

Abstract
Human antigen R (HuR) is a 32 kDa protein with 3 RNA Recognition Motifs (RRMs), which bind to Adenylate and uridylate Rich Elements (AREs) of mRNAs. Whereas the N-terminal and central domains (RRM1 and RRM2) are essential for AREs recognition, little is known on the C-terminal RRM3 beyond its implication in HuR oligomerization and apoptotic signaling. We have developed a detergent-based strategy to produce soluble RRM3 for structural studies. We have found that it adopts the typical RRM fold, does not interact with the RRM1 and RRM2 modules, and forms dimers in solution. Our NMR measurements, combined with Molecular Dynamics simulations and Analytical Ultracentrifugation experiments, show that the protein dimerizes through a helical region that contains the conserved W261 residue. We found that HuR RRM3 binds to 5′-mer U-rich RNA stretches through the solvent exposed side of its β-sheet, located opposite to the dimerization site. Upon mimicking phosphorylation by the S318D replacement, RRM3 mutant shows less ability to recognize RNA due to an electrostatic repulsion effect with the phosphate groups. Our study brings new insights of HuR RRM3 as a domain involved in protein oligomerization and RNA interaction, both functions regulated by 2 surfaces on opposite sides of the RRM domain.
Keywords: dimerization, Human antigen R (HuR), Nuclear Magnetic Resonance (NMR), RNA binding protein (RBP), RNA binding, RNA recognition motif (RRM), Serine Phosphorylation
Abbreviations
- AREs
Adenylate and uridylate Rich Elements
- AU
Analytical Ultracentrifugation
- Chk2
Checkpoint kinase 2
- CD
Circular Dichroism
- CARM1
Coactivator associated Arginine Methyltransferase 1
- Cdk1
Cyclin-dependent kinase 1
- EMSA
Electrophoretic Mobility Shift Assay
- ELAV1
Embryonic Lethal Abnormal Vision system human homolog 1
- FIR
FBP-Interacting Repressor
- hnRNP1
heterogeneous nuclear RiboNucleoprotein C protein
- HSQC
Heteronuclear Single-Quantum Correlation
- HuR
Human antigen R
- FL
Full-Length, HNS, HuR Nucleocytoplasmic Shuttling Sequence
- MD
Molecular Dynamics
- NMR
Nuclear Magnetic Resonance
- NOE
Nuclear Overhauser Effect
- PMSF
PhenylMethylSulfonyl Fluoride
- PTB
Polypyrimidine Tract Binding protein
- PCA
Principal Component Analysis
- PKCα
Protein Kinase C α
- PKCδ
Protein Kinase C δ
- RBPs
RNA Binding Proteins
- RRMs
RNA Recognition Motifs
- SPR
Surface Plasmon Resonance
- WT
Wild-Type
Introduction
HuR is a ubiquitously expressed RNA binding protein from ELAV (Embryonic Lethal Abnormal Vision) – like family which recognizes Adenylate- and uridylate-Rich Elements (ARE)1 of mRNA as it was first reported for the HuB protein.2 HuR stabilizes and regulates the translation of specific ARE-bearing mRNAs coding for cell cycle regulators, growth factors, proto-oncogenes, apoptosis-regulatory proteins, cytokines, among others, and it can alter the cellular response to proliferative,3,4 stress,5-9 apoptotic,9-12 angiogenesis,13 differentiation,14,15 senescence,4,16 inflammatory,17 and immune stimuli.18-20 Overexpression of HuR impairs those HuR-governed gene expression programs with consequences in diseases such as chronic inflammation, cardiovascular pathologies and cancer (for a review, see ref. 21).
HuR is built up by 3 RNA Recognition Motifs (RRMs)22-24 which are well conserved among the ELAV-like proteins25 and the N-terminal domains (RRM1 and RRM2) show the cognate topology β1α1β2β3α2β4 with a β-sheet of 4 antiparallel β-strands packed against 2 α-helices (Fig. 1).26 The RRM1 and RRM2 motifs – separated by a short 310 helix – work as a compact structural unit as described before for other RNA binding proteins,27 whereas the C-terminal RRM domain (RRM3) is separated from RRM2 by a linker region that includes a 60-residue long HuR Nucleocytoplasmic Shuttling Sequence (HNS).28 HNS is mainly responsible for nuclear/cytoplasmic shuttling upon binding adaptor proteins for nuclear export such as pp32/PHAP-I and APRIL29,30 and with import factors transportin-1, -2 and importin α.31-33 Previous Electrophoretic Mobility Shift Assay (EMSA)-based report34 suggests that the RRM23 linker, together with the RRM3 domain, could also have an additional role in stabilizing HuR-AREs complexes. This enhancement of ARE-binding activity was greater than that observed in Surface Plasmon Resonance (SPR) experiments with HuD, a highly homolog protein to HuR,35 where the RRM23 linker showed a negligible effect in RNA binding.
Figure 1.

Schematic Domain Organization of HuR. The positions of the phosphorylation site S318, and of W261, responsible for HuR dimerization, are both marked. The HuR Nucleocytoplasmic Shuttling sequence (HNS) is also represented. The boundaries of RRM3 construct used in this work are from W244 to K326 in reference to the HuR FL protein.
HuR cell localization and HuR-triggered cell functions are controlled by post-translational modifications (for a review see ref. 36). Thereby, caspase-mediated cleavage of HuR at D226 located in its HNS region amplifies the apoptotic signal promoting cell death under extreme stress conditions.11 HuR is phosphorylated by kinases involved in different signaling pathways (for review see ref. 37): Cyclin-dependent kinase 1 (Cdk1),38,39 cell cycle Checkpoint kinase 2 (Chk2),10 Protein Kinase C α (PKCα)40 and PKCδ.41 HuR ability to bind its target RNAs is modulated when Chk2 phosphorylates HuR at either S88 and S100 residues, or when PKCδ phosphorylates S221 and S318, both of them in the RRM3 domain.9,41 Several kinases, including PKCα and Cdk1, favor the nucleocytoplasmic shuttling of HuR by adding a phosphate group to S158, S202, S221 and S242.38–40 Other kinds of post-translational modifications have been reported, such as methylation at R117 by the Coactivator associated Arginine Methyltransferase 1 (CARM1)42 or NEDDylation at K283, K313 and K326, as recently described.43
Cytoplasmic binding of HuR to single-stranded ARE-containing mRNAs is mainly driven by RRM1 and RRM2 domains.34,44-46 RRM3 is involved in binding long poly-A tails of mRNAs47-49 and in catalyzing the 3′-terminal adenosyl modification of non-polyadenylated RNA substrates.50 Besides its role in RNA recognition, RRM3 is also responsible for the formation of HuR multimers,34 as occurs with the homologous Drosophila ELAV protein.51 In fact, mutations in the RRM3 domain of Drosophila ELAV protein lead to a temperature-sensitive phenotype by impairing HuR oligomerization, highlighting the crucial role of this RRM module.52 Given the multifunctional relevance of the HuR RRM3 domain in RNA recognition and modification as well as in HuR oligomerization, we set out to investigate the molecular structure of this motif, which has been hampered by its low solubility when recombinantly produced in large quantities for structural studies. We have overcome this difficulty by developing a successful detergent-based strategy to obtain the pure soluble protein in milligram amounts suitable for NMR analysis. We show that HuR dimerizes through RRM3 α1-helix and the following loop, located opposite to the RNA-binding platform, but independently of the binding to RNA molecules. The W261-by-E261 substitution in α1-helix results in a monomeric HuR RRM3 form. Interestingly, isolated HuR RRM3 preferably binds U-rich versus AU-rich RNA stretches, despite previous studies suggesting that RRM3 has a negligible contribution to ARE-binding events in comparison with the other 2 HuR RRM motifs.34, 44–46 Although the HuR linker between RRM2 and RRM3 could contribute to the ARE stabilization,34,35 it was unfeasible to obtain a linker-bearing RRM3 construct in a soluble manner. Moreover, HuR RRM3 phosphorylation, which was mimicked by a S318-to-D318 substitution, causes no significant structural changes of RRM3, in contrast to a phosphorylation-mediated unfolding described in other types of RNA domains.53 However, mimicking RRM3 phosphorylation reduces the binding to U-rich RNA sequences, suggesting that this post-translational modification acts as a master switch for regulating HuR activity.
Results
The isolated HuR C-terminal RRM3 domain is folded and does not Interact with the RRM1 and RRM2 domains
Studying the structural basis of RRM3 function within the HuR protein has been hampered by the difficulty in producing sufficient amounts of pure soluble material for high resolution studies. We managed to surmount this obstacle by a purification protocol that used an anionic detergent sarkosyl in the initial steps of purification but that could be later removed from the sample buffer. This protocol allowed the preparation of isotope enriched samples for NMR analysis of the domain and its interactions. The NMR spectra of the isolated domain showed a well dispersed set of signals indicating that the RRM module is folded into a defined tertiary structure (Fig. 2, panel A).
Figure 2.
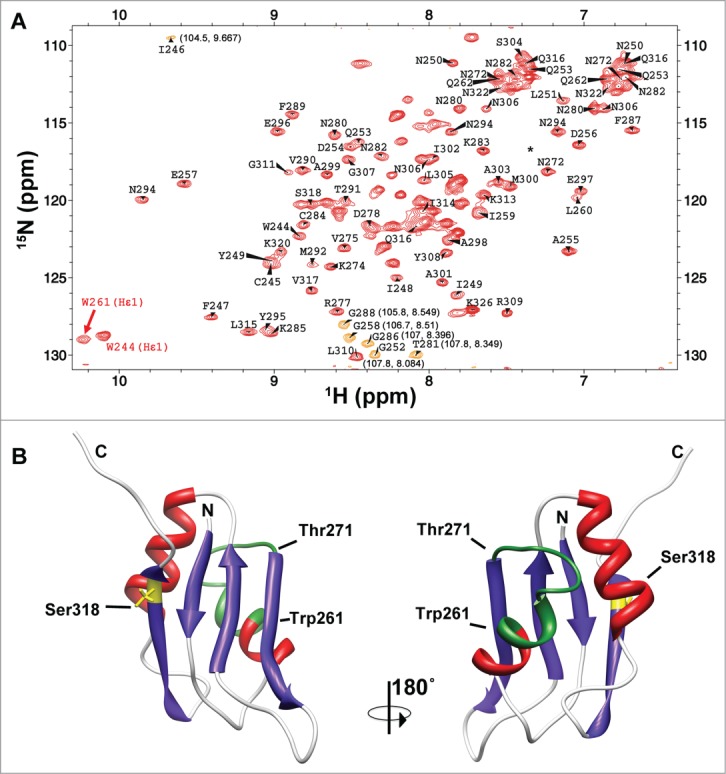
The RRM3 of HuR. (A)1H-15N-HSQC spectrum of HuR RRM3. Labels stand for the amino acids of RRM3 following the numbers of HuR FL protein. The signals in orange are folded in the15N dimension and the correct chemical shifts are indicated in brackets. The asterisk stands for residue T293. Most of the unlabeled signals correspond to residues N-terminal to the HuR RRM3 sequence coming from the cloning strategy. (B) Structural model of HuR RRM3 domain built using chemical shifts of backbone atoms as restraints in the CS23D server. The α-helices are depicted in red and β-strands in blue. The side-chain of S318, which becomes phosphorylated, is highlighted in yellow. Residues between W261 and T271, which are not observed in the NMR spectra, are colored in green. The structure views are rotated 180° around the vertical axis.
Taking into account that HuR RRM1 and RRM2 domains behave as a functional unit,54 we investigated whether RRM3 is also part of this domain arrangement or is independent, so as to characterize the overall structure of HuR full-length (FL). By recording 15N-HSQC spectra of 15N-RRM3 alone or in the presence of 14N-RRM12 (Figure S1, panel A) or vice versa (15N-RRM12 titrated with 14N-RRM3; Figure S1, panel B), no significant changes in the chemical shifts or the line widths of the NMR resonances were detected. This indicates that there is no direct interaction between RRM12 and RRM3 domains or, if any, it is extremely weak and transient in absence of the linker between RRM12 and RRM3. Therefore, the C-terminal RRM module of HuR probably tumbles in solution independently, without a fixed and long-lived orientation with respect to the N-terminal ones. In this sense, our data is consistent with RRM3 being easily removed from the HuR core by a caspase-mediated cleavage, which is a regulatory event that enhances the apoptotic response.11,12
Solution structure of HuR RRM3
We have assigned the backbone and 13Cβ resonances of HuR RRM3 (Fig. 2A) except for the segment from W261 to T271. The signals of these residues were not observed in the spectra, probably due to conformational exchange events with unfavorable kinetics for NMR observation. The NMR samples used were of sufficient quality to measure triple resonance experiments for backbone resonance assignments, however, we observed that increasing the concentration of the samples, as necessary for the measurement of a large number of reliable NOEs, caused a broadening of the signals that dramatically limited the sensitivity of the NMR measurements. Therefore, we built the structure of the RRM3 molecule with the CS23D server, which uses the chemical shifts as restraints and models the non-observed residues based on a database of protein structure fragments (Fig. 2B).55 The structure shows the conserved RRM fold with 2 α-helices packed against 4 anti-parallel β-strands with the canonical β1α1β2β3α2β4 topology characteristic of RRM motifs. Figure 2B also shows how the side-chain of S318 at the β4 strand is exposed to solvent, which is consistent with its accessibility to Protein Kinase C δ (PKCδ) to become phosphorylated.
Dimerization of HuR RRM3
The broadening of the NMR signals of RRM3 with increasing concentrations point to a possible oligomerization of this domain. However, the low ionic strength of the NMR measurements barely shields the charges of ionizable groups, favoring the repulsion between RRM3 domains, so it shifts the self-association equilibrium toward the monomeric state, as inferred from fluorescence assays (see Supplementary Material for more details; Fig. S2). Still, the missing NMR signals in the region W261-T271 may be a related phenomenon with the RRM3 oligomerization. Notably, the oligomerization of ELAV – a homolog to HuR in Drosophila – requires its RRM3 domain.51 Moreover, W419 in ELAV (the residue corresponding to W261 in HuR) seems to be essential for ELAV oligomerization. Thus, we hypothesized that the W261-T271 stretch forms a dimerization epitope that involves the C-terminal end of helix α1.
To test this hypothesis the formation of HuR RRM3 multimers was investigated by analytical ultracentrifugation (AU). AU experiments recorded on RRM3 (Figs. 3A and 3B) determine that the apparent molecular weight is ca. 19.2 kDa, which is higher than the expected value for a monomer (14.3 kDa). The best fit of the measurements was to an exchange model between monomeric and dimeric species, with an association constant KA of 1.8 × 104 M-1. Then, about 70 % of HuR RRM3 is in the monomeric form at protein concentrations used in the AU assays. In addition, a small fraction of the protein (5 %) tends to form aggregates with a molecular weight of ca. 79.8 kDa (Fig. 3B).
Figure 3.

Oligomerization of HuR RRM3. (A) Sedimentation equilibrium measurements of RRM3. The apparent molecular weight (MW) is determined as 19033 ± 1425 Da, larger than the expected for a monomer (14342.2 Da). (B) Sedimentation velocity measurements on RRM3 (S is the sedimentation coefficient). (C) Sedimentation velocity measurements on RRM3 W261E done as in (B). (D) Overlay of the1H-15N-HSQC spectra of RRM3 WT (gray) and RRM3 W261E (black). The resonances of the W261-T271 segment are labeled. A proline residue is at position 266. (E) Comparison of the CS23D structural models of RRM3 WT (light gray) and RRM3 W261E (dark gray). The RMSD for backbone atoms between both models is 1.5 Å, being the main differences at the β2-β3 and α2-β4 flexible loops. The structure views are rotated 180° around the vertical axis.
Within this context, it is worth to mention that the linewidth of the1H-15N correlation peak resonance of the W261 indole group was broader than the corresponding signal for W244, placed at the beginning of β1 strand in RRM3 (with full widths at half-heights of 32.9 Hz in15N and 55.7 Hz in1H vs. 27.7 Hz in15N and 40.8 Hz in1H, respectively; Fig. 2A). This observation is consistent with a monomer-dimer exchange through the W261-T271 region.
To confirm that W261 is involved in the dimerization process, we designed and analyzed the RRM3 W261E mutant, which is expected to destabilize by the introduction of repulsive forces between negatively charged residues at the dimer interface. Under same conditions as used for the wild-type (WT) protein, sedimentation velocity experiments yielded a molecular weight of 14.1 kDa for this mutant, in agreement with the theoretical mass of the monomer (14.2 kDa). Some degree of aggregation could still be observed as a minor peak with a molecular weight of 70.6 kDa, similar as for the WT protein (Fig. 3C).
In agreement with these results, new resonances were observed in the NMR spectra of RRM3 W261E and could be unambiguously assigned to the E261-T271 sequence (Fig. 3D). Moreover, the superposition of the structures of the WT and the W261E proteins, reveals no substantial conformational changes except for the β2-β3 and α2-β4 loops (Fig. 3E). Altogether, these data suggest that the substitution of W261 by a negatively charged glutamic residue inhibits dimerization of the HuR RRM3 domain.
In silicoDynamics
To evaluate the possibility of conformational exchange phenomena in the RRM3 sequence W261-T271, 2 Molecular Dynamics (MD) trajectories of RRM3 monomers at 2 different temperatures were calculated (Supplemental Fig. S3). The computations reckoned 49 ns at 298 K and 20 ns at 310 K. Along the simulations, the HuR RRM3 monomer hardly showed overall structural changes (see Supplementary Material for more details), except for the first helix of the RRM motif, which shows some instability (Supplemental Fig. S3). Indeed, residues G265 to G268 show substantial fluctuations, which are larger at 310 K than at 298 K. This region matches with the NMR gap of HuR RRM3 WT between W261 and T271 (Supplemental Fig. S3). Besides the changes in H-bonding at the first RRM3 helical region, the dynamics of its nearby aromatic residues – W261, F264 and F267 – could contribute to magnetic-field heterogeneity at the W261-T271 stretch. For instance, W261 rotates slowly along the simulations. Indeed, it flips 160 degrees twice along the trajectory at 298 K, dwelling in the flipped conformation along 6 ns (Fig. S3). In the 310 K trajectory, W261 shows an alternative conformation.
To gain insights on how self-association is affecting the behavior of the NMR signals of the W261-T271 residue stretch, we computed 2 MD trajectories with a model of the dimer of RRM3. This model was built aligning 2 of our NMR structures of RRM3 to the coordinates of a crystallographic RRM1 dimer as a starting model (RRM1 shows ca. 31% sequence identity with RRM3).56 The template unit cell comprises 4 RRM1 domains, suggesting 2 putative orientations between RRM domains in the dimer. Both orientations may be biologically relevant since in both conformations the RRM1 dimerization interfaces exclude the residues involve in RNA binding56. Conformation A (Fig. 4A) is consistent with data regarding site-directed mutagenesis of an N-terminal cysteine residue affecting HuR RRM1 dimerization.56 The model of RRM3 dimer in conformation B (Fig. 4B) shows the W261 of each monomer face each other.
Figure 4.
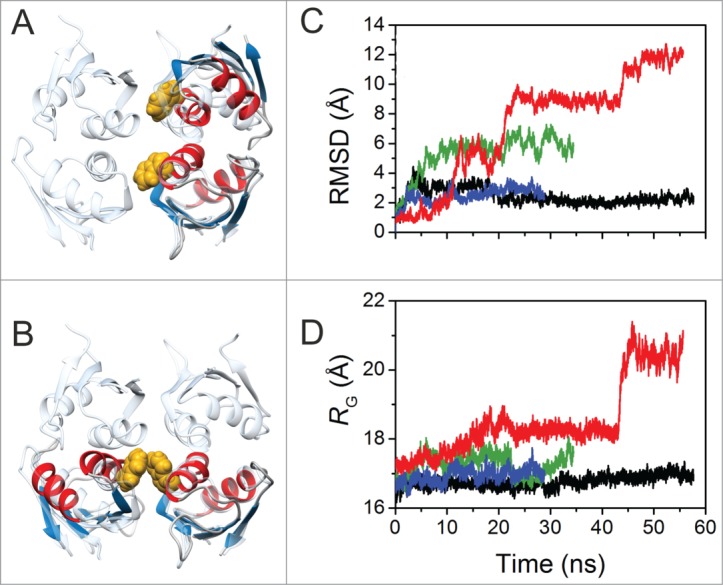
Molecular Dynamic Simulations of Dimeric HuR RRM3. (A) Overlay of RRM3 dimer conformer A model and the unit cell of X-ray structure of HuR RRM1 (pdb code 3hi9). RRM3 ribbons are colored according to secondary structure: α-helixes in red, β-strands in blue. RRM1 is in translucent cyan. The indole rings of W261 in the dimer are represented in yellow spheres. (B) Overlay of RRM3 dimer conformation B model and the HuR RRM1 unit cell. Color code and representations are the same as in (A). (C) Time evolution of overall RMSD values for the RRM3 WT dimer conformers A (black trace) and B (green), and the W261E conformers A (blue) and B (red). W261E initial structures were modeled on those at the end of the corresponding WT trajectories. (D) Time evolution of the radius of gyration of dimers along trajectories. Color code is the same as in (C).
As regards conformation A, the RMSD values show a maximum of 4.4 Å during the first 5 ns. Then, they decay later to a plateau at ca. 2.1 Å, holding this value for the last 37 ns (Fig. 4C). This may agree with an initial relaxation at the dimer interface and a subsequent stabilization of the complex. Residues at the dimer interface (defined as those within 4 Å of any other atom of the partner) belong to 2 regions. The first one comprises I259, Q262, M263, G265, P266 and F267 and involves helix α1. The second contains residues Y308 to D312 at loop α2-β4. The distance between the 2 W261 rings in the dimer is ca.10 Å for this dimer conformation. In contrast (Fig. 4C) conformation B shows a large initial drift. Then, RMSD values fluctuate within the range from 4.5 to 6 Å. During the last 20 ns, however, the dimer dwells in an alternative conformation. The average of the plateau structures shows the W261 ring on a monomer facing its homologous in the second monomer with a distance of 4 Å. Besides W261, the interface involves residues from G268 to V273 in one monomer, and D256 to P266 in the other, so matching the region for which NMR signals are not observed.
Then, we simulated 2 additional MD trajectories for 2 conformations A and B of the W261E mutant but using the final dimer conformations reckoned for the WT species (Fig. 4C). Notably, the structure of conformation A remains unaffected by the mutation along a 30 ns trajectory. The RMSD values with respect by to the original structure oscillate around 2.6 Å. In contrast, RMSD values increase stepwise for the conformation B. A chloride ion bridges the 2 opposed glutamate residues, thereby stabilizing the initial structure for 8 ns. Once the counter ion exits the interface, the RMSD rises step by step to 5, 9, 11 and 12 Å. Such changes are concomitant with increments in the radius of gyration up to 20 Å at the end of the trajectory (Fig. 4D), suggesting that the HuR RRM3 dimerization is impaired by the W261E mutation (Supplemental Fig. S4) in agreement with the experimental data.
Binding of RRM3 to 5 ′-UUUUU-3′ and 5′-AUUUA-3′ RNA Oligos
To test the RNA sequence specificity by HuR RRM3 domain, its interactions with 2 short 5-mer RNA oligonucleotides, namely 5′-AUUUA-3′ and 5′-UUUUU-3′, were studied by NMR (Fig. 5A and C). Average chemical-shift perturbations (Δδavg) measured in the titrations of 15N-labeled RRM3 with RNA reveal that both oligonucleotides are targets of RRM3 (Fig. 5 and Supplemental Fig. S5) although their magnitudes were too small to be used for quantitative assessment of binding affinities by NMR.
Figure 5.
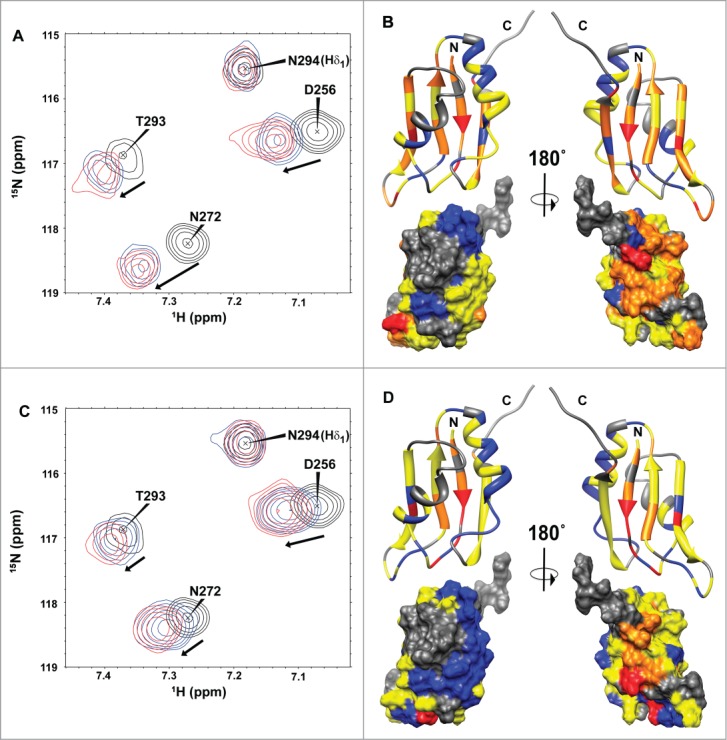
RNA binding of HuR RRM3 with the 5´-UUUUU-3´ (A,B) and 5´-AUUUA-3´ RNAs (C,D) by NMR. (A,C) Overlay of selected regions of the1H-15N-HSQC spectra of free HuR RRM3 (black) and bound to RNA oligos in a RNA:protein ratio of 2:1 (blue) and 4:1 (red). (B,D) Map of HuR RRM3 interface upon binding to RNA. RRM3 surface is rotated 180˚ around the vertical axis in each view. Residues are colored, according to Δδavg (ppm): blue for values< 0.025, yellow for 0.025 ≤ Δδavg ≤ 0.075 and orange for values > 0.075. Resonances broadened beyond the detection limit are colored in red. Prolines and unassigned resonances are indicated in gray.
The residues with the largest perturbations upon RNA addition indicate that the RNA binds to the RRM3 canonical RNA-binding platform, which comprises aromatic residues mainly localized at β1 and β3, the central strands in the β−sheet (Figs. 5B and 5D). In titrations of the proteins with both RNAs,57,58 the largest perturbations occurred in the backbone amides of residues F287 to M292 (at the β3-strand in RNP1) and F247-L251 (at the β1-strand of RNP2). Interestingly, the protein platform with which HuR RRM3 binds to 5′-UUUUU-3′ RNA is extended to the whole β-sheet, with residues at both the β2 and β4 strands experiencing relatively large chemical-shift perturbations (Δδavg > 0.075 ppm; Figure 5B and Supplemental Fig. S5). In addition to perturbations in the chemical shifts, some RRM3 signals at the protein-nucleic acid interface broadened beyond the detection limit independently of the RNA oligonucleotide used. Residues such as I248, L251, V275 and K320 show this behavior for both 5′-UUUUU-3′ and 5′-AUUUA-3′ RNAs.
We next assessed the affinity of HuR RRM3 for 25-mer U-rich and AUUUA RNA oligonucleotides by CD (Fig. 6). The protein shows more affinity toward U-rich RNA than to AUUUA-containing RNA molecules (KD = 61.1 ± 9.5 and KD = 123.8 ± 6.9 μM, respectively), although both complexes show a protein:RNA stoichiometry of 1:4. χ2 values of the fittings were 0.00231 for the RRM3:U-rich RNA system and 0.00312 for the RRM3:AUUUA-containing RNA complex. This demonstrates that HuR RRM3 is a bona fide ARE-RNA interacting domain that preferably binds U-rich stretches, rather than AUUUA motifs.
Figure 6.
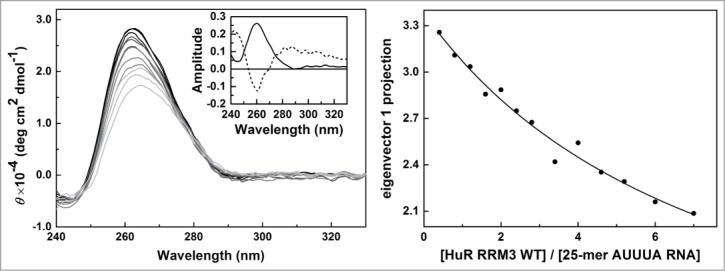
RNA binding of HuR RRM3 with the AUUUA motif 25-mer RNA by CD. Left – Far-UV CD data of the 5′-AUUUAUUUAUUUAUUUAUUUAUUUA-3′ 25-mer RNA molecules at different concentrations of the HuR RRM23 construct. Inset: Normalized first and second principal components resulting from covariance analysis of the CD spectra along the titration. The first one (continuous line) accounts for a 90.7 % of the spectral changes; the second (dashed line) for a 7.9 % of them. Right - Projection of the titration data in left panel on their first principal component obtained by covariance analysis of the whole spectra. Data was fitted according to a model considering 4 binding sites on RNA with similar affinity toward the protein.
The Phosphomimetic HuR RRM3 S318D Mutant
Phosphorylation at S318 residue of HuR RRM3 domain was mimicked by Ser-to-Asp single mutation. The assignment of the NMR spectrum of S318D allowed us to build a model of its structure revealing only minor differences mainly found at the loops. Indeed, the RMSD for backbone atoms between both models is 1.1 Å (data not shown). Similarly to the WT molecule, the signals of the residues W261-T271 were not observed.
HuR RRM3 S318D binding to RNA targets was monitored by NMR in solution (Fig. 7A). Data reveals that the magnitude of the average chemical-shift perturbations for the S318D amide signals upon binding to the 5′-UUUUU-3′ RNA oligonucleotide are slightly smaller than those measured for the RRM3 WT (Fig. 7B), suggesting a diminished binding ability. Indeed, the dissociation equilibrium constant for the complex between the HuR RRM3 S318D mutant and the 25-mer U-rich RNA is slightly larger than that of the WT-involving complex (KD = 90.1 ± 3.7 μM; Fig. 7C) with a protein:RNA stoichiometry of 1:4. χ2 value of the fittings was 0.00035. Perhaps the introduction of a negatively charged group results in electrostatic repulsion with the phosphate groups from RNA (Fig. 7B).
Figure 7.
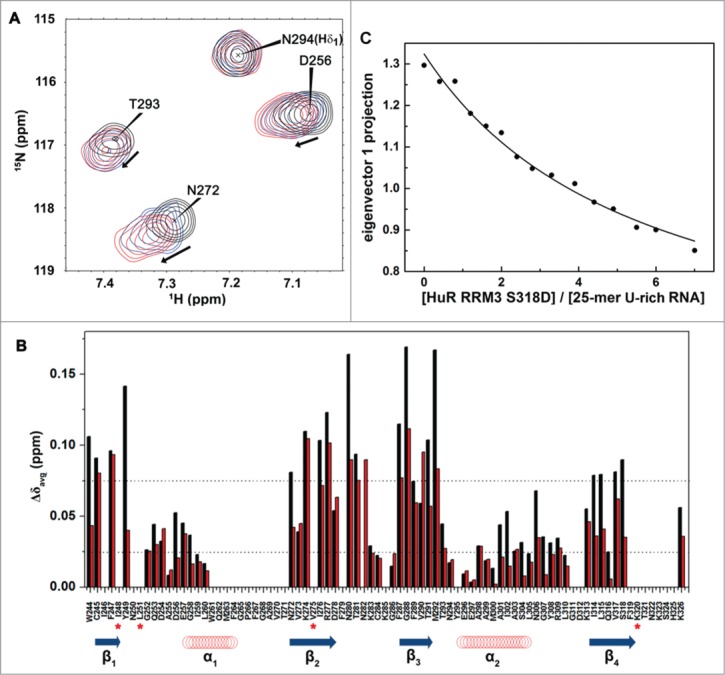
HuR RRM3 S318D: (A) phosphomimetic mutant. (A) Overlay of 1H-15N-HSQC spectra of HuR RRM3 S318D in the absence (black) and presence of 5′-UUUUU-3′ RNA at a RNA:protein ratio of 2:1 (blue) and 4:1 (red). A subset of 4 representative resonances is labeled in black. (B) Comparison of average chemical shift differences (Δδavg) between free and RNA-bound HuR RRM3 WT (black) and HuR RRM3 S318D (red) in a ratio of 4:1 for 5′-UUUUU-3′ RNA. Secondary structure elements of RRM3 are symbolized by blue arrows for β-strands and red coil symbols for α-helices. Asterisks indicate those residues disappearing upon RNA binding to the RRM3 WT or S318D mutant. (C) Changes in the far-UV CD signal of the 25-mer U-rich RNA spectrum during the titration with HuR RRM3 S318D construct.
Discussion
Our results suggest that HuR RRM3 domain tumbles in solution independently of the RRM12 tandem orientation, which is consistent with an unstructured HNS linker ― in particular S226 ― for caspase-dependent cleavage.11 The RRM3-containing cleavage product may acquire new functions in triggering apoptosis as it selectively binds to and stabilizes caspase-9 mRNA in an ARE-dependent manner.12 The NMR titration data herein reported and earlier findings with HuB,59 demonstrate that isolated RRM3 can bind to AREs, although previous studies suggested a negligible role of HuR RRM3 ― as part of HuR FL ― in AREs recognition.34, 44-46
Isolated RRM3 is in a monomer/dimer exchange on a time scale unfavorable for NMR observation of the residues at the dimerization site, notably those in the W261-T271 region at the C-end of helix α1. Indeed, this region shows large fluctuations in MD simulations, which affect the H-bonding pattern at α1 and the dynamics of W261 ring. AU measurements confirm not only the dimerization of RRM3 but also that the W261E mutation shifts the monomer/dimer equilibrium toward the monomeric form. Moreover, the resonances belonging to the E261-T271 region became visible to NMR (Fig. 3D). Thus, we propose that RRM3 dimerization involves its α1-helix and the loop α1-β2, both placed at the opposite side of the RNA-binding platform, as previously proposed for homologous proteins such as ELAV in Drosophila upon mutating W419 (W261 in HuR).51 This suggestion is supported by MD computations explaining the different stability of 2 possible dimeric structures and the effects of the W261E mutation to prevent dimerization. There are other instances of RRM-RRM interactions taking place through their α-helices. For instance, RRM3 and RRM4 from Polypyrimidine Tract Binding protein (PTB) mainly contact through helix α2 of RRM4 and helix α1 and α2 of RRM3, resulting in the perpendicular positioning of both RRM.60 On the other hand, in FBP-Interacting Repressor (FIR) protein, the α-helix face of RRM1 packs onto the β-sheet face of RRM2, creating a stable interface.61,62 Examples of Trp-mediated homodimerization in other RRM domains have been previously reported, as in the Nup35 protein.63 In contrast, the RRM modules of the splicing factor Pub60 form a dimeric interface driven by electrostatic interactions involving a flexible loop.64 Interestingly enough is the crystallographic structure of HuR RRM1 motif, which reveals a tetramer assembled through helices α1 and α2 to form the so-called dimer conformations A and B used as templates for MD simulations on HuR RRM3.56
Multimerization of HuR protein is then dependent on RRM3 motif and more specifically on its well-conserved W261.51 In fact, the whole W261-T271 stretch is highly conserved among Hu proteins as it was described in earlier studies.25 Our structural studies on HuR RRM3 also demonstrate that the monomer/dimer exchange of HuR RRM3 takes place even in absence of RNA, in contrast to previous reports suggesting that RNA promotes HuR FL multimerization.34,65 Of note, no additional resonances corresponding to the W261-T271 gap appear on the 15N-HSQC NMR spectra upon binding to short single-stranded 5-mer RNA oligonucleotides, from which it follows that the oligomerization exchange remains unaltered. The fact that HuR FL forms stable oligomeric complexes with long RNA fragments make necessary additional experiments to explain how far the oligomerization of isolated RRM3 would change by long and highly-structured ARE-bearing RNAs.
RRM-comprising RNA Binding Proteins (RBPs) are usually known to bind single-stranded nucleic acids by stacking with aromatic residues placed at the central β−strands (β1 and β3) of the β-sheet, although a high variety of RNA binding mechanisms have been described for RRM modules (for a review see ref. 66). By NMR experiments we confirm the RNA binding to these secondary structure elements of RRM3 for both 5′-AUUUA-3′ and 5′-UUUUU-3′ oligonucleotides. In addition, we found out that the HuR RRM3 protein platform that interacts with 5′-UUUUU-3′ RNA includes perturbed residues at β2 and β4 strands so as to involve the whole β-sheet, which suggest a preference of RRM3 by U-rich stretches, as was further confirmed by CD. Then, HuR RRM3 might experience a sliding motion on the U-rich 25-mer RNA surface, but not with AUUUA-containing RNA molecules. These differences in affinity were already observed for the heterogeneous nuclear RiboNucleoprotein C protein (hnRNP1)67 and HuR FL.68,69 Later, it has been demonstrated that HuR FL recognizes U-rich mRNAs and that the substitution of U by A or C decreases the HuR binding affinity, whereas an exchange by G has a drastic effect on the interaction.46 Meriting particular interest, the RRM3-containing cleavage product specifically binds ARE1 of caspase-9 mRNA, which lacks of 5′-AUUUA-3′ sequences but it contains a U-rich region.12
HuR is regulated by post-translational modifications. Phosphorylation at RRM3 S318 residue by PKCδ means an important mode in HuR regulation, with consequences in colon carcinoma cells due to HuR dysregulation.41,70 Therefore, the S318 residue of HuR RRM3 was mutated by an aspartate to mimic phosphorylation events. Both RRM3 WT and RRM3 S318D models agree on the structure. The main difference was found at the level of RNA recognition, since the CD data and the chemical-shift perturbations for RRM3 S318D resonances upon binding to the U-rich RNA oligonucleotides indicate that the binding affinity is slightly smaller than that of the RRM3 WT, although RNA docks on both RRM3 species using similar platforms. This result can be explained by an electrostatic repulsion effect between D318 and the negatively charged RNA.46 When the protein is phosphorylated, the negative charge of the protein is further increased (in absolute terms) and the repulsion effect would be even larger. In contrast to our findings, Schulz and coworkers71 have recently reported a higher binding affinity of HuR S318D with U-rich bearing mRNA stretches. Plausible explanations arise from differences in length and degree of structure of U-rich long mRNAs and 5′-UUUUU-3′ short RNA molecules and/or differences in number of RRMs making up HuR FL and RRM3 S318D species.
Our study underlines the exceptional properties of HuR RRM3 as a multi-functional domain which may lead to HuR oligomerization and binding to RNA targets at once, using 2 surfaces on opposite sides of the RRM domain.
Materials and Methods
Design of HuR Constructs
pGEX 5×2 vectors containing the sequences coding for HuR FL protein as well as the C-terminal RRM3 domain, were kindly provided by Dr. M. Gorospe (National Institutes of Health, Baltimore, USA) and Prof. J. A. Steitz (Yale University, New Haven, USA). The HuR RRM12 tandem-construct containing the 2 N-terminal RRM1 and RRM2 modules along with the 6xHis-tag was used as previously described.54 The RRM3 domain comprises the amino acid sequence from W244 to K326, which was cloned into the pETM-11 vector using EcoRI and NotI restriction sites. Further site-directed mutagenesis was performed on the gene coding RRM3 WT to replace the S318 by an alanine (RRM3 S318A) or an aspartate (RRM3 S318D) so as to mimic the phosphorylation of HuR at S318. The mutant replacing W261 by glutamic acid (RRM3 W261E) was obtained by similar means.
Protein Expression and Purification of HuR Constructs
Recombinant proteins were expressed in Escherichia coli BL21 (DE3) strain as follows. Competent cells were transformed with plasmids containing HuR RRM12 or HuR RRM3 constructs, along with its mutants RRM3 W261E, RRM3 S318A and RRM3 S318D. Cells were grown at 37 ˚C in Luria Bertani (LB) medium supplemented with ampicillin (50 g/L) for pGEX vectors or kanamycin (50 g/L) for pETM vectors. Isotopically 15N and 13C-labeled proteins were expressed in minimal medium (M9) supplemented either with 15NH4Cl or 15NH4Cl and 13C-glucose following the Marley protocol.72 Protein expression was induced by addition of 1 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG) when OD600 was 0.6–0.8 and the temperature for RRM3 expression lowered to 30˚C. After 5 h of expression, cells were harvested by centrifugation at 7,000 g and further resuspended in 50 mM Tris-HCl buffer (pH 8.0), 150 mM NaCl for RRM12. For RRM3 species, the ionic strength was increased up to 800 mM NaCl. His-tagged HuR domains were purified by nickel affinity chromatography (Ni Sepharose 6 Fast Flow, GE Healthcare). To make RRM3 soluble, buffers were supplemented with 0.1 % (v/v) N-Lauroylsarcosine (Sarkosyl, SIGMA, St. Louis, USA) detergent. RRM12 and RRM3 samples were concentrated up to 900 and 150 μM, respectively in 10 mM sodium phosphate buffer (pH 7.3) supplemented with 0.5 mM DTT and 0.002 % phenylmethylsulfonyl fluoride (PMSF). In case of RRM3, the excess of detergent was washed out by consecutive cycles of dilution and concentration of the protein using a buffer devoid of sarkosyl and NaCl. Protein concentrations were determined by spectrophotometry with predicted extinction coefficients.
Nuclear Magnetic Resonance Spectroscopy
NMR samples of HuR RRM3 WT and its RRM3 W261E and RRM3 S318D mutants were prepared in 95% H2O / 5% D2O solutions of 10 mM sodium phosphate buffer, 3 mM DTT (pH 7.3) at concentrations in a range from 80 to 175 µM. NMR spectra were recorded at 298 K on a Bruker Avance III 800 MHz spectrometer with a [1H,13C,15N] triple resonance cryoprobe equipped with z gradient coil.1HN,15N,13C′,13Cα,13Cβ and 1Hα assignments were obtained from the analysis of 2D1H-15N-HSQC,1H-13C HSQC and 3D HNCO, HN(CA)CO, HNCACB, HN(CO)CACB, HNCA, HN(CO)CA, HN(CA)HA, HN(COCA)HA. The spectra were processed with TopSpin (Bruker). An initial partial automatic assignment of the backbone and 13Cβ resonances was obtained with the program MARS73 which was confirmed and completed manually. The assignments of the 3 RRM3 molecules studied in this work have been deposited in BioMagResBank with entry IDs 19494, 19499 and 19500 for the WT, W261E and S318D variants, respectively. Structural models were built based on the measured chemical shifts using the CS23D server.55
RNA binding of HuR RRM3 WT and RRM3 S318D was monitored by acquiring 1H-15N-HSQC spectra on a 500 MHz Bruker spectrometer equipped with a cryoprobe at 298 K. Spectra were recorded on a sample of 50 μM 15N-RRM3 in 10 mM sodium phosphate buffer, 3 mM DTT pH 7.3, upon addition of small aliquots of concentrated stocks of the 5′-AUUUA-3′ and 5′-UUUUU-3′ RNA oligonucleotides (IDT, Integrated DNA Technologies) up to RNA:protein ratios of 2:1 and 4:1. The pH value of the sample was verified after each titration step. Weighted average chemical-shift perturbations (Δδavg) of each backbone amide resonance were calculated as follows: Δδavg = (([ΔδH]2 + [ΔδN/5]2)/ 2)1/2, where ΔδH and ΔδN are the differences in the 1H and 15N chemical shifts, respectively.
Analytical Ultracentrifugation
Sedimentation equilibrium experiments of HuR RRM3 domain were performed at 20 °C in an Optima XL-A Analytical Ultracentrifuge (AU, Beckman Instruments) with an AN50-Ti rotor. 80 μl samples at ~27 μM HuR RRM3 were examined in 10 mM phosphate, 50 mM NaCl, 0.02 % NaN3, 3 mM DTT, pH 7.3, at 3 successive speeds (18,500; 22,000 and 32,000 rpm) and absorbance was measured at 280 nm at 12 and 48 h to assess that the equilibrium condition was reached. Base-line signals were determined taking a radial scan at 18,500 rpm, after running the samples 8 h at 45,000 rpm. Mass conservation within the cell was checked in all the experiments. The apparent weight-averaged molecular weights were obtained by fitting individual data sets to a sedimentation equilibrium model for single species, using the program HeteroAnalysis.74 The equilibrium dimerization constant, K2, was calculated by fitting the experimental data to a monomer/dimer sedimentation equilibrium model constraining the monomer molecular mass to 14,342 Da. Sedimentation velocity experiments were performed at 45,000 rpm and 20 °C with 400-μl samples loaded into double sector cells, using the buffer and protein concentrations employed in equilibrium experiments. Radial scans at 280 nm were taken every 10 min and the sedimentation coefficient distribution was calculated by least squares boundary modeling of the sedimentation velocity data using the program SEDFIT.75 The experimental coefficients were converted to standard conditions (s20,w). The partial specific volume of HuR RRM3 (0.728 gL-1), calculated from the amino acid composition, and the buffer density and viscosity were determined with the SEDNTERP program.76
Circular Dichroism
RNA binding was monitoring in the UV range of 240–330 nm by the addition of increasing amounts of the HuR RRM3 constructs to a 3 μM sample of the 25-mer oligonucleotides (5′-UUUUUUUUUUUUUUUUUUUUUUUUU-3′ and 5′-AUUUAUUUAUUUAUUUAUUUAUUUA-3′). A temperature of 10 °C was chosen to optimize the signal change upon protein binding. The Principal Component Analysis (PCA) on CD data was performed using Matlab R2010b (MathWorks, http://www.mathworks.es/). A matrix of 91 × 13–15 points was analyzed, which comprised the values of the CD signal in the 240–330 nm range used in the 13–15 titration points. The percentage of amplitude of each component was calculated. Each component projection was independently analyzed. The first component of the CD signal was plotted against the ratio [RRM3] / [25-mer RNA] and fitted to a 1:4 binding site model as reported previously.54,77
Molecular Dynamics Simulations
MD computations were performed using the AMBER 9 package78 and using the AMBER-2003 force field.79 The NMR-derived coordinates of RRM3 domain in this work was used as the starting structures. To model the RRM3 dimer, the structure was aligned against 2 of the 4 monomers in the HuR RRM1 structure (pdb code 3hi9).56 Simulations were carried out under periodic boundary conditions using an orthorhombic cell geometry (minimum distance between protein and cell faces was initially set to 10 Å) and PME electrostatics with a Ewald summation cut off of 9 Å. The structures were solvated with TIP3P water molecules; and Na+ counterions were added to neutralize the net charge of the full systems. Afterwards, solvent and counter-ions were subjected to 500 steps of steepest descent minimization followed by 500 ps NPT-MD computations using isotropic molecule position scaling and a pressure relaxation time of 2 ps at 298 K. Temperature was regulated using a Langevin thermostat80 with a collision frequency of 5 ps-1. The density of the system reached a plateau during the first 150 ps. Then, the whole system was energy minimized and submitted to NVT MD computations at 298 K. The SHAKE algorithm81 was used to constrain bonds involving hydrogen atoms. The PTRAJ module of AMBER was used for trajectory analysis. Molecular graphics were performed with UCSF Chimera.82
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
I.D.-M. wishes to thank the Andalusian Government (P07-CVI-02896, P11-CVI-07216 and BIO198) for financial support. F.J.B. acknowledges financial support from the Spanish MINECO (CTQ2011–28680). M.L.M.-C. contribution is supported by grants from NIH AT-1576, ETORTEK-2011, Educación Gobierno Vasco 2011, FIS PI11/01588. Ciberehd is funded by the Instituto de Salud Carlos III. L.A.M.-C. thanks the Basque Government (PI2010–17) and the Spanish MICINN (BFU2010–17857) for funding. Dr. Scheiba is grateful to be awarded with a Spanish Scientific Council Fellowship (JAEpre_08_00375).
Acknowledgments
Our special appreciation is addressed to Prof. Menéndez and Dr. Bustamate for the analytical ultracentrifugation experiments performed at the Institute of Physical Chemistry “Rocasolano” (Madrid, Spain). Authors also thank technical assistance at the NMR services in CITIUS (University of Sevilla) and CICBiogune (Derio). The plasmid containing the HuR full-length protein was kindly provided by Dr. M. Gorospe (National Institutes of Health, Baltimore, USA) and Dr. J. A. Steitz (Yale University, New Haven, USA). We are grateful to Prof. Miguel A. De la Rosa for critical reading of the manuscript.
Supplemental Materials
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Ma WJ, Cheng S, Campbell C, Wright A, Furneaux H. Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J Biol Chem. 1996; 271: 8144-51; PMID:8626503; http://dx.doi.org/10.1074/jbc.271.14.8144 [DOI] [PubMed] [Google Scholar]
- 2.Levine TD, Gao F, King PH, Andrews LG, Keene JD. Hel-N1: an autoimmune RNA-binding protein with specificity for 3' uridylate-rich untranslated regions of growth factor mRNAs. Mol Cell Biol 1993; 13: 3494-504; PMID:8497264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang W, Caldwell MC, Lin S, Furneaux H, Gorospe M. HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. EMBO J 2000; 19: 2340-50; PMID:10811625; http://dx.doi.org/10.1093/emboj/19.10.2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang W, Yang X, Cristofalo VJ, Holbrook NJ, Gorospe M. Loss of HuR is linked to reduced expression of proliferative genes during replicative senescence. Mol Cell Biol 2001; 21: 5889-98; PMID:11486028; http://dx.doi.org/10.1128/MCB.21.17.5889-5898.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gallouzi IE, Brennan CM, Stenberg MG, Swanson MS, Eversole A, Maizels N, Steitz JA. HuR binding to cytoplasmic mRNA is perturbed by heat shock Proc Natl Acad Sci USA 2000; 97: 3073-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang W, Furneaux H, Cheng H, Caldwell MC, Hutter D, Liu Y, Holbrook N, Gorospe M. HuR regulates p21 mRNA stabilization by UV light. Mol Cell Biol 2000; 20: 760-769; PMID:10629032; http://dx.doi.org/10.1128/MCB.20.3.760-769.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gallouzi IE, Brennan CM, Steitz JA. Protein ligands mediate the CRM1-dependent export of HuR in response to heat shock. RNA 2001; 7: 1348-61; PMID:11565755; http://dx.doi.org/10.1017/S1355838201016089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mazan-Mamczarz K, Galban S, Lopez de Silanes I, Martindale JL, Atasoy U, Keene JD, Gorospe M. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc Natl Acad Sci USA 2003; 100: 8354-59; PMID:12821781; http://dx.doi.org/10.1073/pnas.1432104100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abdelmohsen K, Pullmann R, Jr, Lal A, Kim HH, Galban S, Yang X, Blethrow JD, Walker M, Shubert J, Gillespie DA, et al. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol Cell 2007; 25: 543-57; PMID:17317627; http://dx.doi.org/10.1016/j.molcel.2007.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdelmohsen K, Lal A, Kim HH, Gorospe M. Posttranscriptional orchestration of an anti-apoptotic program by HuR. Cell Cycle 2007; 6: 1288-92; PMID:17534146; http://dx.doi.org/10.4161/cc.6.11.4299 [DOI] [PubMed] [Google Scholar]
- 11.Mazroui R, Di Marco S, Clair E, von Roretz C, Tenenbaum SA, Keene JD, Saleh M, Gallouzi I.E. Caspase-mediated cleavage of HuR in the cytoplasm contributes to pp32/PHAP-I regulation of apoptosis. J Cell Biol 2008; 180: 113-27; PMID:18180367; http://dx.doi.org/10.1083/jcb.200709030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.von Roretz C, Jin Lian X, Macri AM, Punjani N, Clair E, Drouin O, Dormoy-Raclet V, Ma JF, Gallouzi IE. Apoptotic-induced cleavage shifts HuR from being a promoter of survival to an activator of caspase-mediated apoptosis. Cell Death Differ 2013; 20: 154-68; PMID:22955946; http://dx.doi.org/10.1038/cdd.2012.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tran H, Maurer F, Nagamine Y. Stabilization of urokinase and urokinase receptor mRNAs by HuR is linked to its cytoplasmic accumulation induced by activated mitogen-activated protein kinase-activated protein kinase 2. Mol Cell Biol 2003; 23: 7177-88; PMID:14517288; http://dx.doi.org/10.1128/MCB.23.20.7177-7188.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Figueroa A, Cuadrado A, Fan J, Atasoy U, Muscat GE, Munoz-Canoves P, Gorospe M, Munoz A. Role of HuR in skeletal myogenesis through coordinate regulation of muscle differentiation genes. Mol Cell Biol 2003; 23: 4991-5004; PMID:12832484; http://dx.doi.org/10.1128/MCB.23.14.4991-5004.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cherry J, Karschner V, Jones H, Pekala PH. HuR, an RNA-binding protein, involved in the control of cellular differentiation. In Vivo 2006; 20: 17-23; PMID:16433023 [PubMed] [Google Scholar]
- 16.Yi J, Chang N, Liu X, Guo G, Xue L, Tong T, Gorospe M, Wang W. Reduced nuclear export of HuR mRNA by HuR is linked to the loss of HuR in replicative senescence. Nucleic Acids Res 2009; 38: 1547-58; PMID:20007147; http://dx.doi.org/10.1093/nar/gkp1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katsanou V, Papadaki O, Milatos S, Blackshear PJ, Anderson P, Kollias G, Kontoyiannis DL. HuR as a negative posttranscriptional modulator in inflammation. Mol Cell 2005; 19: 777-89; PMID:16168373; http://dx.doi.org/10.1016/j.molcel.2005.08.007 [DOI] [PubMed] [Google Scholar]
- 18.Atasoy U, Watson J, Patel D, Keene JD. ELAV protein HuA (HuR) can redistribute between nucleus and cytoplasm and is upregulated during serum stimulation and T cell activation. J Cell Sci 1998; 111: 3145-56; PMID:9763509 [DOI] [PubMed] [Google Scholar]
- 19.Atasoy U, Curry SL, Lopez de Silanes I, Shyu AB, Casolaro V, Gorospe M, Stellato C. Regulation of eotaxin gene expression by TNF-α and IL-4 through mRNA stabilization: involvement of the RNA-binding protein HuR. J Immunol 2003; 171: 4369-78; PMID:14530362; http://dx.doi.org/10.4049/jimmunol.171.8.4369 [DOI] [PubMed] [Google Scholar]
- 20.McMullen MR, Cocuzzi E, Hatzoglou M, Nagy LE. Chronic ethanol exposure increases the binding of HuR to the TNFalpha 3'-untranslated region in macrophages. J Biol Chem 2003; 278: 38333-41; PMID:12876290; http://dx.doi.org/10.1074/jbc.M304566200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Srikantan S, Gorospe M. HuR function in disease. Front Biosci 2012; 17: 189-205; http://dx.doi.org/10.2741/3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adam SA, Nakagawa T, Swanson MS, Woodruff TK, Dreyfuss G. mRNA polyadenylate-binding protein: gene isolation and sequencing and identification of a ribonucleoprotein consensus sequence. Mol Cell Biol 1986; 6: 2932-43; PMID:3537727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sachs AB, Bond MW, Kornberg RD. A single gene from yeast for both nuclear and cytoplasmic polyadenylate-binding proteins: domain structure and expression. Cell 1986; 45: 827-35; PMID:3518950; http://dx.doi.org/10.1016/0092-8674(86)90557-X [DOI] [PubMed] [Google Scholar]
- 24.Maris C, Dominguez C, Allain FH. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J 2005; 272: 2118-2131; PMID:15853797; http://dx.doi.org/10.1111/j.1742-4658.2005.04653.x [DOI] [PubMed] [Google Scholar]
- 25.Good PJ. A conserved family of elav-like genes in vertebrates. Proc Natl Acad Sci USA 1995; 92: 4557-61; PMID:7753842; http://dx.doi.org/10.1073/pnas.92.10.4557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H, Zeng F, Liu Q, Liu Z, Niu L, Teng M, Li X. The structure of the ARE-binding domains of Hu antigen R (HuR) undergoes conformational changes during RNA binding. Acta Crystallogr B Biol Crystallogr 2013; 69: 373-80; http://dx.doi.org/10.1107/S0907444912047828 [DOI] [PubMed] [Google Scholar]
- 27.Díaz-Moreno I, Hollingworth D, Kelly G, Martin S, García-Mayoral M, Briata P, Gherzi R, Ramos A. Orientation of the central domains of KSRP and its implications for the interaction with the RNA targets. Nucleic Acids Res 2010; 38: 5193-205; http://dx.doi.org/10.1093/nar/gkq216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fan XC, Steitz JA. HNS, a nuclear-cytoplasmic shuttling sequence in HuR. Proc Natl Acad Sci USA 1998; 95: 15293-98; PMID:9860962; http://dx.doi.org/10.1073/pnas.95.26.15293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brennan CM, Gallouzi IE, Steitz JA. Protein ligands to HuR modulate its interaction with target mRNAs in vivo. J Cell Biol 2000; 151: 1-14; PMID:11018049; http://dx.doi.org/10.1083/jcb.151.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang X, Kim HE, Shu H, Zhao Y, Zhang H, Kofron J, Donnelly J, Burns D, Ng SC, Rosenberg S, et al. Distinctive roles of PHAP proteins and prothymosin-α in a death regulatory pathway. Science 2003; 299: 223-26; PMID:12522243; http://dx.doi.org/10.1126/science.1076807 [DOI] [PubMed] [Google Scholar]
- 31.Rebane A, Aab A, Steitz JA. Transportins 1 and 2 are redundant nuclear import factors for hnRNP A1 and HuR. RNA 2004; 10: 590-99; PMID:15037768; http://dx.doi.org/10.1261/rna.5224304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang W, Yang X, Kawai T, López de Silanes I, Mazan-Mamczarz K, Chen P, Chook YM, Quensel C, Köhler M, Gorospe M. AMP-activated protein kinase-regulated phosphorylation and acetylation of importin alpha1: involvement in the nuclear import of RNA-binding protein HuR. J Biol Chem 2004; 279: 48376-88; PMID:15342649; http://dx.doi.org/10.1074/jbc.M409014200 [DOI] [PubMed] [Google Scholar]
- 33.van der Giessen K, Gallouzi IE. Involvement of transportin 2-mediated HuR import in muscle cell differentiation. Mol Biol Cell 2007; 18: 2619-29; PMID:17475777; http://dx.doi.org/10.1091/mbc.E07-02-0167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fialcowitz-White EJ, Brewer BY, Ballin JD, Willis CD, Toth EA, Wilson GM. Specific protein domains mediate cooperative assembly of HuR oligomers on AU-rich mRNA-destabilizing sequences. J Biol Chem 2007; 282: 20948-59; PMID:17517897; http://dx.doi.org/10.1074/jbc.M701751200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park S, Myszka DG, Yu M, Littler SJ, Laird-Offringa IA. HuD RNA Recognition Motifs Play Distinct Roles in the Formation of a Stable Complex with AU-Rich RNA. Mol Cell Biol 2000; 20: 4765-72; PMID:10848602; http://dx.doi.org/10.1128/MCB.20.13.4765-4772.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doller A, Pfeilschifter J, Eberhardt W. Signalling pathways regulating nucleo-cytoplasmic shuttling of the mRNA-binding protein HuR. Cell Signal 2008; 20: 2165-73; PMID:18585896; http://dx.doi.org/10.1016/j.cellsig.2008.05.007 [DOI] [PubMed] [Google Scholar]
- 37.Eberhardt W, Doller A, Pfeilschifter J. Regulation of the mRNA-binding protein HuR by posttranslational modification: spotlight on phosphorylation. Curr Protein Pept Sci 2012; 13: 380-90; PMID:22708484; http://dx.doi.org/10.2174/138920312801619439 [DOI] [PubMed] [Google Scholar]
- 38.Kim HH, Abdelmohsen K, Lal A, Pullmann RJ, Yang X, Galban S, Srikantan S, Martindale JL, Blethrow J, Shokat KM, et al. Nuclear HuR accumulation through phosphorylation by Cdk1. Genes Dev 2008; 22: 1804-15; PMID:18593881; http://dx.doi.org/10.1101/gad.1645808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim HH, Yang X, Kuwano Y, Gorospe M. Modification at HuR (S242) alters HuR localization and proliferative influence. Cell Cycle 2008; 7: 3371-77; PMID:18948743; http://dx.doi.org/10.4161/cc.7.21.6895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doller A, Huwiler A, Muller R, Radeke HH, Pfeilschifter J, Eberhardt W. Protein kinase C α-dependent phosphorylation of the mRNA-stabilizing factor HuR: implications for posttranscriptional regulation of cyclooxygenase-2. Mol Biol Cell 2007; 18: 2137-48; PMID:17392515; http://dx.doi.org/10.1091/mbc.E06-09-0850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doller A, Akool elS, Huwiler A, Muller R, Radeke HH, Pfeilschifter J, Eberhardt W. Posttranslational modification of the AU-rich element binding protein HuR by protein kinase Cdelta elicits angiotensin II-induced stabilization and nuclear export of cyclooxygenase 2 mRNA. Mol Cell Biol 2008; 28: 2608-25; PMID:18285462; http://dx.doi.org/10.1128/MCB.01530-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H, Park S, Kilburn B, Jelinek MA, Henschen-Edman A, Aswad DW, Stallcup MR, Laird-Offringa IA. Lipopolysaccharide-induced methylation of HuR, an mRNA-stabilizing protein, by CARM1. Coactivator-associated arginine methyltransferase. J Biol Chem 2002; 277: 44623-30; PMID:12237300; http://dx.doi.org/10.1074/jbc.M206187200 [DOI] [PubMed] [Google Scholar]
- 43.Embade N, Fernandez-Ramos D, Varela-Rey M, Beraza N, Sini M, de Juan VG, Woodhoo A, Martinez-Lopez N, Rodriguez-Iruretagoyena B, Bustamante FJ, et al. Murine double minute 2 regulates Hu antigen R stability in human liver and colon cancer through NEDDylation. Hepatology 2012; 55: 1237-48; PMID:22095636; http://dx.doi.org/10.1002/hep.24795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chung S, Jiang L, Cheng S, Furneaux H. Purification and properties of HuD, a neuronal RNA-binding protein. J Biol Chem 1996; 271: 11518-24; PMID:8626712; http://dx.doi.org/10.1074/jbc.271.19.11518 [DOI] [PubMed] [Google Scholar]
- 45.Yeap BB, Voon DC, Vivian JP, McCulloch RK, Thomson AM, Giles KM, Czyzyk-Krzeska MF, Furneaux H, Wilce MC, Wilce JA, et al. Novel binding of HuR and poly(C)-binding protein to a conserved UC-rich motif within the 3'-untranslated region of the androgen receptor messenger RNA. J Biol Chem 2002; 277: 27183-92; PMID:12011088; http://dx.doi.org/10.1074/jbc.M202883200 [DOI] [PubMed] [Google Scholar]
- 46.Barker A, Epis MR, Porter CJ, Hopkins BR, Wilce MC, Wilce JA, Giles KM, Leedman PJ. Sequence requirements for RNA binding by HuR and AUF1. J Biochem 2012; 151: 423-37; PMID:22368252; http://dx.doi.org/10.1093/jb/mvs010 [DOI] [PubMed] [Google Scholar]
- 47.Abe R, Sakashita E, Yamamoto K, Sakamoto H. Two Different RNA Binding Activities for the AU-Rich Element and the Poly(A) Sequence of the Mouse Neuronal Protein mHuC. Nucleic Acids Res 1996; 24: 4895-901; PMID:9016658; http://dx.doi.org/10.1093/nar/24.24.4895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma WJ, Chung S, Furneaux H. The Elav-like proteins bind to AU-rich elements and to the poly (A) tail of mRNA. Nucleic Acids Res 1997; 25: 3564-69; PMID:9278474; http://dx.doi.org/10.1093/nar/25.18.3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anderson KD, Morin MA, Beckel-Mitchener A, Mobarak CD, Neve RL, Furneaux HM, Burry R, Perrone-Bizzozero NI. Overexpression of HuD, but not of its truncated form HuD I+II, promotes GAP-43 gene expression and neurite outgrowth in PC12 cells in the absence of nerve growth factor. J Neurochem 2000; 75: 1103-14; PMID:10936192; http://dx.doi.org/10.1046/j.1471-4159.2000.0751103.x [DOI] [PubMed] [Google Scholar]
- 50.Meisner NC, Hintersteiner M, Seifert JM, Bauer R, Benoit RM, Widmer A, Schindler T, Uhl V, Lang M, Gstach H, et al. Terminal adenosyl transferase activity of posttranscriptional regulator HuR revealed by confocal on-bead screening. J Mol Biol 2009; 386: 435-50; PMID:19109971; http://dx.doi.org/10.1016/j.jmb.2008.12.020 [DOI] [PubMed] [Google Scholar]
- 51.Toba G, White K. The third RNA recognition motif of Drosophila ELAV protein has a role in multimerization. Nucleic Acids Res 2008; 36: 1390-99; PMID:18203745; http://dx.doi.org/10.1093/nar/gkm1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Samson ML, Lisbin MJ, White K. Two distinct temperature-sensitive alleles at the elav locus of Drosophila are suppressed nonsense mutations of the same tryptophan codon. Genetics 1995; 141: 1101-11; PMID:8582616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Díaz-Moreno I, Hollingworth D, Frenkiel TA, Kelly G, Martin S, Howell S, García-Mayoral M, Gherzi R, Briata P, Ramos A. Phosphorylation-mediated unfolding of a KH domain regulates KSRP localization via 14-3-3 binding. Nat Struct Mol Biol 2009; 16: 238-46; http://dx.doi.org/10.1038/nsmb.1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scheiba RM, Aroca A, Díaz-Moreno I. HuR thermal stability is dependent on domain binding and upon phosphorylation. Eur Biophys J 2012; 41: 597-605; PMID:22706953; http://dx.doi.org/10.1007/s00249-012-0827-3 [DOI] [PubMed] [Google Scholar]
- 55.Wishart DS, Arndt D, Berjanskii M, Tang P, Zhou J, Lin G. CS23D: a web server for rapid protein structure generation using NMR chemical shifts and sequence data. Nucleic Acids Res 2008; 36: W496-W502; PMID:18515350; http://dx.doi.org/10.1093/nar/gkn305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Benoit RM, Meisner NC, Kallen J, Graff P, Hemmig R, Cebe R, Ostermeier C, Widmer H, Auer M. The x-ray crystal structure of the first RNA recognition motif and site-directed mutagenesis suggest a possible HuR redox sensing mechanism. J Mol Biol 2010; 397: 1231-44; PMID:20219472; http://dx.doi.org/10.1016/j.jmb.2010.02.043 [DOI] [PubMed] [Google Scholar]
- 57.Query CC, Bentley RC, Keene JD. A common RNA recognition motif identified within a defined U1 RNA binding domain of the 70K U1 snRNP protein. Cell 1989; 57: 89-101; PMID:2467746; http://dx.doi.org/10.1016/0092-8674(89)90175-X [DOI] [PubMed] [Google Scholar]
- 58.Shamoo Y, Krueger U, Rice LM, Williams KR, Steitz TA. Crystal structure of the two RNA binding domains of human hnRNP A1 at 1.75 A resolution. Nat Struct Biol 1997; 4: 215-222; PMID:9164463; http://dx.doi.org/10.1038/nsb0397-215 [DOI] [PubMed] [Google Scholar]
- 59.Gao FB, Keene JD. Hel-N1/Hel-N2 proteins are bound to poly(A)+ mRNA in granular RNP structures and are implicated in neuronal differentiation. Journal of Cell Science 1996; 109: 579-89; PMID:8907704 [DOI] [PubMed] [Google Scholar]
- 60.Oberstrass FC, Auweter SD, Erat M, Hargous Y, Henning A, Wenter P, Reymond L, Amir-Ahmady B, Pitsch S, Black DL, et al. Structure of PTB bound to RNA: specific binding and implications for splicing regulation. Science 2005; 309: 2054-57; PMID:16179478; http://dx.doi.org/10.1126/science.1114066 [DOI] [PubMed] [Google Scholar]
- 61.Crichlow GV, Zhou H, Hsiao HH, Frederick KB, Debrosse M, Yang Y, Folta-Stogniew EJ, Chung HJ, Fan C, De la Cruz EM, et al. Dimerization of FIR upon FUSE DNA binding suggests a mechanism of c-myc inhibition. EMBO J 2008; 27: 277-89; PMID:18059478; http://dx.doi.org/10.1038/sj.emboj.7601936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cukier CD, Hollingworth D, Martin SR, Kelly G, Díaz-Moreno I, Ramos A. Molecular basis of FIR-mediated c-myc transcriptional control. Nat Struct Mol Biol 2010; 17: 1058-64; PMID:20711187; http://dx.doi.org/10.1038/nsmb.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Handa N, Kukimoto-Niino M, Akasaka R, Kishishita S, Murayama K, Terada T, Inoue M, Kigawa T, Kose S, Imamoto N, et al. The crystal structure of mouse Nup35 reveals atypical RNP motifs and novel homodimerization of the RRM domain. J Mol Biol 2006; 363: 114-24; PMID:16962612; http://dx.doi.org/10.1016/j.jmb.2006.07.089 [DOI] [PubMed] [Google Scholar]
- 64.Corsini L, Hothorn M, Stier G, Rybin V, Scheffzek K, Gibson TJ, Sattler M. Dimerization and protein binding specificity of the U2AF homology motif of the splicing factor Puf60. J Biol Chem 2008; 284: 630-39; PMID:18974054; http://dx.doi.org/10.1074/jbc.M805395200 [DOI] [PubMed] [Google Scholar]
- 65.David PS, Tanveer R, Port JD. FRET-detectable interactions between the ARE binding proteins, HuR and p37AUF1. RNA 2007; 13: 1453-68; PMID:17626845; http://dx.doi.org/10.1261/rna.501707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Daubner GM, Clery A, Allain FH. RRM-RNA recognition: NMR or crystallography…and new findings. Curr Opin Struct Biol 2013; 23: 100-8; PMID:23253355; http://dx.doi.org/10.1016/j.sbi.2012.11.006 [DOI] [PubMed] [Google Scholar]
- 67.Sokolowski M, Schwartz S. Heterogeneous nuclear ribonucleoprotein C binds exclusively to the functionally important UUUUU-motifs in the human papillomavirus type-1 AU-rich inhibitory element. Virus Res 2001; 73: 163-75; PMID:11172920; http://dx.doi.org/10.1016/S0168-1702(00)00238-0 [DOI] [PubMed] [Google Scholar]
- 68.Sokolowski M, Furneaux H, Schwartz S. The inhibitory activity of the AU-rich RNA element in the human papillomavirus type 1 late 3' untranslated region correlates with its affinity for the elav-like HuR protein. J Virol 1999; 73: 1080-91; PMID:9882309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lopez de Silanes I, Zhan M, Lal A, Yang X, Gorospe M. Identification of a target RNA motif for RNA-binding protein HuR. Proc Natl Acad Sci USA 2004; 101: 2987-92; PMID:14981256; http://dx.doi.org/10.1073/pnas.0400352101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Doller A, Winkler C, Azrilian I, Schulz S, Hartmann S, Pfeilschifter J, Eberhardt W. High constitutive HuR phosphorylation at Ser 318 by PKCd propagates tumor relevant functions in colon carcinoma cells. Carcinogenesis 2011; 32: 676-85; PMID:21310943; http://dx.doi.org/10.1093/carcin/bgr024 [DOI] [PubMed] [Google Scholar]
- 71.Schulz S, Doller A, Pendini NR, Wilce JA, Pfeilschifter J, Eberhardt W. Domain-specific phosphomimetic mutation allows dissection of different protein kinase C (PKC) isotype-triggered activities of the RNA binding protein HuR. Cell Signal 2013; 25: 2485-95; PMID:23978401; http://dx.doi.org/10.1016/j.cellsig.2013.08.003 [DOI] [PubMed] [Google Scholar]
- 72.Marley J, Lu M, Bracken C. A method for efficient isotopic labeling of recombinant proteins. J Biomol NMR 2001; 20: 71-75; PMID:11430757; http://dx.doi.org/10.1023/A:1011254402785 [DOI] [PubMed] [Google Scholar]
- 73.Jung YS, Zweckstetter M. Mars - robust automatic backbone assignment of proteins. J Biomol NMR 2004; 30: 11-23; PMID:15452431; http://dx.doi.org/10.1023/B:JNMR.0000042954.99056.ad [DOI] [PubMed] [Google Scholar]
- 74.Cole J, Lary J. 2009. HeteroAnalysis, Analytical Ultracentrifugation Facility, Bioservices Center, University of Connecticu; t. [Google Scholar]
- 75.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 2000; 78: 1606-19; PMID:10692345; http://dx.doi.org/10.1016/S0006-3495(00)76713-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Laue TM, Shah BD, Ridgeway TM, Pelletier SL. Computer-aided interpretation of analytical sedimentation data for proteins. In Analytical Ultracentrifugation in Biochemistry and Polymer Science (eds. SE Harding, AJ Rowe, JC Horton.), 1992; pp. 90−125 Royal Society of Chemistry, Cambridge. [Google Scholar]
- 77.Cruz-Gallardo I, Aroca A, Persson C, Karlsson BG, Díaz-Moreno I. RNA Binding of T-cell Intracellular Antigen-1 (TIA-1) C-terminal RNA Recognition Motif Is Modified by pH.Conditions. J. Biol. Chem. 2013; 288: 25986-94; PMID:23902765; http://dx.doi.org/10.1074/jbc.M113.489070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Case DA, Darden TA, Cheatham TE, Simmerling CL, III, Wang J, Duke RE, Luo R, Merz KM, Pearlman DA, Crowley M, et al. 2006. AMBER 9 University of California, San Francisco. [Google Scholar]
- 79.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Rong Y, Cieplak P, Luo R, Lee T, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem 2003; 24: 1999-2012. [DOI] [PubMed] [Google Scholar]
- 80.Andersen HC. Molecular dynamics simulations at constant pressure and/or temperature. J Chem Phys 1980; 72: 2384-93; http://dx.doi.org/10.1063/1.439486 [Google Scholar]
- 81.Ryckaert JP, Ciccoti G, Berendsen HJC. Numerical integration of the Cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Computational Phys 1977; 23: 327-41; http://dx.doi.org/10.1016/0021-9991(77)90098-5 [Google Scholar]
- 82.Pettersen EFG, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera - A Visualization System for Exploratory Research and Analysis. J Comput Chem 2004; 25: 1605-12; PMID:15264254; http://dx.doi.org/10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.