

Abstract
The microbiome is now widely recognized as being important in health and disease, and makes up a substantial subset of the biome within the ecosystem of the vertebrate body. At the same time, multicellular, eukaryotic organisms such as helminths are being recognized as an important component of the biome that shaped the evolution of our genes. The absence of these macroscopic organisms during the early development and life of humans in Western culture probably leads to a wide range of human immunological diseases. However, the interaction between the microbiome and macroscopic components of the biome remains poorly characterized. In this study, the microbiome of the cecum in rats colonized for 2 generations with the small intestinal helminth Hymenolepis diminuta was evaluated. The introduction of this benign helminth, which is of considerable therapeutic interest, led to several changes in the cecal microbiome. Most of the changes were within the Firmicutes phylum, involved about 20% of the total bacteria, and generally entailed a shift from Bacilli to Clostridia species in the presence of the helminth. The results point toward ecological relationships between various components of the biome, with the observed shifts in the microbiome suggesting potential mechanisms by which this helminth might exert therapeutic effects.
Keywords: clostridia, helminths, Hymenolepis, microbiome, rat
Abbreviations
- ELB
enzymatic lysis buffer
- GIT
gastrointestinal tract
- LEfSe
Linear discriminant analysis Effect Size
- OTU
operational taxonomic unit
- PCoA
principle coordinate analysis
- QIIME
Quantitative Insights Into Microbial Ecology
Introduction
The biome of the vertebrate body contains a vast array of microorganisms, termed the microbiome, as well as a variety of multicellular, macroscopic organisms. Importantly, the microbiome, which by definition contains only microscopic organisms, is a subset of the body's biome, which includes multicellular, macroscopic organisms that often reach several millimeters in length and sometimes even meters in length. Such multicellular organisms include a variety of arthropod ectoparasites, as well as nematodes and cestodes that colonize various internal organs. Although multicellular organisms living within the vertebrate body have been largely lost in humans living in post-industrial society, these organisms have been an integral part of the biome of vertebrates for hundreds of millions of years1 and were key components of the human biome during the evolution of our genome.2 That is, these organisms were an integral component of our “environment of evolutionary adaptedness,”3,4 an important concept in the field of evolutionary medicine. Briefly, emerging evidence strongly suggests that loss of this compartment of our biome has led, in large part, to pandemics of allergic, autoimmune, and other inflammatory related diseases in modern society.1,5-7 Further, the emerging view of the vertebrate body is that of an ecosystem, with numerous interconnected components, including the immune system and various parts of the biome. However, how these components interact is only now beginning to be understood.
The effect of helminths on disease is well documented, with a large number of studies finding that helminths attenuate allergic, autoimmune, and other inflammation-related conditions.1,6,8,9 It is thus not surprising that “helminth therapy” has proven very successful in both animal studies as well as some clinical studies, and is currently receiving considerable attention as a means of normalizing immune function in Western culture, thus eliminating allergies and autoimmune conditions.7 However, given the lack of knowledge regarding the effect of helminths on the microbiome, it is not surprising that the role of the microbiome in the treatment of disease using helminths is entirely unclear. Work in non-human primates, using helminths to ameliorate idiopathic chronic diarrhea, showed several changes in the mucosal-associated microbiome during the course of treatment,10 and it seems likely that changes in the microbiome were influenced strongly by inflammation in the mucosal surface and the effect of the helminths on that inflammation.11 Perhaps more telling was the observation that Schistosoma japoni simultaneously ameliorated trinitrobenzenesulfonic acid-induced colitis in mice and reduced bacterial translocation across the gut epithelial barrier.12 Further, Walk et al.13 found that colonization with the roundworm Heligmosomoides polygyrus led to concomitant changes in the microbiome and alleviation of colitis in interleukin-10 deficient mice. Whether these changes in the microbiome as a result of helminth colonization are important in the amelioration of disease remains unknown, and will undoubtedly be a subject of intense future study. Further, studies will undoubtedly be required to individually examine the effect on the microbiome of each helminth-host relationship of interest.
The helminth Hymenolepis diminuta (the rat tapeworm) is of considerable interest. Its natural hosts include various species of rat, including Rattus norvegicus, but it has been known to colonize other rodents with varying success, and, on rare occasions, humans.14 The animal has a lifespan comparable to that of the rat, and lacks a digestive tract, adsorbing nutrients through its outer epithelial surface. Despite being one of the most widely studied helminths in the laboratory,15 its effect on the microbiome remains unknown. The helminth, a cestode which lives exclusively in the small intestine, blocks experimentally induced colitis in mice more effectively than daily immunosuppression with steroids.16 More compelling is the observation that 2 companies, WormTherapy based in Tijuana, Mexico, and Biome Restoration based in Lancaster, UK, have recently begun production of this helminth for use in humans as a means of treating immune disease and enriching the body's biome, respectively. The cestode has some attractive features when compared to other helminths, all nematodes, which are used for helminth therapy. For example, since Hymenolepis diminuta does not effectively colonize humans and must be introduced artificially into the body, it may be more attractive from a regulatory perspective than organisms such as the human hookworm (Necator americanus) and human whipworm (Trichuris trichiura), which can colonize humans and, under some circumstances, be transmitted from human-to-human. Further, unlike the hookworm, the tapeworm is non-invasive in its primary host, restricted in distribution to the lumen of the small bowel. Importantly, the therapeutic stage of the Hymenolepis diminuta is readily cultivated in arthropod intermediate hosts and thus, unlike hookworms and whipworms, is not harvested from the feces of the primary host. This last factor may have benefits in terms of cost effectiveness, which may prove especially important as prophylactic treatment of human populations with helminths1 is considered. Although these factors merit consideration, the most important issues regarding the selection of helminth species for human therapy and disease prevention relate to the relative effectiveness of the species on human disease, of course. Studies to evaluate these most important issues are, unfortunately, not yet underway.
Given the interest in Hymenolepis diminuta as a potential therapeutic agent, and given the known role of the microbiome in immune health, we evaluated the helminth's effect on the colonic microbiome of its native host, Rattus norvegicus. Animals were colonized for 2 generations to ensure that the effects of helminth colonization were present from the time of birth via transmittal of immune components through the milk. Further, the second generation of animals was colonized with helminths at the time of weaning to ensure a continuous influence of the helminth on the rodent's biome. The animals were further divided by exposing half of the animals, both with and without helminths, to lipopolysaccharide (LPS) 4 days prior to analysis of the microbiome. This experiment was designed to evaluate the effects of Hymenolepis diminuta on the rat's microbiome following exposure to and recovery from a mild inflammatory challenge: LPS-induced inflammation dissipates within hours and is not apparent 4 days post-exposure. The analysis of the microbiome was conducted with the view that tapeworm-induced changes in the microbiome may shed light on the ability of the helminth to alleviate inflammation in vertebrate hosts.
Results
Effect of colonization with Hymenolepis diminuta on the rat microbiome
The overall experimental design, described briefly in the Introduction and in detail in the Methods, is summarized in Figure 1. In Group S (all animals not exposed to LPS), colonization with Hymenolepis diminuta did not affect α or β diversity in a statistically significant manner (Fig. 2). However, differences in community composition associated with helminth colonization were evident (Fig. 3). The most prominent differences were the relatively fewer amounts of Turicibacter and the relatively greater amounts of Peptostreptococcaceae (unknown genus) in the presence of helminths. On average, Turicibacter accounted for more than 22% of the sequences in animals without helminths, but only about 4% of the sequences in the samples from animals with helminths (an 82% decrease in the presence of helminths). In contrast, colonization with helminths was associated with a 2.7-fold increase in the relative number of Peptostreptococcaceae sequences found. Similar results were observed in Group L (all animals exposed to LPS), with, on average, a 90% decrease in Turibacter and a 2.4-fold increase in Peptostreptococcaceae in the presence of helminths.
Figure 1.
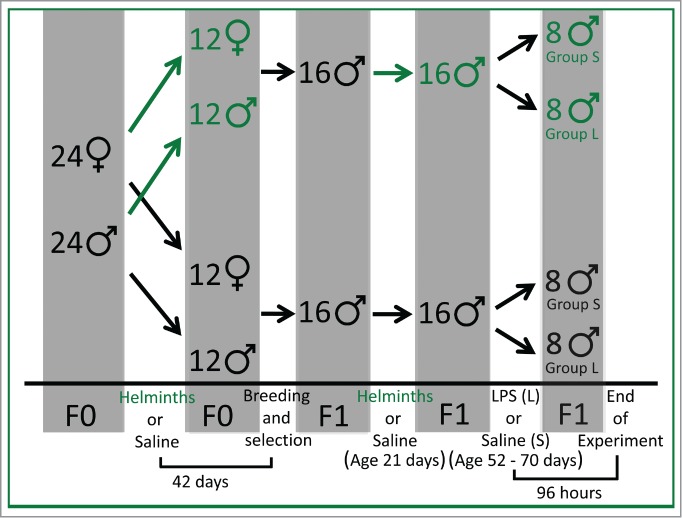
The experimental design. The top portion of the diagram shows the division of animals during the study, and the bottom portion shows the timeline for specific events. Animals colonized with helminths are shown in green throughout the diagram, and green arrows indicate the feeding of helminths to the animals. Forty-two days after colonization of F0 rats (F0) with helminths or sham (saline), the animals were bred. At the time of weaning (age 21 days), 32 F1 males (F1) were randomly selected from the offspring, and were colonized with and without helminths as shown. Finally, when the animals reached an average age of 62 days (range 56–74), 96 hours before termination of the experiment, the animals were again divided further and received immune stimulation with LPS or with sham (saline) as shown in the diagram.
Figure 2.
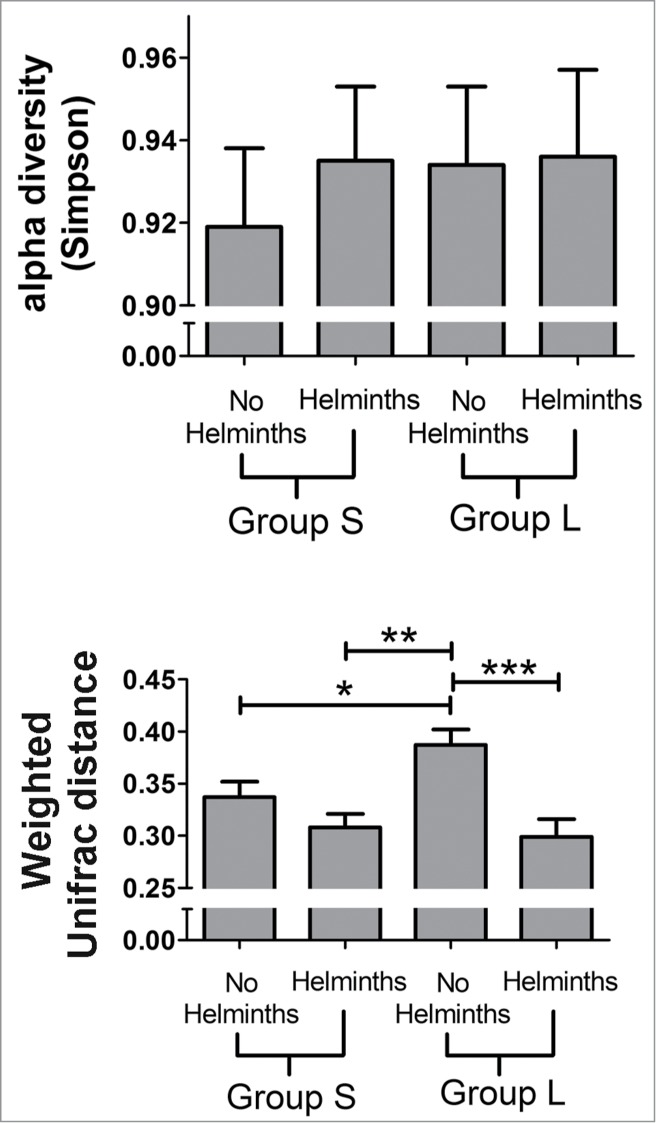
Alpha (top) and β (bottom) diversity measures in animals from Groups S colonized with (n = 8) and without (n = 7) helminths and Group L colonized with (n = 6) and without (n = 7) helminths. Weighted pairwise UniFrac distances were averaged within each group to calculate an average β diversity value (a proxy for inter-individual variation). Analysis by 2-way ANOVA revealed no statistically significant changes in the α diversity. Results from the Simpson diversity metric are shown, and are similar to diversity measures using Chao1, Shannon-Weaver, and Faith's Phylogenetic Diversity indices. Significant changes in the β diversity (UniFrac) were observed for helminth treatment (p = 0.0001) and for the interaction between helminths and LPS treatment (p = 0.047). The means and standard errors are shown. The bars show the result of the post-hoc t-tests (p < 0.0001***; p < 0.005**; p < 0.05*) and indicate that the predominant characteristic associated with the β diversity measure is a relatively large value for the β diversity of animals without helminths in group L.
Figure 3.

Microbiome composition in animals from Groups S colonized with (n = 8) and without (n = 7) helminths and Group L colonized with (n = 6) and without (n = 7) helminths. The composition is based on 16S libraries isolated from digesta taken from the ceca when the animals reached an average age of 62 days (range 56–74 days). Results are shown at the (A) phylum and (B) genus level.
Quantitative analyses of the microbiome in Group S indicated that the different amounts of Turibacter to Peptostreptococcaceae in the presence and absence of helminths was statistically significant and indeed the major change observed as a result of the helminths, with a linear discriminant analysis (LDA) effect size (log 10) of approximately 5 (Fig. 4A). However, a number of other less substantial changes with lower LDA effect sizes were observed in Group S. The same quantitative assessment of Group L (Fig. 4B) with and without helminths again revealed major differences in the amount of Turibacter to Peptostreptococcaceae and a number of less substantial changes with lower LDA effect sizes. However, the less substantial changes were, with few exceptions, not shared between Group S and Group L. Whether these minor changes represent differences between Groups S and L resulting from differences between their respective experimental protocols (i.e., exposure to LPS versus saline) is unknown.
Figure 4.

Bacterial lineages with significantly different representation in rats inoculated with or without helminths in (A) Group S or (B) Group L. The log linear discriminant analysis (LDA) effect size quantifies the degree to which each lineage contributes to the uniqueness of each sample class.
Figure 4.

(Continued)
Phylogenetic assessment of changes in the microbiome as a result of colonization with Hymenolepis diminuta
Organizing the results shown in Figure 4 into cladograms (Fig. 5) provides an easily appreciated view of the shifts in the microbiome as a result of colonization with helminths. In Group S, all microbial lineages significantly distinguishing rats treated with Hymenolepis diminuta from those receiving saline belonged to Firmicutes (Fig. 5A). In Group L, the isolated impact on Firmicutes was less dramatic, but the clade was still heavily affected by colonization with Hymenolepis diminuta: 67% (10 out of 15) of the differential abundances detected in the microbiome at the operational taxonomic unit (OTU) level as a result of colonization with Hymenolepis diminuta involved differences in Firmicutes (Fig. 5B).
Figure 5.

Cladograms of bacterial lineages with significantly different representation in rats with or without helminths in (A) Group S or (B) Group L. Lineages on the bacterial trees are color-coded to indicate whether the taxon does (red or green) or does not (yellow) significantly differ between sample classes.
The effect of exposure to LPS on the microbiome
Because the only difference between Group S and Group L was the exposure of Group L to LPS as described in the methods, the effect of LPS exposure on the microbiome could be assessed by comparing the microbiome in Group S with that in Group L. Injection with LPS did not affect α diversity measures, but LPS significantly impacted the β diversity (β diversity between experimental groups, see Methods) in rats without helminths (Fig. 2) as assessed by a 2-way ANOVA and post-hoc t-tests. However, in rats with helminths, the β diversity was unaffected by LPS. Further, as shown in Figure 6, increased prevalence of some lineages was observed in the presence of LPS. However, the LDA effect sizes were relatively small, and the changes associated with LPS exposure were not consistent when comparing animals with and without helminths.
Figure 6.

Bacterial lineages with significantly different representation in rats treated with LPS or without LPS (saline). The results from animals (A) without and (B) with helminths are shown. The log linear discriminant analysis (LDA) effect size quantifies the degree to which each lineage contributes to the uniqueness of each sample class.
Principal Coordinate Analysis (PCoA) of the effects of colonization with Hymenolepis diminuta on the microbiome
The PCoAs of the microbiome in Groups S and L illustrate principal components (PCs) which showed a distinction between animals colonized with helminths and those without helminths (Fig. 7). The two groups showed similar component contributions to the observed variation, with PC1 accounting for 35–40%, PC2 accounting for 21–23%, and PC3 accounting for 12%. However, the presence of helminths caused a shift along principal component 2 in Group S (PC2S), but not PC2 of Group L (PC2L). In Group L, the presence of helminths caused a shift along principal component 3 (PC3L) rather than principal component 2.
Figure 7.
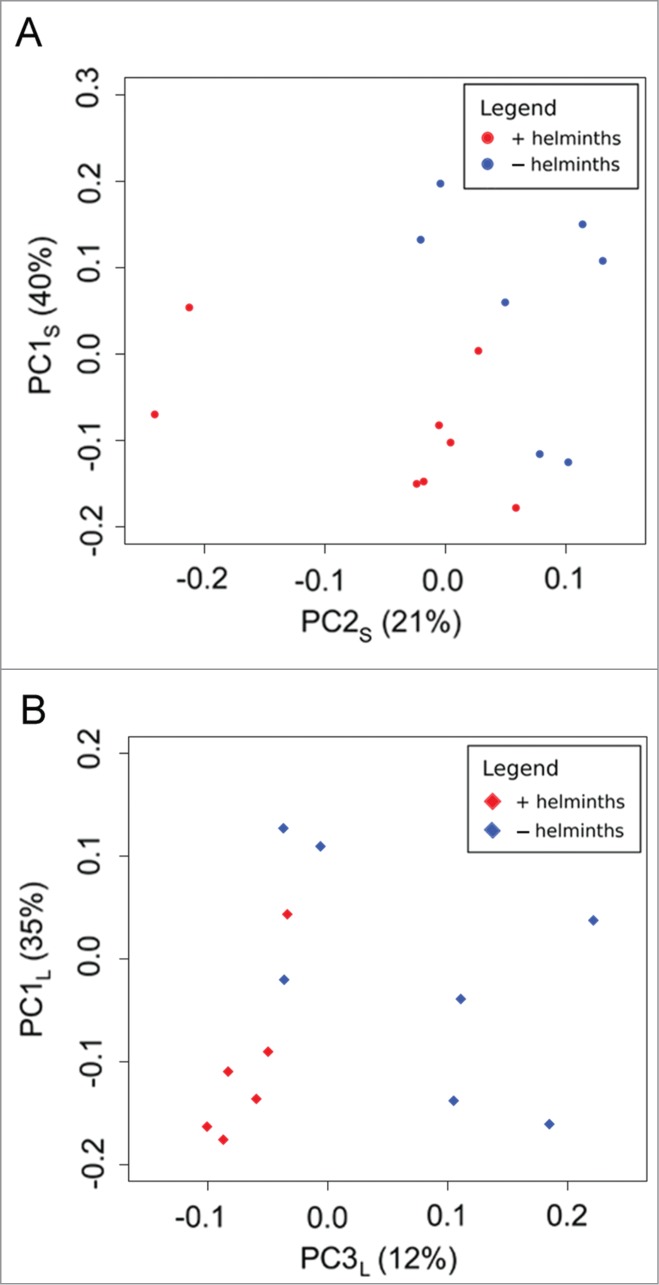
Principle Coordinate Analysis (PCoA) of weighted Unifrac distances between libraries isolated from rats with (red) or without (blue) helminths in (A) Group S or (B) Group L. Each data point represents a library from a single animal (i.e., a single bar in Figure 3). Calculations were performed using the UniFrac method; values are weighted to account for differences in lineage frequencies. The distance between points represents unique branches on a phylogenetic tree (i.e. evolutionary history not shared between libraries in Figure 3), as well as differences in the relative abundance of lineages. Closer points share more branch length and have similar frequencies, while points more distant from one another have more unique or disparate gut microbiomes.
Discussion
The results presented herein point toward profound changes in the microbiome in response to enriching the gut ecosystem with a helminth having therapeutic potential. Colonization of the rats used in this study with Hymenolepis diminuta causes a substantial shift in the microbial community, primarily characterized by changes in the relative contributions from species within the Firmicutes phylum. Specifically, colonization with the helminth is associated with increased Clostridia and decreased Bacilli. The contribution of Bacilli to the microbiome is higher with a Western diet characterized by processed sugars and high fat content,17 whereas some species of Clostridia are known to tighten the epithelial barrier and decrease propensity for allergy.18-21 Thus, the changes in microbiome composition observed following helminth colonization offer a potential explanation for the therapeutic effects of helminths under some circumstances. However, it remains unknown whether these changes in the microbiome are responsible, in part, for the known therapeutic activity of Hymenolepis diminuta. Given the potential for helminth therapy to resolve a number of immunological problems heretofore resistant to medical efforts,1,6 this area of investigation promises to be very active and informative in the future.
The use of 2 generations of laboratory animals in this study ensured that the rats eventually tested had been exposed to the effects of helminth colonization since their conception in utero. It remains unknown whether exposure to helminths later in life would have had similar effects on the microbiome. Thus, these studies point toward a potentially positive effect of biome enrichment for couples wishing to have children with a healthy microbiome, if biome enrichment is practiced prior to conceiving children. On the other hand, although the effects of biome enrichment in human adults are well documented,22-24 this study did not aim to probe the effects of biome enrichment later in life on the microbiome.
The experimental protocol for Group L was designed to evaluate the effects of Hymenolepis diminuta on the rat's microbiome following exposure to and recovery from a mild inflammatory challenge. The LPS was administered in a manner that (a) produced few if any overt physical responses in the animals, and (b) was timed before assessment of the microbiome such that any effect of the LPS on the animals at the time of microbiome assessment was likely, for the most part, dissipated. Some changes in the microbiome were observed under these conditions, especially an increased disparity between microbial communities in animals without helminths after exposure to LPS, as indicated by the increased β diversity in those animals. However, it might be expected that a greater intensity and duration of inflammation might more profoundly affect the microbiome, and further studies are warranted. At the same time, the relatively minor changes in the microbiome seen when comparing animals with and without exposure to LPS serve as basis for comparison with the more dramatic changes seen with exposure to helminths. Further, the results seen with and without colonization with helminths in Group L serve as a validation for the results seen in Group S. In summary, the results supports the assertion that colonization with Hymenolepis diminuta does indeed have a profound effect on the microbiome of the laboratory rat, resulting in shifts in community structure that affects approximately 20% of the total organisms present, despite the fact that the helminth lives in the small intestine and the microbiome was assessed in the cecum.
Of interest is the mechanism by which Hymenolepis diminuta alters niche space in the cecum and affects the microbiome. Experiments, perhaps using immunodeficient animal models, might be useful in distinguishing between direct alteration of metabolic niche space by the helminth vs. alteration of niche space via alteration of the host immune function.
Knowledge regarding the microbiome and its function is far from complete at the present time. This fact is illustrated by the observation that the OTU (family Peptostreptococcaceae) most dramatically increased in the presence of helminths is not found in the Greengenes database (default reference in QIIME). Evaluation using SeqMatch of the sequences binned into that OTU by QIIME resulted in 44% of the sequences being classified as Clostridium XI, another 22% as unclassified Clostridia, and the rest spread between 43 other taxa. However, only one of the 657 sequences was an identical match to anything in the database (uncultured bacterium; HFDE2688FD12; JQ893212), confirming the QIIME results that the sequences in question were from uncharacterized bacteria. This observation is particularly striking because that OTU accounts for a substantial fraction of the microbiome in the animals: roughly 7% and almost 20% of the total sequences in animals without and with helminth colonization, respectively. These studies point toward a great need for further research in the basic rules that govern the microbiome and interactions between the microbiome and other organisms such as helminths within the biome.
Materials and Methods
Study design and colonization of animals with Hymenolepis diminuta
All experiments were approved by the Duke University Institutional Animal Care and Use Committee (A096-13-04). Using a Hund Wetzlar Wilovert dissecting microscope, Hymenolepis diminuta cysticercoids (rat tapeworm larvae) were harvested from meal bugs previously inoculated with the organisms (original stock purchased from Carolina Biological Supply, Burlington, NC, USA). Male (n = 24) and female (n = 24) Sprague Dawley rats from Harlan Sprague Dawley (Indianapolis, IN, USA) were housed in a standard (hygienic) laboratory setting and fed Purina Mills® LabDiet® 5001 ad libitum. Half of the animals were inoculated with 4 Hymenolepis diminuta cysticercoids (rat tapeworm larvae between 5 weeks and 5 months of age) in a drop of 0.6% saline, and the other half received a sham inoculation with saline only. Animals with helminths were kept in separate cages from animals without helminths, but all animals were kept in the same room.
This approach resulted in confirmed colonization (confirmed using a modified version of the McMaster technique25) with Hymenolepis diminuta in all of the inoculated animals. Periodic screening of non-colonized animals and of sentinel animals in the facility showed no colonization, as is expected since Hymenolepis diminuta requires an intermediate host and thus cannot be transmitted directly rat-to-rat. Forty-two days after inoculation, the animals were bred, resulting in 22 pregnant females. The breeding yielded 125 male pups, which were arbitrarily selected for study in lieu of the female animals. Thirty-two of the male pups, 16 from colonized animals and 16 from uncolonized animals, were selected for use in this study. Colonized animals were selected from 4 litters, and uncolonized animals were selected from 6 litters to minimize any cage effects. The animals were housed in pairs, further minimizing cage effects.
At 21 days of age, the 16 F1 males born to parents colonized with Hymenolepis diminuta were themselves colonized with Hymenolepis diminuta, and the colonization was confirmed as described above for the parent generation. The 16 F1 males born to parents not colonized with Hymenolepis diminuta were used as controls and received a sham inoculation with saline only. At 58 days of age on average (range 52–70), the 32 animals were divided arbitrarily into 2 groups, each containing 8 colonized animals and 8 uncolonized animals. The first group (Group S) received an intraperitoneal injection of 0.4 ml saline, and the second group (Group L) received an intraperitoneal injection of 25 μg/kg lipopolysaccharide (LPS; derived from E. coli Serotype 0111:B4, Sigma, St. Louis, MO) in 0.4 ml phosphate buffered saline. This injection without (Group S) or with (Group L) LPS was the only difference between the experimental protocol for the 2 groups. Animals were sacrificed 4 days after receiving treatment with LPS or sham, at an average age of 62 days (range 56–74). Samples from both groups were collected and processed during the same time period and in the same facilities by the same personnel, and samples from both groups were analyzed at the same time in the same “batch.” Animals that received LPS were housed in separate cages from animals that were not exposed to LPS.
Procurement of samples for microbiome analysis
Ninety-six hours after saline or LPS injection, F1 males were sacrificed by exsanguination under anesthesia, and the ceca were collected. A longitudinal incision was made in each cecum, from the ileocecal junction to the apex of the cecum, and then the cecum was submerged in 5 mL of sterile filtered phosphate buffered saline. Each cecum was vortexed for 60 seconds to release cecal contents, and the cecal tissue was removed using sterile forceps. The remaining mixture was separated into 3 aliquots, and each was centrifuged at 16,000 × g for 10 minutes. The supernatant was removed and the pellets were snap frozen in liquid nitrogen, then stored at −80°C until use. These pellets were used as a source of DNA for microbiome analysis.
Microbial DNA extraction
DNA was extracted with the DNeasy kit (QIAGEN, Hilden, Germany), using the following protocol. Fifty mL of sterile enzymatic lysis buffer (ELB) were prepared fresh for each reaction by combining 1 mL 1 M Tris-Cl (pH 8.0), 200 μL 0.5 M EDTA (pH 8.0), 2.5 mL 20% Triton X-100 (Sigma-Aldrich, St. Louis, Missouri, USA), and 46.3 mL H20. Lysozyme (Sigma-Aldrich) was also prepared fresh for each reaction, at a concentration of 200 mg/mL in ELB.
A DuraTube (BioExpress, Kaysville, Utah, USA) containing one aliquot from each rat (above) was removed from −80°C storage, and 0.4 g silica beads and 400 μL ELB were added to each tube. DuraTubes containing sample, beads, and ELB were thawed quickly at 65°C, then shaken for 2 minutes at 1500 rpm on a GenoGrinder 2010 (SPEX Sample Prep, Metugen, New Jersey, USA) to disrupt microbial cell walls. Forty μL lysozyme were added to each tube (to a final concentration of 20 mg/mL); tubes were vortexed briefly, then incubated at 30°C for 45 minutes to achieve enzymatic disruption of cells. After incubation, tubes were vortexed for 15 seconds, then centrifuged for 1 minute at 20,000 × g to pellet stool particles. Supernatant (containing DNA) was pipetted to a new 2-mL tube containing 20 μL Proteinase K (QIAGEN, Hilden, Germany). 440 μL Buffer AL (QIAGEN) were added, and each tube was vortexed briefly before incubation at 56°C for 30 minutes. After incubation, 440 μL 100% ethanol were added to each reaction and mixed thoroughly by vortexing. Seven hundred μL of the liquid were added to a spin column (QIAGEN) and centrifuged for 1 minute at 12,000 × g. Flowthrough was removed, and this step was repeated until all liquid was processed. The column was then rinsed with 200 μL of a 1:1:1 premade mix of ELB:AL:ethanol and centrifuged for 1 minute at 12,000 × g. Flowthrough was discarded, 500 μL Buffer AW1 (QIAGEN) were added, and the column was spun for 1 minute at 12,000 × g. Flowthrough was discarded, 500 μL Buffer AW2 (QIAGEN) were added and the column was spun for 3 minutes at 20,000 × g. The column was moved to a sterile 1.5-mL microfuge tube for eluate collection. Two hundred μL Buffer AE (QIAGEN) were added to the column, which was incubated at room temperature for 1 minute before centrifugation for 1 minute at 12,000 × g. The eluate was added back to the column and centrifuged a second time for 1 minute at 12,000 × g to increase DNA yields. All samples were quantified on a Nanodrop-1000 (Thermo Fischer Scientific, Waltham, Massachusetts, USA).
Illumina sequencing and analysis
Samples containing 27–50 ng DNA from each extraction were sent to Argonne National Laboratory for downstream amplification and sequencing, as described in McKenney et al.26 The PCR primers 515F (GTG-CCA-GCM-GCC-GCG-GTA-A) and 806R (GGA-CTA-CHV-GGG-TWT-CTA-AT) were used to amplify the v4 region of 16S rRNA with length ∼300 bp for 150 bp paired-end sequencing on the Illumina MiSeq platform according to the methods of Caporaso et al.27 Forward and reverse Illumina reads were joined using ea-utils.28 A total of 10,296,305 16S rRNA sequences were joined from the forward and reverse reads; only the joined reads were used in our analysis. A fastq file containing these joined reads was deposited in the NCBI Sequence Read Archive under BioProject ID PRJNA270622. Joined 16S rRNA sequence reads were demultiplexed and analyzed using Quantitative Insights Into Microbial Ecology (QIIME, v1.8.0)29 to classify microbial constituents and compare membership between samples.30 Quality filtering was performed using default settings and the input sequence file was split into libraries using 12 bp barcodes. A total of 2,637,412 sequences (an average of 82,419 per sample) passed quality filtering thresholds. The range of sequences that passed quality filtering thresholds was 68,491 to 139,715 for 28 of the samples. However, the remaining 4 samples had less than 30 sequences each (range 5 to 23; average = 14.25), and these 4 samples were excluded from the study. Elimination of these samples from the study was the only measure taken to control for the effects of differing sequencing depth. This left Group S with 8 animals colonized with helminths and 7 uncolonized, and Group L with 6 animals colonized with helminths and 7 uncolonized.
The remaining sequences were classified into operational taxonomic units (OTUs, a proxy for taxa based on 97% sequence similarity) using the Uclust method31 and identified using the Greengenes 13_8 database.29 Sequences from some OTU's of interest which were not identified at the genus level in the Greengenes database were subsequently evaluated using SeqMatch.32 QIIME was then used to calculate the number and abundances of OTUs (using the Greengenes identifications) to summarize the total unique bacterial lineages per sample and their relative frequencies (richness and α diversity, respectively), as well as the net lineage difference between any 2 or more experimental groups (β diversity). Beta diversity was quantified using weighted UniFrac,33 which measures the proportion of branch lengths shared between samples. Here we report the β diversity between treatment groups. Diversity data was analyzed using a 2-way ANOVA and post-hoc t-tests in JMP® Pro (Version 11, SAS Institute Inc., Cary, NC, 1989–2013) to test for the effect of treatment variables on microbial diversity. An α of 0.05 was used as the threshold for significance. Principle Coordinate Analyses (PCoA, in QIIME) was also performed on the gut microbiome β diversity to detect underlying relationships between the microbiome and treatment variables. Finally, Linear discriminant analysis Effect Size (LEfSe)34 was calculated to identify bacterial lineages whose frequencies differ significantly as a function of treatment variables. LEfSe detects differentially distributed lineages with the Kruskall-Wallis test, then checks the consistency of subclass distinctions with the pairwise Wilcoxon text. The final linear discriminant analysis was used to rank all differentiating lineages by their effect size.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Melanie Wiley for technical assistance. The authors thank Zoie Holzknecht for assistance with the preparation of the manuscript.
Funding
This study was funded in part by a grant from The Coalition for SafeMinds.
References
- 1.Parker W, Ollerton J.. Evolutionary biology and anthropology suggest biome reconstitution as a necessary approach toward dealing with immune disorders. Evol Med Public Health 2013; 2013:89-103; PMID:24481190; http://dx.doi.org/ 10.1093/emph/eot008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fumagalli M, Pozzoli U, Cagliani R, Comi GP, Riva S, Clerici M, Bresolin N, Sironi M.. Parasites represent a major selective force for interleukin genes and shape the genetic predisposition to autoimmune conditions. J Exp Med 2009; 206:1395-408; PMID:19468064; http://dx.doi.org/ 10.1084/jem.20082779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crompton DWT. How much human helminthiasis is there in the world? J Parasitol 1999; 85:397-403; PMID:10386428; http://dx.doi.org/ 10.2307/3285768 [DOI] [PubMed] [Google Scholar]
- 4.Bowlby J. Attachment and Loss. Vol. 1. Attachment. New York: Basic Books, 1969. [Google Scholar]
- 5.Jackson JA, Friberg IM, Little S, Bradley JE.. Review series on helminths, immune modulation and the hygiene hypothesis: Immunity against helminths and immunological phenomena in modern human populations: coevolutionary legacies? Immunology 2009; 126:18-27; PMID:19120495; http://dx.doi.org/ 10.1111/j.1365-2567.2008.03010.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bilbo SD, Wray GA, Perkins SE, Parker W.. Reconstitution of the human biome as the most reasonable solution for epidemics of allergic and autoimmune diseases. Med Hypotheses 2011; 77:494-504; PMID:21741180; http://dx.doi.org/ 10.1016/j.mehy.2011.06.019 [DOI] [PubMed] [Google Scholar]
- 7.Parker W, Perkins SE, Harker M, Muehlenbein MP.. A prescription for clinical immunology: the pills are available and ready for testing. Curr Med Res Opin 2012; 28:1193-202; PMID:22612580; http://dx.doi.org/ 10.1185/03007995.2012.695731 [DOI] [PubMed] [Google Scholar]
- 8.Capron A, Dombrowicz D, Capron M.. Helminth infections and allergic diseases. Clin Rev Allergy Immunol 2004; 26:25-34; PMID:14755073; http://dx.doi.org/ 10.1385/CRIAI:26:1:25 [DOI] [PubMed] [Google Scholar]
- 9.Rook GAW. Review series on helminths, immune modulation and the hygiene hypothesis: the broader implications of the hygiene hypothesis. Immunology 2009; 126:3-11; PMID:19120493; http://dx.doi.org/ 10.1111/j.1365-2567.2008.03007.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Broadhurst MJ, Ardeshir A, Kanwar B, Mirpuri J, Gundra UM, Leung JM, Wiens KE, Vujkovic-Cvijin I, Kim CC, Yarovinsky F, et al.. Therapeutic helminth infection of macaques with idiopathic chronic diarrhea alters the inflammatory signature and mucosal microbiota of the colon. PLoS Pathog 2012; 8:e1003000; PMID:23166490; http://dx.doi.org/ 10.1371/journal.ppat.1003000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leung JM, Loke PN.. A role for IL-22 in the relationship between intestinal helminths, gut microbiota and mucosal immunity. Int J Parasitol 2013; 43:253-7; PMID:23178750; http://dx.doi.org/ 10.1016/j.ijpara.2012.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Y, Zhang S, Jiang L, Jiang J, Liu H.. Preventive effects of Schistosoma japonicum ova on trinitrobenzenesulfonic acid-induced colitis and bacterial translocation in mice. J Gastroenterol Hepatol 2009; 24:1775-80; PMID:20136961; http://dx.doi.org/ 10.1111/j.1440-1746.2009.05986.x [DOI] [PubMed] [Google Scholar]
- 13.Walk ST, Blum AM, Ewing SA, Weinstock JV, Young VB.. Alteration of the murine gut microbiota during infection with the parasitic helminth Heligmosomoides polygyrus. Inflamm Bowel Dis 2010; 16:1841-9; PMID:20848461; http://dx.doi.org/ 10.1002/ibd.21299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wiwanitkit V. Overview of hymenolepis diminuta infection among Thai patients. MedGenMed 2004; 6:7; PMID:15266234 [PMC free article] [PubMed] [Google Scholar]
- 15.Arai HP. Biology of the Tapeworm Hymenolepis Diminuta. New York: Academic Press, 1980. [Google Scholar]
- 16.Melon A, Wang A, Phan V, McKay DM.. Infection with Hymenolepis diminuta is more effective than daily corticosteroids in blocking chemically induced colitis in mice. J Biomed Biotechnol 2010; 2010:384523; PMID:20011066; http://dx.doi.org/ 10.1155/2010/384523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Trans Med 2009; 1:6ra14; PMID:20368178; http://dx.doi.org/ 10.1126/scitranslmed.3000322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stefka AT, Feehley T, Tripathi P, Qiu J, McCoy K, Mazmanian SK, Tjota MY, Seo GY, Cao S, Theriault BR, et al.. Commensal bacteria protect against food allergen sensitization. Proc Natl Acad Sci 2014; 111:13145-50; PMID:25157157; http://dx.doi.org/ 10.1073/pnas.1412008111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, et al.. Induction of colonic regulatory T cells by indigenous clostridium species. Science 2011; 331:337-41; PMID:21205640; http://dx.doi.org/ 10.1126/science.1198469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al.. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013; 500:232-6; PMID:23842501; http://dx.doi.org/ 10.1038/nature12331 [DOI] [PubMed] [Google Scholar]
- 21.Geuking Markus B, Cahenzli J, Lawson Melissa AE, Ng Derek CK, Slack E, Hapfelmeier S, McCoy KD, Macpherson AJ.. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity 2011; 34:794-806; PMID:21596591; http://dx.doi.org/ 10.1016/j.immuni.2011.03.021 [DOI] [PubMed] [Google Scholar]
- 22.Correale J, Farez M.. Association between parasite infection and immune responses in multiple sclerosis. Ann Neurol 2007; 61:97-108; PMID:17230481; http://dx.doi.org/ 10.1002/ana.21067 [DOI] [PubMed] [Google Scholar]
- 23.Summers RW, Elliott DE, Qadir K, Urban JF Jr., Thompson R, Weinstock JV.. Trichuris suis seems to be safe and possibly effective in the treatment of inflammatory bowel disease. Am J Gastroenterol 2003; 98:2034-41; PMID:14499784; http://dx.doi.org/ 10.1111/j.1572-0241.2003.07660.x [DOI] [PubMed] [Google Scholar]
- 24.Cheng AM, Jaint D, Thomas S, Wilson J, Parker W.. Overcoming evolutionary mismatch by self-treatment with helminths: current practices and experience. J Evolut Med 2015; In Press. [Google Scholar]
- 25.Sloss MW, Kemp RL, Zajac AM. Veterinary Clinical Parasitology. Journal Investment Dermatol. Ames, Iowa: Iowa State Press, 1994:9. [Google Scholar]
- 26.McKenney EA, Rodrigo A, Yoder, AD. Patterns of gut bacterial colonization in three primate species. PLoS One 2015; 10(5): e0124618; http://dx.doi.org/ 10.1371/journal.pone.0124618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, et al.. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 2012; 6:1621-4; PMID:22402401; http://dx.doi.org/ 10.1038/ismej.2012.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aronesty E. ea-utils: Command-line tools for processing biological sequencing data. 2011. [Google Scholar]
- 29.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al.. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010; 7:335-6; PMID:20383131; http://dx.doi.org/ 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuczynski J, Stombaugh J, Walters WA, Gonzalez A, Caporaso JG, Knight R.. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr Protoc Bioinformatics 2011; Chapter 10:Unit 10.7; PMID:NOT_FOUND [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010; 26:2460-1; PMID:20709691; http://dx.doi.org/ 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 32.Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM.. Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 2014; 42:D633-42; PMID:24288368; http://dx.doi.org/ 10.1093/nar/gkt1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lozupone C, Knight R.. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 2005; 71:8228-35; PMID:16332807; http://dx.doi.org/ 10.1128/AEM.71.12.8228-8235.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C.. Metagenomic biomarker discovery and explanation. Genome Biol 2011; 12:R60; PMID:21702898; http://dx.doi.org/ 10.1186/gb-2011-12-6-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]