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993; 364:362; PMID:8332197 [DOI] [PubMed] [Google Scholar]
- 3. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008; 319:1668-72; PMID:18309045; http://dx.doi.org/ 10.1126/science.1154584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF, Stewart H, Kelley BJ, Kuntz K, Crook RJ, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet 2008; 4:e1000193; PMID:18802454; http://dx.doi.org/ 10.1371/journal.pgen.1000193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kwiatkowski TJ, Jr., Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009; 323:1205-8; PMID:19251627; http://dx.doi.org/ 10.1126/science.1166066 [DOI] [PubMed] [Google Scholar]
- 6. Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010; 466:1069-75; PMID:20740007; http://dx.doi.org/ 10.1038/nature09320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011; 72:245-56; PMID:21944778; http://dx.doi.org/ 10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011; 72:257-68; PMID:21944779; http://dx.doi.org/ 10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. J Cell Biol 2013; 201:361-72; PMID:23629963; http://dx.doi.org/ 10.1083/jcb.201302044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kwong LK, Neumann M, Sampathu DM, Lee VM, Trojanowski JQ. TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol 2007; 114:63-70; PMID:17492294; http://dx.doi.org/ 10.1007/s00401-007-0226-5 [DOI] [PubMed] [Google Scholar]
- 11. Farg MA, Soo KY, Warraich ST, Sundaramoorthy V, Blair IP, Atkin JD. Ataxin-2 interacts with FUS and intermediate-length polyglutamine expansions enhance FUS-related pathology in amyotrophic lateral sclerosis. Hum Mol Genet 2013; 22:717-28; PMID:23172909; http://dx.doi.org/ 10.1093/hmg/dds479 [DOI] [PubMed] [Google Scholar]
- 12. Huertas P, Aguilera A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol Cell 2003; 12:711-21; PMID:14527416; http://dx.doi.org/ 10.1016/j.molcel.2003.08.010 [DOI] [PubMed] [Google Scholar]
- 13. Roy D, Zhang Z, Lu Z, Hsieh CL, Lieber MR. Competition between the RNA transcript and the nontemplate DNA strand during R-loop formation in vitro: a nick can serve as a strong R-loop initiation site. Mol Cell Biol 2010; 30:146-59; PMID:19841062; http://dx.doi.org/ 10.1128/MCB.00897-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alzu A, Bermejo R, Begnis M, Lucca C, Piccini D, Carotenuto W, Saponaro M, Brambati A, Cocito A, Foiani M, et al. Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell 2012; 151:835-46; PMID:23141540; http://dx.doi.org/ 10.1016/j.cell.2012.09.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Salvi JS, Chan JN, Szafranski K, Liu TT, Wu JD, Olsen JB, Khanam N, Poon BP, Emili A, Mekhail K. Roles for Pbp1 and Caloric Restriction in Genome and Lifespan Maintenance via Suppression of RNA-DNA Hybrids. Dev Cell 2014; 30:177-91; PMID:25073155; http://dx.doi.org/ 10.1016/j.devcel.2014.05.013 [DOI] [PubMed] [Google Scholar]
- 16. Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 2014; 507:195-200; PMID:24598541; http://dx.doi.org/ 10.1038/nature13124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aguilera A, Garcia-Muse T. R loops: from transcription byproducts to threats to genome stability. Mol Cell 2012; 46:115-24; PMID:22541554; http://dx.doi.org/ 10.1016/j.molcel.2012.04.009 [DOI] [PubMed] [Google Scholar]
- 18. Mischo HE, Gomez-Gonzalez B, Grzechnik P, Rondon AG, Wei W, Steinmetz L, Aguilera A, Proudfoot NJ. Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol Cell 2011; 41:21-32; PMID:21211720; http://dx.doi.org/ 10.1016/j.molcel.2010.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Luna R, Jimeno S, Marin M, Huertas P, Garcia-Rubio M, Aguilera A. Interdependence between transcription and mRNP processing and export, and its impact on genetic stability. Mol Cell 2005; 18:711-22; PMID:15949445; http://dx.doi.org/ 10.1016/j.molcel.2005.05.001 [DOI] [PubMed] [Google Scholar]
- 20. Roy D, Lieber MR. G clustering is important for the initiation of transcription-induced R-loops in vitro, whereas high G density without clustering is sufficient thereafter. Mol Cell Biol 2009; 29:3124-33; PMID:19307304; http://dx.doi.org/ 10.1128/MCB.00139-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Duquette ML, Handa P, Vincent JA, Taylor AF, Maizels N. Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes Dev 2004; 18:1618-29; PMID:15231739; http://dx.doi.org/ 10.1101/gad.1200804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Capra JA, Paeschke K, Singh M, Zakian VA. G-quadruplex DNA sequences are evolutionarily conserved and associated with distinct genomic features in Saccharomyces cerevisiae. PLoS Comput Biol 2010; 6:e1000861; PMID:20676380; http://dx.doi.org/ 10.1371/journal.pcbi.1000861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hershman SG, Chen Q, Lee JY, Kozak ML, Yue P, Wang LS, Johnson FB. Genomic distribution and functional analyses of potential G-quadruplex-forming sequences in Saccharomyces cerevisiae. Nucleic Acids Res 2008; 36:144-56; PMID:17999996; http://dx.doi.org/ 10.1093/nar/gkm986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu Y, Suzuki Y, Ito K, Komiyama M. Telomeric repeat-containing RNA structure in living cells. Proc Natl Acad Sci U S A 2010; 107:14579-84; PMID:20679250; http://dx.doi.org/ 10.1073/pnas.1001177107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sun Q, Csorba T, Skourti-Stathaki K, Proudfoot NJ, Dean C. R-loop stabilization represses antisense transcription at the Arabidopsis FLC locus. Science 2013; 340:619-21; PMID:23641115; http://dx.doi.org/ 10.1126/science.1234848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rudra S, Skibbens RV. Chl1 DNA helicase regulates Scc2 deposition specifically during DNA-replication in Saccharomyces cerevisiae. PLoS One 2013; 8:e75435; PMID:24086532; http://dx.doi.org/ 10.1371/journal.pone.0075435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bermejo R, Lai MS, Foiani M. Preventing replication stress to maintain genome stability: resolving conflicts between replication and transcription. Mol Cell 2012; 45:710-8; PMID:22464441; http://dx.doi.org/ 10.1016/j.molcel.2012.03.001 [DOI] [PubMed] [Google Scholar]
- 28. Santos-Pereira JM, Herrero AB, Garcia-Rubio ML, Marin A, Moreno S, Aguilera A. The Npl3 hnRNP prevents R-loop-mediated transcription-replication conflicts and genome instability. Genes Dev 2013; 27:2445-58; PMID:24240235; http://dx.doi.org/ 10.1101/gad.229880.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wahba L, Amon JD, Koshland D, Vuica-Ross M. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol Cell 2011; 44:978-88; PMID:22195970; http://dx.doi.org/ 10.1016/j.molcel.2011.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Paeschke K, Bochman ML, Garcia PD, Cejka P, Friedman KL, Kowalczykowski SC, Zakian VA. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature 2013; 497:458-62; PMID:23657261; http://dx.doi.org/ 10.1038/nature12149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Paeschke K, Capra JA, Zakian VA. DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell 2011; 145:678-91; PMID:21620135; http://dx.doi.org/ 10.1016/j.cell.2011.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou R, Zhang J, Bochman ML, Zakian VA, Ha T. Periodic DNA patrolling underlies diverse functions of Pif1 on R-loops and G-rich DNA. Elife 2014; 3:e02190; PMID:24843019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu Y, Shin-ya K, Brosh RM, Jr. FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol Cell Biol 2008; 28:4116-28; PMID:18426915; http://dx.doi.org/ 10.1128/MCB.02210-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bhatia V, Barroso SI, Garcia-Rubio ML, Tumini E, Herrera-Moyano E, Aguilera A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014; PMID:24896180 [DOI] [PubMed] [Google Scholar]
- 35. Gray LT, Vallur AC, Eddy J, Maizels N. G quadruplexes are genomewide targets of transcriptional helicases XPB and XPD. Nat Chem Biol 2014; 10:313-8; PMID:24609361; http://dx.doi.org/ 10.1038/nchembio.1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Uringa EJ, Youds JL, Lisaingo K, Lansdorp PM, Boulton SJ. RTEL1: an essential helicase for telomere maintenance and the regulation of homologous recombination. Nucleic Acids Res 2011; 39:1647-55; PMID:21097466; http://dx.doi.org/ 10.1093/nar/gkq1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 2013; 14:248-64; PMID:23463272; http://dx.doi.org/ 10.1038/nrn3430 [DOI] [PubMed] [Google Scholar]
- 38. Van Hoecke A, Schoonaert L, Lemmens R, Timmers M, Staats KA, Laird AS, Peeters E, Philips T, Goris A, Dubois B, et al. EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat Med 2012; 18:1418-22; PMID:22922411; http://dx.doi.org/ 10.1038/nm.2901 [DOI] [PubMed] [Google Scholar]
- 39. Eschbach J, Schwalenstocker B, Soyal SM, Bayer H, Wiesner D, Akimoto C, Nilsson AC, Birve A, Meyer T, Dupuis L, et al. PGC-1alpha is a male-specific disease modifier of human and experimental amyotrophic lateral sclerosis. Hum Mol Genet 2013; 22:3477-84; PMID:23669350; http://dx.doi.org/ 10.1093/hmg/ddt202 [DOI] [PubMed] [Google Scholar]
- 40. van Blitterswijk M, Mullen B, Heckman MG, Baker MC, DeJesus-Hernandez M, Brown PH, Murray ME, Hsiung GY, Stewart H, Karydas AM, et al. Ataxin-2 as potential disease modifier in C9ORF72 expansion carriers. Neurobiol Aging 2014; 35:2421.e13-7; PMID:24866401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chiò A, Restagno G, Nicolaou N, Simon-Sanchez J, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 2012; 11:323-30; PMID:22406228; http://dx.doi.org/ 10.1016/S1474-4422(12)70043-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 2013; 79:416-38; PMID:23931993; http://dx.doi.org/ 10.1016/j.neuron.2013.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fratta P, Mizielinska S, Nicoll AJ, Zloh M, Fisher EM, Parkinson G, Isaacs AM. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci Rep 2012; 2:1016; PMID:23264878; http://dx.doi.org/ 10.1038/srep01016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jamonnak N, Creamer TJ, Darby MM, Schaughency P, Wheelan SJ, Corden JL. Yeast Nrd1, Nab3, and Sen1 transcriptome-wide binding maps suggest multiple roles in post-transcriptional RNA processing. RNA 2011; 17:2011-25; PMID:21954178; http://dx.doi.org/ 10.1261/rna.2840711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hazelbaker DZ, Marquardt S, Wlotzka W, Buratowski S. Kinetic competition between RNA Polymerase II and Sen1-dependent transcription termination. Mol Cell 2013; 49:55-66; PMID:23177741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Moreira MC, Klur S, Watanabe M, Nemeth AH, Le Ber I, Moniz JC, Tranchant C, Aubourg P, Tazir M, Schöls L, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet 2004; 36:225-7; PMID:14770181; http://dx.doi.org/ 10.1038/ng1303 [DOI] [PubMed] [Google Scholar]
- 47. Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, Dierick I, Abel A, Kennerson ML, Rabin BA, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet 2004; 74:1128-35; PMID:15106121; http://dx.doi.org/ 10.1086/421054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Skourti-Stathaki K, Proudfoot NJ, Gromak N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell 2011; 42:794-805; PMID:21700224; http://dx.doi.org/ 10.1016/j.molcel.2011.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mekhail K, Moazed D. The nuclear envelope in genome organization, expression and stability. Nat Rev Mol Cell Biol 2010; 11:317-28; PMID:20414256; http://dx.doi.org/ 10.1038/nrm2894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 1999; 13:2570-80; PMID:10521401; http://dx.doi.org/ 10.1101/gad.13.19.2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chan JN, Poon BP, Salvi J, Olsen JB, Emili A, Mekhail K. Perinuclear Cohibin Complexes Maintain Replicative Life Span via Roles at Distinct Silent Chromatin Domains. Dev Cell 2011; 20:867-79; PMID:21664583; http://dx.doi.org/ 10.1016/j.devcel.2011.05.014 [DOI] [PubMed] [Google Scholar]
- 52. Salvi JS, Chan JN, Pettigrew C, Liu TT, Wu JD, Mekhail K. Enforcement of a lifespan-sustaining distribution of Sir2 between telomeres, mating-type loci, and rDNA repeats by Rif1. Aging Cell 2013; 12:67-75; PMID:23082874; http://dx.doi.org/ 10.1111/acel.12020 [DOI] [PubMed] [Google Scholar]
- 53. Houseley J, Kotovic K, El Hage A, Tollervey D. Trf4 targets ncRNAs from telomeric and rDNA spacer regions and functions in rDNA copy number control. EMBO J 2007; 26:4996-5006; PMID:18007593; http://dx.doi.org/ 10.1038/sj.emboj.7601921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vasiljeva L, Kim M, Terzi N, Soares LM, Buratowski S. Transcription termination and RNA degradation contribute to silencing of RNA polymerase II transcription within heterochromatin. Mol Cell 2008; 29:313-23; PMID:18280237; http://dx.doi.org/ 10.1016/j.molcel.2008.01.011 [DOI] [PubMed] [Google Scholar]
- 55. Kobayashi T, Ganley AR. Recombination regulation by transcription-induced cohesin dissociation in rDNA repeats. Science 2005; 309:1581-4; PMID:16141077; http://dx.doi.org/ 10.1126/science.1116102 [DOI] [PubMed] [Google Scholar]
- 56. Imbert G, Saudou F, Yvert G, Devys D, Trottier Y, Garnier JM, Weber C, Mandel JL, Cancel G, Abbas N, et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet 1996; 14:285-91; PMID:8896557; http://dx.doi.org/ 10.1038/ng1196-285 [DOI] [PubMed] [Google Scholar]
- 57. Pulst SM, Nechiporuk A, Nechiporuk T, Gispert S, Chen XN, Lopes-Cendes I, Pearlman S, Starkman S, Orozco-Diaz G, Lunkes A, et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 1996; 14:269-76; PMID:8896555; http://dx.doi.org/ 10.1038/ng1196-269 [DOI] [PubMed] [Google Scholar]
- 58. Sanpei K, Takano H, Igarashi S, Sato T, Oyake M, Sasaki H, Wakisaka A, Tashiro K, Ishida Y, Ikeuchi T, et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet 1996; 14:277-84; PMID:8896556; http://dx.doi.org/ 10.1038/ng1196-277 [DOI] [PubMed] [Google Scholar]
- 59. Ralser M, Albrecht M, Nonhoff U, Lengauer T, Lehrach H, Krobitsch S. An integrative approach to gain insights into the cellular function of human ataxin-2. J Mol Biol 2005; 346:203-14; PMID:15663938; http://dx.doi.org/ 10.1016/j.jmb.2004.11.024 [DOI] [PubMed] [Google Scholar]
- 60. Swisher KD, Parker R. Localization to, and effects of Pbp1, Pbp4, Lsm12, Dhh1, and Pab1 on stress granules in Saccharomyces cerevisiae. PLoS One 2010; 5:e10006; PMID:20368989; http://dx.doi.org/ 10.1371/journal.pone.0010006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 1995; 69:3584-96; PMID:7745706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Buratti E, Baralle FE. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem 2001; 276:36337-43; PMID:11470789; http://dx.doi.org/ 10.1074/jbc.M104236200 [DOI] [PubMed] [Google Scholar]
- 63. Wang X, Arai S, Song X, Reichart D, Du K, Pascual G, Tempst P, Rosenfeld MG, Glass CK, Kurokawa R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008; 454:126-30; PMID:18509338; http://dx.doi.org/ 10.1038/nature06992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SS, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A, et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 2014; 81:536-43; PMID:24507191; http://dx.doi.org/ 10.1016/j.neuron.2013.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 2006; 351:602-11; PMID:17084815; http://dx.doi.org/ 10.1016/j.bbrc.2006.10.093 [DOI] [PubMed] [Google Scholar]
- 66. Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling SC, Liang TY, Mazur C, et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci 2012; 15:1488-97; PMID:23023293; http://dx.doi.org/ 10.1038/nn.3230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Colombrita C, Zennaro E, Fallini C, Weber M, Sommacal A, Buratti E, Silani V, Ratti A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem 2009; 111:1051-61; PMID:19765185; http://dx.doi.org/ 10.1111/j.1471-4159.2009.06383.x [DOI] [PubMed] [Google Scholar]
- 68. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314:130-3; PMID:17023659; http://dx.doi.org/ 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- 69. Buchan JR, Muhlrad D, Parker R. P bodies promote stress granule assembly in Saccharomyces cerevisiae. J Cell Biol 2008; 183:441-55; PMID:18981231; http://dx.doi.org/ 10.1083/jcb.200807043 [DOI] [PMC free article] [PubMed] [Google Scholar]