

Abstract
TDP-43 is an RNA-binding protein involved in several steps of mRNA metabolism including transcription, splicing and stability. It is also involved in ALS and FTD, neurodegenerative diseases characterized by TDP-43 nuclear depletion. We previously identified TDP-43 as a binder of the downstream element (DSE) of the β-Adducin (Add2) brain-specific polyadenylation site (A4 PAS), suggesting its involvement in pre-mRNA 3′ end processing. Here, by using chimeric minigenes, we showed that TDP-43 depletion in HeLa and HEK293 cells resulted in down-regulation of both the chimeric and endogenous Add2 transcripts. Despite having confirmed TDP-43-DSE in vitro interaction, we demonstrated that the in vivo effect was not mediated by the TDP-43-DSE interaction. In fact, substitution of the Add2 DSE with viral E-SV40 and L-SV40 DSEs, which are not TDP-43 targets, still resulted in decreased Add2 mRNA levels after TDP-43 downregulation. In addition, we failed to show interaction between TDP-43 and key polyadenylation factors, such as CstF-64 and CPSF160 and excluded TDP-43 involvement in pre-mRNA cleavage and regulation of polyA tail length. These evidences allowed us to exclude the pre-hypothesized role of TDP43 in modulating 3′ end processing of Add2 pre-mRNA. Finally, we showed that TDP-43 regulates Add2 gene expression levels by increasing Add2 mRNA stability. Considering that Add2 in brain participates in synapse assembly, synaptic plasticity and their stability, and its genetic inactivation in mice leads to LTP, LTD, learning and motor-coordination deficits, we hypothesize that a possible loss of Add2 function by TDP-43 depletion may contribute to ALS and FTD disease states.
Keywords: Add2, DSE, mRNA stability, TDP-43, 3′ end processing, 3′UTR
Abbreviations
- RBP
RNA binding protein
- DSE
downstream element
- PAS
polyadenylation site
- ActD
actinomycin D
Introduction
In mammalian cells, mRNA maturation, stability, and translation are highly dynamic and complex processes that modulate gene expression and protein synthesis in both physiological and pathological conditions. During the mRNA lifecycle, important steps are regulated by RNA-binding proteins (RBPs).1 Among the huge number of RBPs playing crucial roles in mRNA metabolism, AUF1, TTP, KSRP, BRF1, the Hu protein family, TDP-43 and QKI are some of the factors involved in modulating mRNA stability and/or its decay.2,3 TDP-43 is a nuclear ubiquitously expressed RBP belonging to the heterogeneous nuclear ribonucleoprotein (hnRNP) protein family. It is implicated in several transcriptional and post-transcriptional mechanisms of gene expression regulation. TDP-43 is mainly localized in the nucleus, where it regulates transcription, pre-mRNA splicing and microRNA processing. However, it also shuttles between the nucleus and cytoplasm, where it is involved in other cellular processes including mRNA stability, mRNA transport and translation.4–6 It mainly recognizes in its target transcripts GU/UG repeat motifs preferentially localized in long intronic regions, splice sites and 3′UTRs.7-11 TDP-43 has recently been associated with neurodegenerative diseases, such as amyotrophic lateral sclerosis (also called Luo-Ghering disease) and frontotemporal lobular dementia (ALS and FTLD, respectively).12 In these diseases TDP-43 is found as the major component of pathological cytoplasmic inclusions.13 In these pathological states, TDP-43 aggregation and mislocalization in the cytoplasm of affected neurons is believed to be associated with neurodegeneration by loss of its biological functions, by acquisition of a toxic effect, or by a combination of both.14 Therefore, the identification of TDP-43 target transcripts and the understanding of the molecular mechanism by which their expression is regulated by TDP-43 are essential to elucidate the pathogenic pathways in ALS and FTLD.
In a previous work aimed to characterize the molecular mechanisms regulating Add2 pre-mRNA polyadenylation, we identified by RNA pull-down analysis potential factors interacting with Add2 A4 PAS DSE. One important candidate was TDP-43,15 suggesting a possible involvement of this protein factor in pre-mRNA 3′ end processing of the Add2 transcript. The potential role of TDP-43 in polyadenylation has been recently proposed in a model of TDP-43 autoregulation of gene expression levels.16
Adducins are cytoskeletal proteins that cap and stabilize the fast growing end of actin filaments. They are homo- and hetero-dimers composed by association of 3 related subunits: α−, β−, and γ-adducins (Add1, Add2 and Add3). While Add1 and Add3 are expressed ubiquitously, Add2 is restricted to erythroid tissues and the nervous system.17-19 In the brain, the adducin heterodimers are present in the growth cones of axons, presynaptic nerve terminals and post-synaptic dendritic spines.20,21 It is very abundant in hippocampal dendritic spines. It is involved in the assembly of synapses, in the control of synaptic plasticity and synapses stability.22,23 Add2 genetic inactivation in mice leads to motor coordination, behavioral and learning deficits, associated with impairment in LTP and LTD.24,25 Erythroid and neuronal Add2 expression is correlated with the use of tissue specific alternative promoters and polyadenylation sites (PAS). In particular, proximal polyadenylation sites (2 or 3, depending on the species) are active in erythroid tissues while one distal PAS (A4 PAS) is exclusively used in brain. Both short and long mRNA forms encode for the same protein. As a result of alternative polyadenylation, neuronal Add2 mRNA form contains an unusually long 3′UTR of 5-6 kb. This brain-specific Add2 mRNA shows dendritic localization.24,26
In this work we confirmed that TDP-43 interacts with the Add2 PAS DSE in vitro and observed that depletion of TDP-43 in tissue culture cells results in the down-regulation of both the chimeric and endogenous Add2 transcripts. Apparently, this effect was not mediated by the Add2 DSE, as substituting the Add2 DSE with viral E-SV40 and L-SV40 DSEs, which are not TDP-43 targets, still resulted in a decrease of Add2 mRNA levels in vivo after TDP-43 depletion. These data were supported by the failure to observe an in vitro interaction between TDP-43 and some of the main polyadenylation factors. Therefore, we excluded the pre-supposed role of TDP43 in pre-mRNA Add2 polyadenylation and finally demonstrated that the downregulation of Add2 transcripts after TDP-43 depletion was due to a decrease in Add2 mRNA stability, suggesting that TDP-43 stabilizes the Add2 mRNA.
Results
TDP-43 binds to A4 PAS DSE of Add2 transcript in vitro
In a previous work we identified TDP-43 as one of the proteins that bind the DSE of the brain-specific A4 PAS of the Add2 pre-mRNA.15 Since Add2 DSE contains a stretch of 4 GU repeats located immediately downstream of the PAS cleavage site (Fig. 1A), which may act as landing pad for TDP-43, we performed RNA-EMSA experiments using purified recombinant TDP-43 and radioactive RNA probes of the A4 PAS region. TDP-43 was able to interact with the wt A4 PAS RNA probe, but not with the A4 PAS probe in which the DSE was deleted (Fig. 1B). These data confirm that TDP-43 was able to bind in vitro the Add2 A4 DSE and suggest that it might participate in the formation of the 3'end processing complex at the distal brain-specific PAS.
Figure 1.
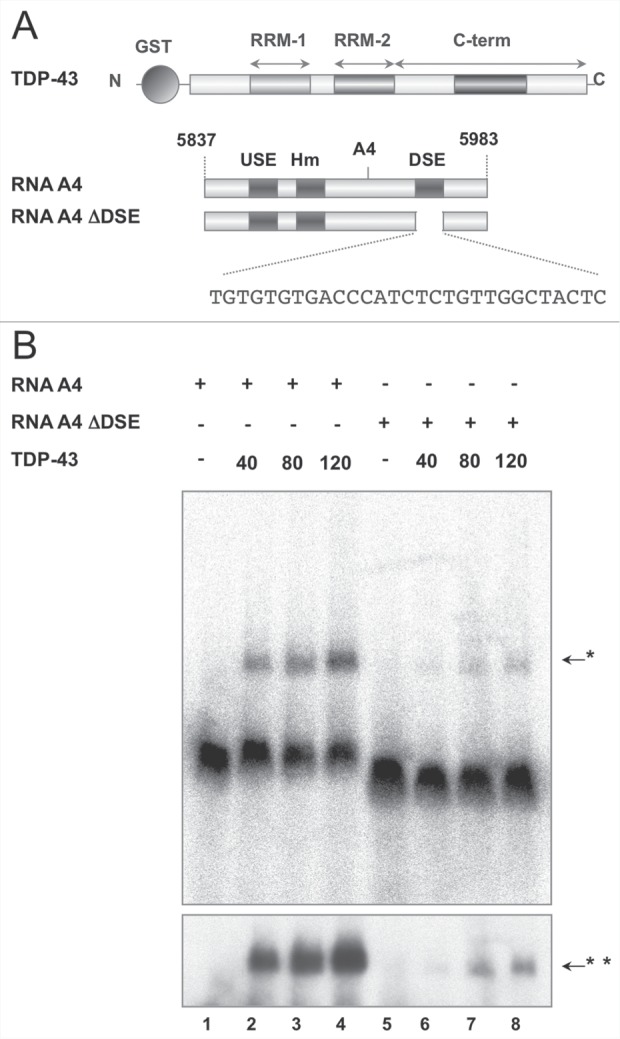
Recombinant TDP-43 binds to A4 PAS DSE of Add2 RNA in vitro. Panel A: schematic representations of recombinant GST-TDP-43 and wt and mutant (ΔDSE) RNA probes covering the A4 PAS used in Panel B are shown. The upstream element (USE), the exanucleotide motif (Hm) and the downstream element (DSE), together with the DSE nucleotide sequence are indicated. The numbering on the RNA scheme (5837-5983) refers to the first base of the last mouse Add2 exon. Panel B: wt (Lane 1-4) and ΔDSE (Lane 5-8) hot RNAs were incubated with increasing amounts of GST-TDP-43 (40-80-120 ng) in a final volume of 20 µl, complexes were separated in polyacrylamide gel and autoradiographed. The upper and lower panels correspond to 4 hours and 24 hours of exposure, respectively. The arrows indicate the RNA protein complexes at 4 hours and 24 hours (one and two asterisks, respectively).
Down-regulation of TDP-43 results in a reduction of Add2 mRNA in vivo
In order to determine whether TDP-43 had a functional role in regulating Add2 mRNA levels in vivo, presumably by modulating pre-mRNA processing of the transcript, we performed siRNA knock-down of the endogenous TDP-43 in HeLa cells and analyzed the effects by transfecting the A1-A23-A4 construct. As we have previously shown,15 the chimeric minigene A1-A23-A4 (Fig. 2A), which contains all polyadenylation regions of the Add2 gene, reproduces the use of the distal brain specific A4 PAS when transiently transfected into HeLa cells (Fig. 2B and C). Interestingly, when total mRNA, purified after depletion of the endogenous TDP-43 by siRNA and minigene transfection, was analyzed by Northern Blot, we observed a 60% decrease in the levels of mRNA derived from the A1-A23-A4 plasmid (Luciferase siRNA was used as control, Figure 2B lanes 1-2). To rule out possible differences in mRNA expression of the A1-A23-A4 construct as a consequence of variations in transfection efficiency, we co-transfected a plasmid expressing GFP (pEGFP-C2) and analyzed its mRNA levels in the same membrane by Northern blot. A similar result was obtained in HEK293 cells after siRNA knock down of TDP-43 (Fig. 2D, lanes 4-5), excluding the possibility of a cell type-specific effect.
Figure 2.
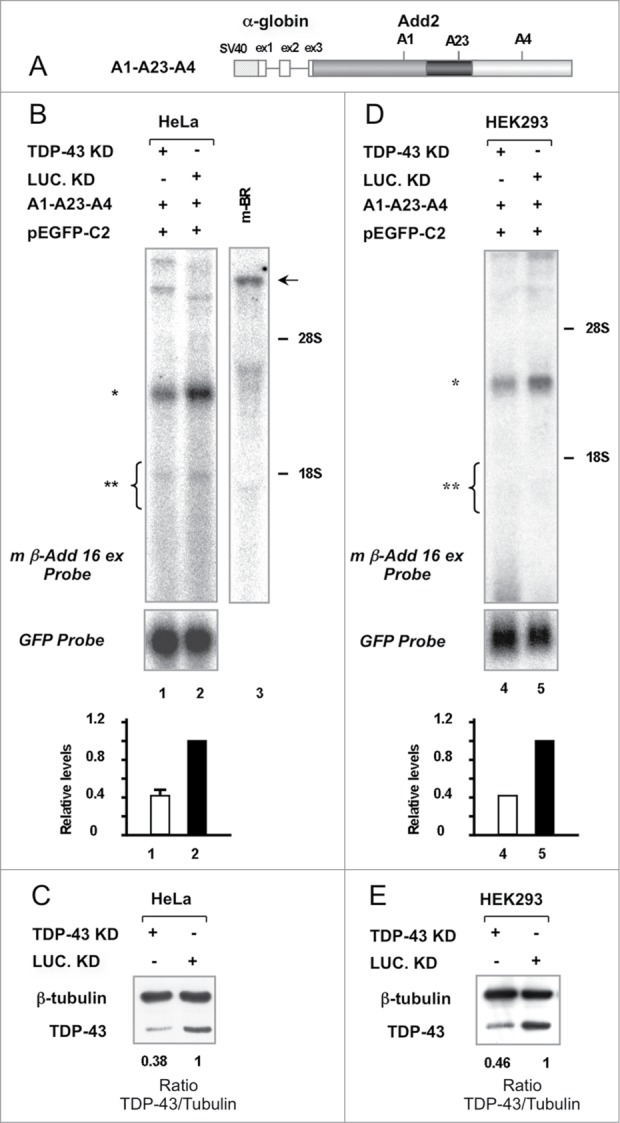
Depletion of TDP-43 results in a decrease in the levels of Add2 minigene derived mRNA in HeLa and HEK293 cells. Panel A: Scheme of the chimeric construct containing the polyadenylation regions of Add2. Panels B and D: Northern blot analysis of total RNA prepared from HeLa (Lanes 1 and 2) and HEK293 (Lanes 4 and 5) cells transfected with the A1-A23-A4/pEGFP-C2 constructs, previously treated with TDP-43 siRNA (Lanes 1 and 4) or Luciferase siRNA (Lane 2 and 5) as a control. The pEGFP-C2 plasmid encoding for GFP was used to normalize for differences in transfection efficiency. Add2 minigene-derived mRNA and GFP transcripts were detected with the m β-Add 16 ex (top panel) and GFP probes (bottom panel), respectively. The relative levels are shown in the bar graph (bottom; Add2/GFP ratio, mean ± SD of 3 different experiments for Panel A and mean of 2 different experiments for Panel C). The position of the expected Add2 minigene-derived transcripts (A4 PAS selection: ∗, ∼2.9 kb; proximal PASs selection: ∗∗, ∼1.95–1.45 kb) and rRNAs (28S and 18S) are indicated. The arrow indicates the position of the Add2 mRNA of mouse brain, used as positive control of the assay (∼8.2 kb) (Lane 3). Panels C and E: Western blot analysis of total protein extracts prepared from HeLa (Panel C) and HEK293 (Panel E) cells treated with TDP-43 or Luciferase siRNA, and then transfected with the A1-A23-A4/pEGFP-C2 constructs. The TDP-43/β-tubulin ratio (bottom) indicates the level of TDP-43 down-regulation.
In addition, we analyzed by Northern blot and qPCR the expression levels of the endogenous Add2 mRNA (normalized by the endogenous GAPDH and RPL13A mRNAs) and, similarly to the results obtained with the A1-A23-A4 reporter plasmid, we observed a ~60-80% decrease in Add2 mRNA levels in both HeLa and HEK293 cells (Fig. 3B and C–F). In HEK293 cells, we observed that the expression level decrease was observed in both the short and long 3′UTR isoforms (Fig. 3B, 3D and 3E). These results underscore a role of TDP-43 in gene expression regulation of the Add2 pre-mRNA, probably related to pre-mRNA 3′ end-processing.
Figure 3.
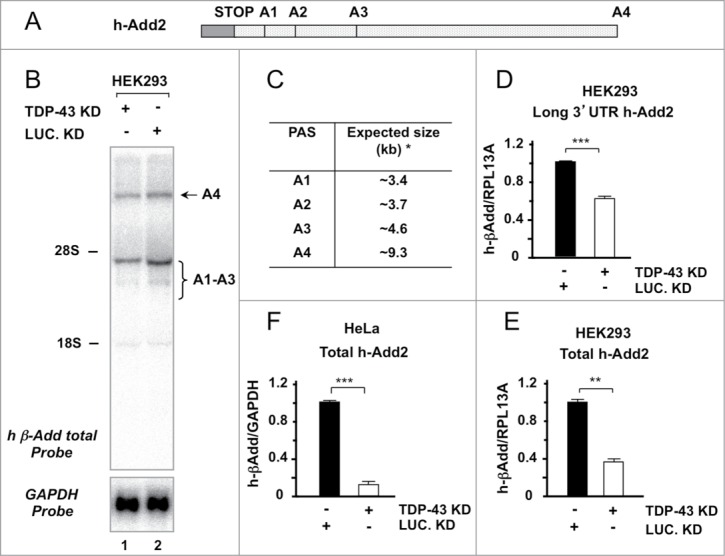
TDP-43 downregulation results in a reduction of endogenous Add2 transcript in HeLa and HEK293 cells. Panel A: A schematic representation of the last exon of the human Add2 gene is shown. The ORF (gray box), the stop codon and the different PASs in the 3′UTR are indicated. Panel B: Total RNA (25 μg) from HEK293 treated with TDP-43 (Lane 1) and Luciferase siRNA (Lane 2) was analyzed by Northern blot using the h β-Add total (top panel) and GAPDH probes (bottom panel). The position of the rRNAs is indicated, as well as the position of the expected endogenous mRNA bands (A4 PAS selection: ∼9.3 kb; proximal A1-A2 PASs selection: ∼3.4–4.6 kb). Panel C: The Table indicates the expected sizes of the mRNAs generated using the different human Add2 PAS (the asterisk indicates the sizes without considering the pA tail). Panels D, E and F: qRT-PCR of the endogenous Add2 transcript in HEK293 and HeLa cells after TDP-43 depletion. The relative amounts of all human Add2 mRNA forms (total) in HEK293 and HeLa cells are shown in Panels E and F, respectively, while that of the brain specific transcript in HEK293 is reported in Panel D. The relative levels are expressed as Add2/GAPDH (Panel F) or Add2/RPL13A ratio (Panels D and E) (mean ±SD of 3 different experiments).
The decrease of Add2 mRNA levels is not mediated by the direct interaction of TDP-43 with the DSE
In order to obtain direct evidence supporting the hypothesis that the decrease in the expression levels of the chimeric transcript obtained from the reporter plasmid (Fig. 2B and D) could be determined by the interaction between TDP-43 and A4 PAS DSE, we first tested the ability of TDP-43 to interact with some of the key factors involved in 3′ end processing. We transiently transfected HeLa cells with a flagged version of TDP-43 (plasmid pFLAG-TDP-43), the HeLa cell lysate was immunoprecipitated with anti-FLAG antibody and the binding between TDP-43 and the polyadenylation factors was tested by Western blot with antibodies anti Cstf-64 and CSPF-160 (Fig. S1). The IP reaction was performed in the absence or presence to RNase A, to evidence whether the potential interaction may require the binding to RNA. We failed to detect any interaction between TDP-43 and Cstf-64 or CSPF-160 polyadenylation factors. In addition, we were not able to detect a supershift indicating protein interactions did not occur, when we performed super-shift mobility assays using the Add2 DSE RNA probe and antibody anti Cstf-64 and CSPF-160 (data not shown). These data suggest that TDP-43 was not able to interact, directly or indirectly, with any of the tested proteins of the polyadenylation machinery.
Since these negative results obtained in the in vitro experiments could be related to weak interactions in the reconstituted in vitro system or to low sensitivity of the assay, we performed an in vivo approach to determine the role of TDP-43 in Add2 DSE-dependent polyadenylation. To this aim, we generated DSE mutant minigenes by substituting the Add2 DSE by the E-SV40 and L-SV40 DSEs in the A1-A23-A4 construct (Fig. 4A). These viral DSE sequences do not contain GU repeats, the most common binding site of TDP-43.7-11 In addition, the L-SV40 polyadenylation signal RNA was used as a model in a functional proteomic approach to identify the components of the pre-mRNA 3′ end processing complex and TDP-43 was not identified among the isolated factors,27 strongly suggesting it does not bind the viral DSE. Therefore, the mutant constructs were transfected into HeLa cells after siRNA knock-down of the endogenous TDP-43, and total RNA analyzed by Northern blot. We observed that the viral DSE enhanced the expression of the chimeric transcript (Fig. 4B and 4C, compare lanes 2 vs. 4 and 6). When we reduced endogenous TDP-43 levels by siRNA, we also observed a decrease in mRNA levels in both constructs having the viral DSEs (Fig. 4B, lanes 3 and 5). The decrease in mRNA levels compared to controls treated with Luciferase siRNA was of the same magnitude (~60%) of the one observed for the wt A1-A23-A4 construct (Fig. 4 B and 4C, lanes 1-2 vs. 3-4 and 5-6).
Figure 4.
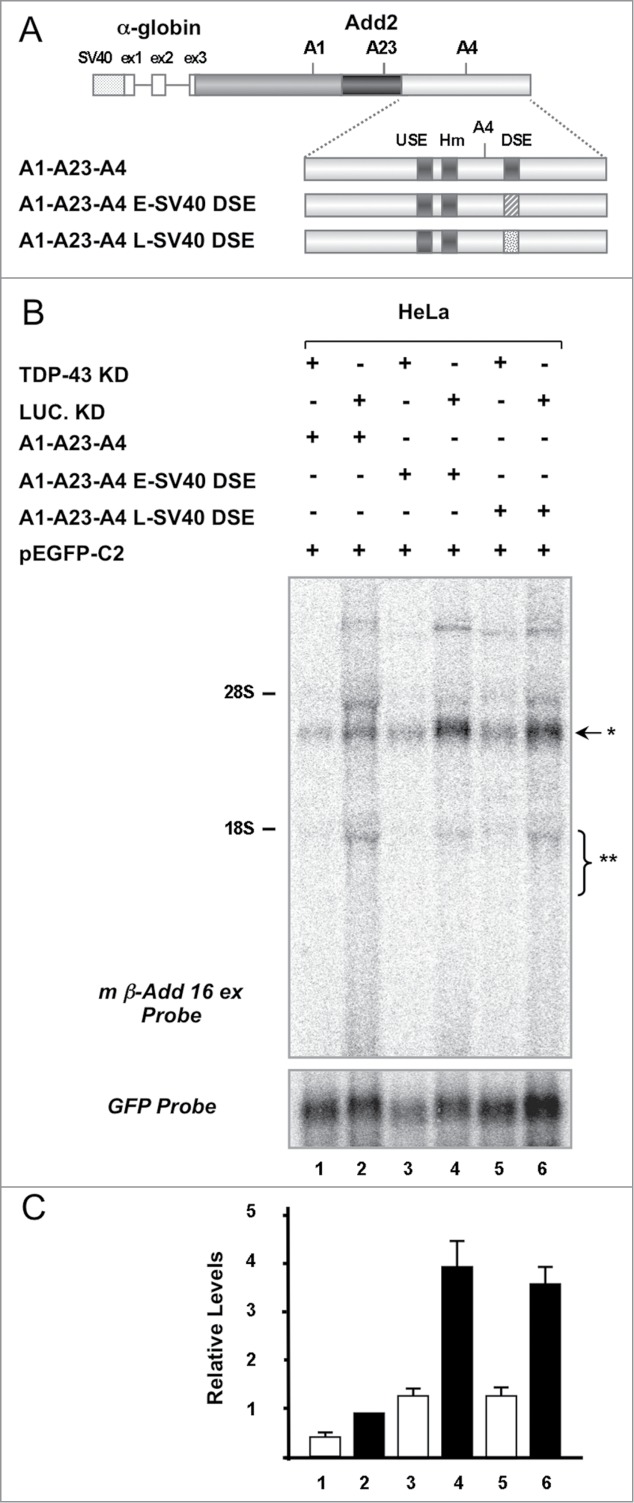
The effect of TDP-43 on Add2 expression is not mediated by its direct interaction with the A4 PAS DSE. Panel A: Scheme of the constructs used in Panel B. The Add2 DSE sequence was substituted with that of E-SV40 and L-SV40 PASs as indicated in detail in the Materials and Methods section. Panel B: Northern blot of RNA prepared from HeLa cells treated with TDP-43 (Lane 1, 3 and 5) or Luciferase siRNA (Lane 2, 4 and 6), co-transfected with the constructs shown in Panel A and pEGFP-C2 plasmid, and hybridized with the m β-Add 16 ex (top panel) and the GFP probes (bottom panel). The position of the rRNAs is indicated, as well as the position of the expected minigene-derived RNA bands (A4 PAS selection: ∗, ∼2.9 kb; proximal PASs selection: ∗∗, ∼1.95–1.45 kb). Panel C: The bar graph indicates the relative levels of expression normalized to the GFP signal (Add2/GFP signal ratio, mean ±SD of 3 different experiments).
Therefore, these data strongly suggest that the effect of TDP-43 on Add2 expression was not related to its physical interaction with the A4 PAS DSE and that TDP-43 was not directly involved in modulating the 3′ end processing of Add2 pre-mRNA at the A4 PAS.
Knock-down of TDP-43 does not affect Add2 pre-mRNA cleavage efficiency and polyA tail length
To rule out that the absence of TDP-43 could reduce the efficiency of cleavage, which would likely result in increased levels of unprocessed transcripts and a reduction in the amount of mature Add2 mRNA, we performed a RNAse protection assay (RPA).28 We utilized a 324 nt-long input RNA probe having 289 nt complementary to the pre-mRNA transcript (205 nt upstream and 84 nt downstream of the A4 PAS cleavage site) (Fig. 5A). Cleavage efficiency was calculated as the ratio between the signals obtained for the protected product of the cleaved mRNA and that of the uncleaved pre-mRNA band. The same cleavage efficiency values were obtained in siLuciferase and siTDP-43-treated samples (Fig. 5B, Lanes 1 and 2, Cleavage efficiency: 1.95 and 1.90, respectively), suggesting that TDP-43 depletion did not affect cleavage efficiency at the A4 PAS.
Figure 5.
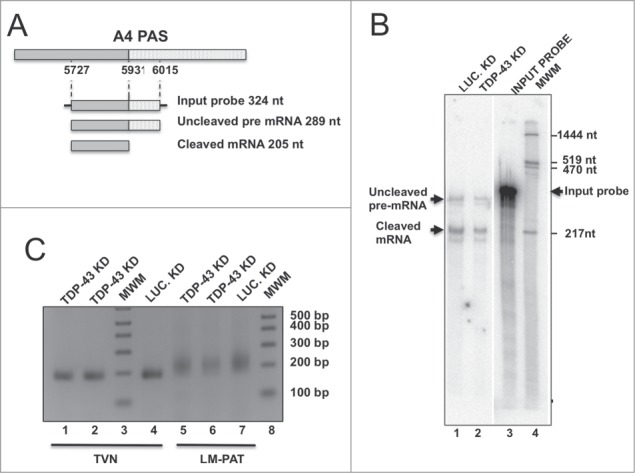
Cleavage efficiency and polyA tail length are not affected by TDP-43 knock-down. Panel A: Schematic representation of the Add2 A4 region containing A4 PAS and the probe used in the RNase protection experiment shown in Panel B; the expected protected fragments, with their expected sizes are shown; nucleotide positions are numbered respective to the first base of the Add2 last exon. Panel B: RNase protection analysis of total RNA prepared from HeLa cells transfected with the A1-A23-A4/pEGFP-C2 constructs, treated with TDP-43 (Lane 2) or Luciferase siRNA (Lane 1). The Input probe (Lane 3), uncleaved pre-mRNA and cleaved mRNA are indicated by black arrows. MWM, molecular weight markers (Lane 4); Panel C: Shortest possible LT-PAT transcript (TVN) and Ligation-mediated Poly A tail (LM-PAT) assays of total RNA samples of HeLa cells transfected with the A1-A23-A4/pEGFP-C2 constructs and treated with with TDP-43 (Lanes 1 and 2, TVN; Lanes 5 and 6, LM-PAT) or Luciferase siRNA (Lane 4, TVN; Lane 7, LM-PAT) is shown. MWM indicates Molecular Weight Markers.
Since polyA tail length could be reduced in the absence of TDP-43, with an impact on mRNA stability, we performed the ligation-mediated polyA length test (LM-PAT) assay to determine polyA tail length in the same siLUC and siTDP-treated RNA samples. As shown in Fig. 5C (Lanes 5 and 6 vs. 7), no differences were observed in the length of the polyA tails of the Add2 chimeric transcript in the 2 different conditions, that appeared as a short smear on the agarose gel electrophoresis. We also analyzed the polyadenylation status of the endogenous β-actin mRNA in the same samples and observed no differences in poly A tail length (data not shown). These data indicate that the reduction in Add2 mRNA was not the consequence of reduced pre-mRNA cleavage or differences in the polyA tail length, supporting the hypothesis that TDP-43 could influence Add2 mRNA half-life through the binding to the 3′UTR.
Downregulation of TDP-43 results in a reduction of Add2 mRNA stability
Several previous findings have shown that TDP-43 interacts not only with introns but also with regulatory 3′UTR sequences participating in controlling mRNA stability and/or translation.29-31 The observation that the TDP-43 modulation of Add2 expression levels was detected in both the endogenous transcript and minigene derived mRNAs, which contains only the PAS and their respective 3′UTR flanking regions of Add2, and in minigene constructs in which the Add2 DSE was replaced by DSE sequences that do not contain GU repeats (Fig. 4A), further supported the possibility that TDP-43 could influence Add2 mRNA half-life through the binding to the 3′UTR.
Thus, we tested TDP-43 involvement in maintaining mRNA stability by blocking transcription and determining mRNA levels by Northern blot analysis. We co-transfected HeLa cells with the A1-A23-A4 construct/pEGFP-C2, performed TDP-43 or Luciferase (control) siRNA knock-down and treated the cells with actinomycin D (ActD) to block transcription. Then, total RNA was collected at different time points and analyzed by Northern Blot. To confirm the efficacy of ActD treatment we used probes for the c-myc and GAPDH transcripts, which are endogenous mRNAs known to have short and long half-life, respectively.32 As observed in Fig. S2, no signal was observed for c-myc after 180 min of ActD treatment, while no significant change was detected for the GAPDH mRNA, confirming the effect of the ActD treatment.
The degradation kinetics of the chimeric transcript was evaluated by normalizing its expression levels with that of the control GFP mRNA at the different time points. As shown in Fig. 6, ActD treatment resulted in a significant reduction of the A1-A23-A4 derived mRNA half-life (Fig. 6A and 6B, lanes 1-4 vs 5-8). However, when TDP-43 was knocked down by siRNA, Add2 mRNA decay was clearly visible after 30 min (Fig. 6, lane 2) suggesting that TDP-43 was involved in stabilizing Add2 chimeric transcript.
Figure 6.
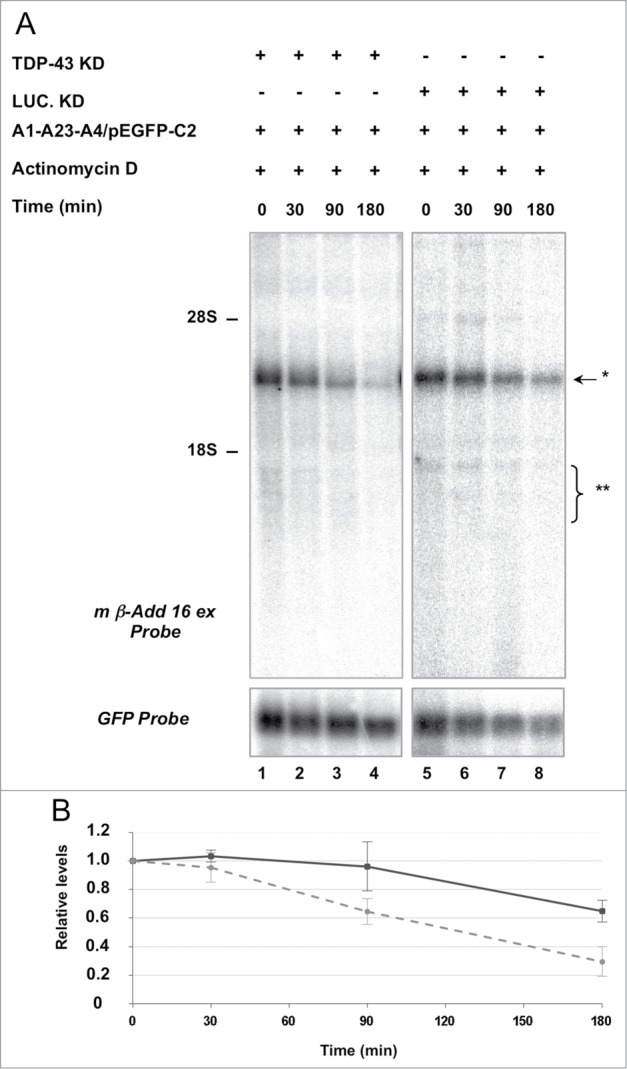
Add2 chimeric mRNA stability is affected by TDP-43. Panel A: After TDP-43 and Lucifearse siRNA treatments and co-transfection with the A1-A23-A4 minigene/pEGFP-C2 plasmid, total mRNA from HeLa cells was collected after different times (0, 30, 90 and 180 minutes) of Actinomycin D (ActD) incubation and analyzed by Northern blot using m β-Add 16 ex (top panel) or the GFP probes (bottom panel). The positions of rRNAs (28S and 18S) and that of the expected Add2 minigene-derived transcripts are indicated. Panel B: Minigene-derived transcript degradation kinetics curve is shown. The graph represents the relative levels of the chimeric mRNA (Add2/GFP signal ratio, mean ±SD of 4 different experiments) at the different ActD treatment times in the TDP-43 knock-down (gray dash lane) and control (black solid lane) conditions.
Discussion
In this work we investigated the potential role of TDP-43 in Add2 transcript 3′ end processing. The involvement of TDP-43 in polyadenylation was initially suggested by its identification as a strong brain-specific Add2 PAS DSE binding factor.15 We confirmed that TDP-43 interacts in vitro with an Add2 RNA probe containing the complete Add2 A4 PAS, and that this interaction requires the presence of the DSE (Fig. 1). The Add2 DSE is an essential element for the efficient and correct cleavage and polyadenylation at the distal A4 PAS.15 In the present work, the functional role of TDP-43 on Add2 mRNA metabolism was supported by the observation that TDP-43 depletion in HeLa and HEK293 cells results in the down-regulation of Add2 chimeric transcript and endogenous Add2 gene (Fig. 2 and 3). However, this effect appeared to be related to the involvement of TDP-43 in maintaining the stability of the Add2 mRNA, and not to a role in 3′ end processing.
TDP-43 was initially discovered as a regulator of HIV transcription33 but since then it was implicated in multiple nuclear and cytoplasmic functions ranging from alternative splicing,7,34 miRNA biogenesis,35 mRNA translation,36 mRNA stability,30,31 to TDP-43 mRNA levels autoregulation,16,29 Recently, it was demonstrated its involvement in 3′ end processing of its own pre-mRNA. High levels of TDP-43 block the recognition of the first PAS present on its transcript by competing with the polyadenylation factor CstF-64 for DSE binding.16 Several global scale analyses based on RIP-seq and CLIP-seq have been performed to identified TDP-43 mRNA targets in neuronal, non-neuronal cells, mouse brain, healthy and diseased human brain tissues.8-11,30 These studies showed that the most significantly enriched pentamer-binding sites were GUGUG and UGUGU, confirming previous in vitro data.7,37 Add2 brain specific PAS DSE contains 4 GU repeats in its sequence (Fig. 1A), so it is an optimal TDP-43 target sequence that can be recognized by TDP-43. In vitro experiments showed that the Kd of TDP-43 for oligos containing GU repeats range between 3600 nM and 2.8 nM, for (UG)3 and (UG)8, respectively.38 In the case of the Add2 DSE, there are 4 UG repeats and for this UG length the reported Kd value is 115 nM.38 Curiously, expression analysis, upon TDP-43 depletion, of chimeric transcripts from the Add2 minigene in which the Add2 DSE was replaced by the SV40 DSE, which did not contain GU repeats, allowed stating that TDP-43 in vivo role was not correlated to the direct interaction with the Add2 A4 PAS DSE (Fig. 4). These results also suggested that TDP-43 was not directly involved in 3′ end processing at the A4 PAS in vivo and that the identification of TDP-43 as an Add2 DSE binding factor in vitro was probably determined only by the high binding affinity of TDP-43 by the 4 GU motif. Furthermore, the observation that neither cleavage activity nor polyA tail length were affected by TDP-43 depletion, together with the inability of some of the main polyadenylation factors to co-precipitate with TDP-43, strongly supported the idea that TDP-43 modulates the levels of the Add2 transcript by another mechanism.
To date, modest evidences sustained the possible existence of interactions between TDP-43 and polyadenylation factors. Only some global approaches have been performed to identify the TDP-43 “interactome”.9,39 These analyses discovered nuclear TDP-43 binding factors proteins involved in several aspects of RNA metabolism (pre-mRNA splicing, mRNA stability, and transport) and, curiously, among the numerous members of the polyadenylation machinery only CPSF160 was identified as a weak TDP-43 interacting protein, but no functional experiments were presented.39 Moreover, TDP-43 was not found among the component of the pre-mRNA 3′end-processing complex of the L-SV40 and adenovirus L3 transcripts.27 In our work, we also failed to detect interactions between TDP-43 and polyadenylation factors.
Having discarded a link between TDP-43 and 3′ end processing, we looked for another potential TDP-43 function on Add2 gene expression. The differences identified in the kinetic degradation curves of the chimeric mRNA in the control and TDP-43 downregulation conditions allowed us to conclude that TDP-43 is involved in preserving Add2 mRNA stability. The role of TDP-43 in influencing mRNA stability was showed for several genes. Strong et al. suggested that TDP-43 stabilized the hNFL (human low molecular weight neurofilament) mRNA while an opposite destabilizing effect was observed on Vegf (vascular endothelial growth factor) and Grn (progranulin) transcripts.30,31 In both cases TDP-43 acts by a direct interaction with a (UG)n motif or (UG)/(UGUG) motifs in stem loop structures located in their 3′UTR.30,40 It was also demonstrated that TDP-43 is able to regulate the half-life of its own mRNA: in fact, overexpression of the protein promotes instability of its transcript.29 TDP-43 auto-regulation is mediated by multiple binding sites located in a 615 nucleotide long sequence in the 3′UTR, that did not include classical UG/UG repeats motif.16,29 These specific examples show that TDP-43 performs some of its functions by interacting with its target motifs in the 3′UTR of the transcript. The relevance of this interaction was also highlighted by large-scale CLIP studies: even if TDP-43 binding sites are preferentially located in long intronic regions and near splice site acceptors, a significant percentage of its target sequences mapped in the 3′UTR.8,10,30 Interestingly, Tollervey et al. analyzing the interaction with TDP-43 and RNA from the cytoplasmic fraction, observed that the binding of TDP-43 to 3′UTR was tenfold enriched in comparison to the nuclear fraction, supporting the shuttling of the protein in this cellular compartment and its role in modulating mRNA fate in the cytoplasm by interacting with the 3′UTR.10 Considering that the TDP-43 in vivo effect was shown in both endogenous and minigene derived Add2 mRNAs (Fig. 2 and 3) and that only the PASs and their flanking regions were in common in the 2 transcripts, TDP-43 may perform its function by recognizing some elements in the Add2 3′UTR. Further studies are necessary to identify the Add2 elements recognized by TDP-43. From previously analyses also emerged that in a minor subset of targets (UG)nTA(UG)m, A-rich and U-rich elements, often coexisting with (UG)n motif, are 3′UTR motif recognizes by the TDP-43.9,11,30 Curiously, in the Add2 3′UTR several U-rich stretch elements flanked by GU/UG-rich sequences, highly conserved between mouse and human, are located in the 450 nucleotides upstream A4 PAS, that could be potential multiple low affinity TDP-43 binding sites.
However, Add2 transcript was not found as a specific TDP-43 target by RIP-seq and CLIP-seq approaches. Its absence among the CLIP hits could be associated to the fact that TDP-43 probably binds to elements of the 3′UTR present in the long Add2 mRNA and this variant was poorly annotated in databases.26,41,42 Nonetheless, it is important to take into account that these global analyses revealed that TDP-43 targets are enriched in transcripts associated to synaptic functions, neuronal development and RNA metabolism.8,9,30 Add2 mRNA can be included to these classes. In fact, Add2 has a key role in the assembly of synapses, control of synaptic plasticity and stability, associated to LTP, LTD, motor abilities and learning.22-25
The evidenced involvement of TDP-43 in Add2 mRNA stability can be considered relevant also at the light of its possible impact on the neurodegenerative processes associated to ALS and FTLD. The exact molecular mechanisms associated to ALS and FTLD are not completely understood, since TDP-43 cytoplasmic inclusions might have a toxic gain of function or loss of function by sequestering nuclear TDP-43 (or both), disrupting its role in RNA metabolism. Thus, one of the pathological processes associated to disease could be alteration of the stability of specific transcripts and, consequently, variation in the expression levels of the corresponding genes.14 In the specific case of Add2, considering its role at synapti c-terminals,22–25 alteration of Add2 levels due to changes in TDP-43 levels and/or activity may directly affect neurological functions in disease states. However, the understanding of the fine mechanism involving TDP-43 in Add2 mRNA stability and its possible connection to disease states still need additional studies.
To summarize, in the present report we excluded TDP-43 involvement in Add2 A4 PAS 3′ end processing and finally highlighted its role in modulating Add2 transcript stability. Despite the precise mechanism of controlling Add2 stability remains to be elucidated we believe that the identification of new TDP-43 mRNA targets and the full definition of its biological role may help in understanding why its dysfunction, associated to its mislocalization and aggregation in the cytoplasm, leads to selective neuronal death in neurodegenerative diseases.
Materials and Methods
Construction of minigene constructs
The A1-A23-A4 minigene used for the transfection experiments was previously described.15 A1-A23-A4 E-SV40 DSE and A1-A23-A4 L-SV40 DSE were created substituting the sequence of Add2 DSE (5′-TGTGTGTGACCCATCTCTGTTGGCTACTC-3′) with the ones of the viral PAS DSE (L-SV40 DSE: 5′-ACAATTGCATTCATTTTATGTTTCAGGTTCA-3′; E-SV40 DSE: 5′-TTCTAGTTGTGGTTTGTCCAAACTCATCA-3′) by in vitro site directed mutagenesis. The final chimeric minigenes were used to transfect HeLa and HEK293 cells.
In vitro site directed mutagenesis
Oligonucleotides primers containing the desiderate mutations and DNA vector of interest (50 ng) were added to the reaction mix (2.5 U/µl PfuTurbo DNA polymerase, 1X reaction buffer, 200 μM dNTP mix). PfuTurbo DNA polymerase (Stratagene) extended the primers during temperature cycling. The amplification product was digested with Dpn I to eliminate the initial DNA template allowing the selection of mutation-containing synthesized DNA. The mutated vector is then transformed into competent bacterial cells.
Cell culture transient transfections
Plasmid DNA liposome-mediated transfections in HeLa and HEK293 cell lines were performed using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer's protocol. Plasmid pEGFP-C2 (Clontech) was used as transfection efficiency control. In addition, to stimulate the activity of the SV40 promoter contained in A1-A23-A4 wt and mutant minigenes, a plasmid expressing SV40 T antigen was co-trasfected. The plasmid DNA used for transfections was purified with JetStar columns (Genomed).
Oligonucleotides siRNA were transiently transfected using Oligofectamine (Invitrogen) and HyPerFect (Qiagen) transfection reagents for HeLa and HEK293 cells, respectively. To obtain a significant grade of protein downregulation a 70 nM siRNA final concentration was used and the transfection was performed 2 times according to the manufacturer's protocols. The siRNA sequences were as follows: TDP-43 siRNA 5′- GCAAAGCCAAGAUGAGCCU-3′, Luciferase siRNA 5′- UAAGGCUAUGAAGAGAUAC-3′.43,44
RNA preparation, synthesis of cDNA, PCRs
RNA preparation. Total cellular RNA was isolated with TRI REAGENT (Sigma) following manufacturer's instructions.
cDNA synthesis. cDNA was prepared from total RNA (1-2 μg in the final volume of 20 µl) by using M-MLV reverse transcriptase (RT) (Invitrogen). RNA was previously treated with DNase (Promega) when the cDNA was amplified by qPCR. The manufacturer' protocol was followed by the RT and DNase reactions.
PCR. The polymerase chain reaction was performed on plasmid DNA (5-10 ng) following the basic protocol accompanying the Taq DNA Polymerase enzyme (Roche or NEB). Table 1 shows the oligonucleotide primers used to perform the different amplifications.
qPCR. mRNA levels were determined by real time PCR. The reaction was performed using 1/20 of the cDNA volume, Syber green mix (BioRad) and the primers indicated in Table 1. Amplification products were detected by the iCycler system (Bio-Rad). For an internal standard control, the expression level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and ribosomal protein L13a (RPL13A) was simultaneously quantified. Each sample was analyzed in duplicate and triplicate of each condition were used for the statistical analysis. Amplification efficiencies of target and standard control were used for calculations of normalized relative (ΔΔCT) expression levels and the statistical analysis were done using GraphPad Prism software.
Northern blot analysis
The Northern Blot analysis was performed as described.26 RNA was separated in 1.5% agarose formaldehyde gels, transferred to nylon membranes, and hybridized with the appropriate DNA probe labeled with [α−32P] dCTP following pre-hybridization in ULTRAhyb Ultrasensitive Hybridization Buffer (Ambion). The different DNA fragments used as probes were obtained by PCR from genomic DNA (m β-Add 16 ex Probe), cDNA (c-myc Probe, GAPDH Probe), and pEGFP-C2 commercial plasmids (GFP Probe), labeled with Rediprime II Random Prime Labeling System (Amersham Bioscience) and then purified from unincorporated nucleotides by Nick columns (Amersham bioscience).
mRNA stability assay
After transient siRNA silencing and transfection of the chimeric minigene and control vector, cells were treated with Actinomycin D (5 μg/ml final concentration) or DMSO as a control. Total RNA was collected at different time points and analyzed by Northern Blot. The results were quantified by with OptiQuant software (Bio-Rad Laboratories).
In vitro transcription
RNA was in vitro synthesized with T7 RNA polymerase (Stratagene) from a DNA template (PCR amplified) containing the T7 RNA polymerase promoter sequence (oligonucleotides are indicated Table 1) and purified as previously described.15
RNase protection assay (RPA)
The RNase protection assays were performed as described previously.28 Briefly, RNA samples were prepared from total RNA of transfected HeLa cells, treated with siRNA for Luciferase or TDP-43, as described above. Total RNA was treated with DNase, acid phenol-chloroform purified, ethanol precipitated and resuspended in R-loop buffer. Four hundred ng of one RNA sample were used for the assay. The radioactive RNA probe was prepared as described.28 Approximately 500 cps of the probe were used for the hybridization reactions. pUC19 plasmid, digested with TaqI and NdeI, was labeled with (α−32P)-dCTP and used to estimate the size of the observed bands.
Cleavage site analysis (TVN) and ligation mediated poly(A) test (LM-PAT)
The Ligation mediated Poly(A) test assay (LM-PAT) was essentially performed as described.45 Briefly, 0.6-1.2 µg of total RNA (prepared from transfected HeLa cells, treated with siRNA for Luciferase or TDP-43, as described above, were incubated with 20 ng of posphorylated dT(12-18) oligos. Samples were denatured at 65°C, renatured at 42°C, incubated for 60 min in the presence of T4-DNA ligase. The Adapter-(dT)15 oligo was added, incubated for 2 h at 12°C. Then, Superscript III Transcriptase (Invitrogen) was added and samples were incubated for further 2 hours at 42°C. The cDNA generated was used for the PCR reactions.
To generate cDNAs starting at the cleavage site for the shortest possible LM-PAT product, we synthesized additional cDNAs with an anchor-(dT)12VN primer. The PCR product generated from this “clamped” cDNA is labeled “TVN” in Figure 5C.
For the TVN and LM-PAT Add2 PCR reactions, the mBRA4short dir 2 and Adapter or Anchor primers were used. The PCR reaction conditions were as follows: 35-45 cycles, 94°C, 30 sec, 59°C, 30 sec; 72°C, 1 min. For the β-actin polyA tail length assay, we used the same anchor-(dT)12VN and Adapter-(dT)15 primers used above, with the h_β-ACTIN dir primer.
RNA-EMSA
RNA-EMSAs were performed according to procedures already described.15 Radioactively labeled RNA fragments were incubated for 20 minutes at room temperature in EMSA binding buffer (5.2 mM Hepes pH 7.9, 1 mM MgCl2, 0.8 mM MgAc, 0.52 mM DTT, 3.8% glycerol, 0.75 mM ATP, 1 mM GTP, heparin 5-15 µg/µl) with the recombinant protein (final volume, 20 µl). After incubation, the RNA-protein complexes were run on native polyacrylamide gels (5%). Gels were dried before X-OMAT film or Cyclone (Packard) exposure. Plasmid encoding GST-TDP-43 recombinant protein was a gift from Prof. Baralle, ICGEB, Italy.29 Recombinant proteins were expressed and purified with glutathione S–Sepharose 4B beads (Pharmacia) according to the manufacturer's instructions.
Immunoprecipitation
HeLa cells were transfected with a pFLAG-TDP-43 construct and original pFLAG vector as a control. Transfected cell lysates (400 μg) were pre-incubated with agarose beads and with or without HeLa nuclear extract (120 μg, Cilbiotech) for 1 hour at 4°C. HeLa nuclear extract was added to enrich the amount of polyadenylation factors and TDP-43 and so to enhance the interactions. After centrifugation, the obtained pre-absorbed lysate was mixed with or without RNAse A for 30 min at 4°C (mix A). In parallel a second mix (mix B) was obtained incubating agarose beads, BSA (100 μg) and anti FLAG antibody or anti tubulin as a control for 30 min at 4°C. The mix A was combined to mix B and incubated 24 hours at 4°C. After centrifugation and washing steps, elution was performed with the FLAG peptide in PBS, 1 hour at 4°C. The eluted samples were separate by SDS-PAGE and the complexes formation in the different conditions were analyzed by immunoblotting.
Protein preparation from cells, SDS-PAGE and western blot
Total protein extract from cells and Western blot analyses were performer as previously described.15 For detection of proteins of interest the following primary antibodies were used: anti TDP-43 (ProteinTech), anti-β-tubulin mAb E7 (Developmental Studies Hybridoma Bank, University of Iowa), and monoclonal anti-FLAG (Sigma-Aldrich). Secondary HRP-coupled antibodies (DAKO) were used for chemiluminescence detection (ECL Western Blot Substrates (Thermo Scientific)]. The results were quantified by with Quantity One software (Bio-Rad Laboratories).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We wish to thank Drs. Francisco Baralle and the Molecular Pathology Group for experimental suggestions and comments, and for kindly providing us with the GST-TDP-43 pFLAG-TDP-43 construct and TDP-43 siRNA protocols.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Keene JD. RNA regulons: coordination of post-transcriptional events. Nat Rev Genet 2007; 8:533-43; PMID:17572691; http://dx.doi.org/10.1038/nrg2111 [DOI] [PubMed] [Google Scholar]
- 2.Wu X, Brewer G. The regulation of mRNA stability in mammalian cells: 2.0. Gene 2012; 500:10-21; PMID:22452843; http://dx.doi.org/10.1016/j.gene.2012.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lenzken SC, Achsel T, Carri MT, Barabino SM. Neuronal RNA-binding proteins in health and disease. Wiley Interdiscip Rev RNA 2014; PMID:24687864; 10.1002/wrna.1231 [DOI] [PubMed] [Google Scholar]
- 4.Buratti E, Baralle FE. The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol 2010; 7:420-9; PMID:20639693; http://dx.doi.org/10.4161/rna.7.4.12205 [DOI] [PubMed] [Google Scholar]
- 5.Hanson KA, Kim SH, Tibbetts RS. RNA-binding proteins in neurodegenerative disease: TDP-43 and beyond. Wiley Interdiscip Rev RNA 2012; 3:265-85; PMID:22028183; http://dx.doi.org/10.1002/wrna.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fiesel FC, Kahle PJ. TDP-43 and FUS/TLS: cellular functions and implications for neurodegeneration. FEBS J 2011; 278:3550-68; PMID:21777389; http://dx.doi.org/10.1111/j.1742-4658.2011.08258.x [DOI] [PubMed] [Google Scholar]
- 7.Buratti E, Baralle FE. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem 2001; 276:36337-43; PMID:11470789; http://dx.doi.org/10.1074/jbc.M104236200 [DOI] [PubMed] [Google Scholar]
- 8.Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C, et al. . Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 2011; 14:459-68; PMID:21358643; http://dx.doi.org/10.1038/nn.2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sephton CF, Cenik C, Kucukural A, Dammer EB, Cenik B, Han Y, Dewey CM, Roth FP, Herz J, Peng J, et al. . Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J Biol Chem 2011; 286:1204-15; PMID:21051541; http://dx.doi.org/10.1074/jbc.M110.190884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, Konig J, Hortobagyi T, Nishimura AL, Zupunski V, et al. . Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci 2011; 14:452-8; PMID:21358640; http://dx.doi.org/10.1038/nn.2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao S, Sanelli T, Dib S, Sheps D, Findlater J, Bilbao J, Keith J, Zinman L, Rogaeva E, Robertson J. RNA targets of TDP-43 identified by UV-CLIP are deregulated in ALS. Mol Cell Neurosci 2011; 47:167-80; PMID:21421050; http://dx.doi.org/10.1016/j.mcn.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 12.Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurology 2010; 9:995-1007; PMID:20864052; http://dx.doi.org/10.1016/S1474-4422(10)70195-2 [DOI] [PubMed] [Google Scholar]
- 13.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. . Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314:130-3; PMID:17023659; http://dx.doi.org/10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- 14.Lee EB, Lee VM, Trojanowski JQ. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci 2012; 13:38-50; PMID:22127299; http://dx.doi.org/10.1038/nrn3121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Costessi L, Porro F, Iaconcig A, Nedeljkovic M, Muro AF. Characterization of the distal polyadenylation site of the ss-adducin (Add2) pre-mRNA. PloS one 2013; 8:e58879; PMID:23554949; http://dx.doi.org/10.1371/journal.pone.0058879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Avendano-Vazquez SE, Dhir A, Bembich S, Buratti E, Proudfoot N, Baralle FE. Autoregulation of TDP-43 mRNA levels involves interplay between transcription, splicing, and alternative polyA site selection. Genes Dev 2012; 26:1679-84; PMID:22855830; http://dx.doi.org/10.1101/gad.194829.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilligan DM, Lozovatsky L, Gwynn B, Brugnara C, Mohandas N, Peters LL. Targeted disruption of the β adducin gene (Add2) causes red blood cell spherocytosis in mice. Proc Natl Acad Sci U S A 1999; 96:10717-22; PMID:10485892; http://dx.doi.org/10.1073/pnas.96.19.10717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muro AF, Marro ML, Gajovic S, Porro F, Luzzatto L, Baralle FE. Mild spherocytic hereditary elliptocytosis and altered levels of α- and gamma-adducins in β-adducin-deficient mice. Blood 2000; 95:3978-85; PMID:10845937 [PubMed] [Google Scholar]
- 19.Tripodi G, Piscone A, Borsani G, Tisminetzky S, Salardi S, Sidoli A, James P, Pongor S, Bianchi G, Baralle FE. Molecular cloning of an adducin-like protein: evidence of a polymorphism in the normotensive and hypertensive rats of the Milan strain. Biochem Biophys Res Commun 1991; 177:939-47; PMID:2059221; http://dx.doi.org/10.1016/0006-291X(91)90629-L [DOI] [PubMed] [Google Scholar]
- 20.Matsuoka Y, Li X, Bennett V. Adducin is an in vivo substrate for protein kinase C: phosphorylation in the MARCKS-related domain inhibits activity in promoting spectrin-actin complexes and occurs in many cells, including dendritic spines of neurons. J Cell Biol 1998; 142:485-97; PMID:9679146; http://dx.doi.org/10.1083/jcb.142.2.485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seidel B, Zuschratter W, Wex H, Garner CC, Gundelfinger ED. Spatial and sub-cellular localization of the membrane cytoskeleton-associated protein α-adducin in the rat brain. Brain Res 1995; 700:13-24; PMID:8624703; http://dx.doi.org/10.1016/0006-8993(95)00962-P [DOI] [PubMed] [Google Scholar]
- 22.Bednarek E, Caroni P. β-Adducin is required for stable assembly of new synapses and improved memory upon environmental enrichment. Neuron 2011; 69:1132-46; PMID:21435558; http://dx.doi.org/10.1016/j.neuron.2011.02.034 [DOI] [PubMed] [Google Scholar]
- 23.Pielage J, Bulat V, Zuchero JB, Fetter RD, Davis GW. Hts/Adducin controls synaptic elaboration and elimination. Neuron 2011; 69:1114-31; PMID:21435557; http://dx.doi.org/10.1016/j.neuron.2011.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Porro F, Rosato-Siri M, Leone E, Costessi L, Iaconcig A, Tongiorgi E, Muro AF. β-adducin (Add2) KO mice show synaptic plasticity, motor coordination and behavioral deficits accompanied by changes in the expression and phosphorylation levels of the α- and gamma-adducin subunits. Genes, Brain Behav 2010; 9:84-96; PMID:19900187; http://dx.doi.org/10.1111/j.1601-183X.2009.00537.x [DOI] [PubMed] [Google Scholar]
- 25.Rabenstein RL, Addy NA, Caldarone BJ, Asaka Y, Gruenbaum LM, Peters LL, Gilligan DM, Fitzsimonds RM, Picciotto MR. Impaired synaptic plasticity and learning in mice lacking β-adducin, an actin-regulating protein. J Neurosci 2005; 25:2138-45; PMID:15728854; http://dx.doi.org/10.1523/JNEUROSCI.3530-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Costessi L, Devescovi G, Baralle FE, Muro AF. Brain-specific promoter and polyadenylation sites of the β-adducin pre-mRNA generate an unusually long 3′-UTR. Nucleic Acids Res 2006; 34:243-53; PMID:16414955; http://dx.doi.org/10.1093/nar/gkj425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JR, 3rd, Frank J, Manley JL. Molecular architecture of the human pre-mRNA 3' processing complex. Molecular cell 2009; 33:365-76; PMID:19217410; http://dx.doi.org/10.1016/j.molcel.2008.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nedeljkovic M, Costessi L, Iaconcig A, Porro F, Muro AF. Long-distance regulation of Add2 pre-mRNA3′end processing. RNA Biol 2013; 10:516-27; PMID:23411391; http://dx.doi.org/10.4161/rna.23855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ayala YM, De Conti L, Avendano-Vazquez SE, Dhir A, Romano M, D'Ambrogio A, Tollervey J, Ule J, Baralle M, Buratti E, et al. . TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J 2011; 30:277-88; PMID:21131904; http://dx.doi.org/10.1038/emboj.2010.310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colombrita C, Onesto E, Megiorni F, Pizzuti A, Baralle FE, Buratti E, Silani V, Ratti A. TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J Biol Chem 2012; 287:15635-47; PMID:22427648; http://dx.doi.org/10.1074/jbc.M111.333450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strong MJ, Volkening K, Hammond R, Yang W, Strong W, Leystra-Lantz C, Shoesmith C. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol Cell Neurosci 2007; 35:320-7; PMID:17481916; http://dx.doi.org/10.1016/j.mcn.2007.03.007 [DOI] [PubMed] [Google Scholar]
- 32.Dani C, Blanchard JM, Piechaczyk M, El Sabouty S, Marty L, Jeanteur P. Extreme instability of myc mRNA in normal and transformed human cells. Proc Natl Acad Sci U S A 1984; 81:7046-50; PMID:6594679; http://dx.doi.org/10.1073/pnas.81.22.7046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 1995; 69:3584-96; PMID:7745706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J 2001; 20:1774-84; PMID:11285240; http://dx.doi.org/10.1093/emboj/20.7.1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buratti E, De Conti L, Stuani C, Romano M, Baralle M, Baralle F. Nuclear factor TDP-43 can affect selected microRNA levels. FEBS J 2010; 277:2268-81; PMID:20423455; http://dx.doi.org/10.1111/j.1742-4658.2010.07643.x [DOI] [PubMed] [Google Scholar]
- 36.Wang IF, Wu LS, Chang HY, Shen CK. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem 2008; 105:797-806; PMID:18088371; http://dx.doi.org/10.1111/j.1471-4159.2007.05190.x [DOI] [PubMed] [Google Scholar]
- 37.Ayala YM, Pantano S, D'Ambrogio A, Buratti E, Brindisi A, Marchetti C, Romano M, Baralle FE. Human, Drosophila, and C.elegans TDP43: nucleic acid binding properties and splicing regulatory function. J Mol Biol 2005; 348:575-88; PMID:15826655; http://dx.doi.org/10.1016/j.jmb.2005.02.038 [DOI] [PubMed] [Google Scholar]
- 38.Bhardwaj A, Myers MP, Buratti E, Baralle FE. Characterizing TDP-43 interaction with its RNA targets. Nucleic Acids Res 2013; 41:5062-74; PMID:23519609; http://dx.doi.org/10.1093/nar/gkt189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Freibaum BD, Chitta RK, High AA, Taylor JP. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res 2010; 9:1104-20; PMID:20020773; http://dx.doi.org/10.1021/pr901076y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Volkening K, Leystra-Lantz C, Yang W, Jaffee H, Strong MJ. Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res 2009; 1305:168-82; PMID:19815002; http://dx.doi.org/10.1016/j.brainres.2009.09.105 [DOI] [PubMed] [Google Scholar]
- 41.Morgan M, Iaconcig A, Muro AF. Identification of 3′ gene ends using transcriptional and genomic conservation across vertebrates. BMC Genomics 2012; 13:708; PMID:23244605; http://dx.doi.org/10.1186/1471-2164-13-708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ozsolak F, Kapranov P, Foissac S, Kim SW, Fishilevich E, Monaghan AP, John B, Milos PM. Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell 2010; 143:1018-29; PMID:21145465; http://dx.doi.org/10.1016/j.cell.2010.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ayala YM, Pagani F, Baralle FE. TDP43 depletion rescues aberrant CFTR exon 9 skipping. FEBS Lett 2006; 580:1339-44; PMID:16458894; http://dx.doi.org/10.1016/j.febslet.2006.01.052 [DOI] [PubMed] [Google Scholar]
- 44.Ayala YM, Zago P, D'Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci 2008; 121:3778-85; PMID:18957508; http://dx.doi.org/10.1242/jcs.038950 [DOI] [PubMed] [Google Scholar]
- 45.Beilharz TH, Preiss T. Transcriptome-wide measurement of mRNA polyadenylation state. Methods 2009; 48:294-300; PMID:19233282; http://dx.doi.org/10.1016/j.ymeth.2009.02.003 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.