

Abstract
The bacterial genus Polaribacter is distributed widely in marine environments; however, there have been no reports of phages infecting Polaribacter strains. Here, we describe the isolation and genome sequencing of two lytic siphophages, P12002L and P12002S, that infect Polaribacter sp. strain IMCC12002. The two phages and host strain were isolated from coastal seawater of Korea. Complete genome sequences of the two phages were similar to each other and about 50 kb in length, with a G + C content of 28.9 %. The two genomes showed typical characteristics of phage genomes: a modular structure and high proportion of hypothetical proteins. The genome sequences have been deposited in GenBank under accession numbers KR136259 (P12002L) and KR136260 (P12002S).
Electronic supplementary material
The online version of this article (doi:10.1186/s40793-015-0076-z) contains supplementary material, which is available to authorized users.
Keywords: Bacteriophage, Polaribacter, Bacteroidetes, Genome, Siphoviridae
Introduction
Viruses are the most numerous biological entities in the marine water column. The number of virus-like particles is known to exceed that of prokaryotes by approximately 10-fold on average [1]. Recent metagenomic studies on marine viruses have added an immense amount of novel sequence data to public databases [2]. However, the functional and phylogenetic assignment of these novel viral sequences is daunting, partly due to the shortage of available reference genomes [3]. Since most viruses in marine environments are believed to be bacteriophages [4], isolation and genome-based characterization of phages infecting widespread marine bacterial groups are important for a thorough understanding of marine viral diversity.
The genus Polaribacter, affiliated with the family Flavobacteriaceae of the phylum Bacteroidetes, currently contains 14 validly published species identified from marine environments. Several studies have shown that the genus is widely distributed in marine ecosystems, including polar seas and the North Sea [5–7], indicating that the genus Polaribacter may play important ecological roles and also suggesting the importance of any Polaribacter phages.
To our knowledge, however, no phages infecting the genus Polaribacter have been reported. In fact, phages infecting the family Flavobacteriaceae, to which Polaribacter belongs, have rarely been isolated, and currently only a limited number of bacterial groups within the Flavobacteriaceae are known to have phages infecting them [8]. This situation has posed a conundrum in virome studies of marine environments, since the family Flavobacteriaceae is one of the major bacterial assemblages found in the marine water column [9, 10].
Here, we report the genome sequences of two lytic bacteriophages, P12002L and P12002S, that infect strain IMCC12002, a marine bacterium phylogenetically assigned to the genus Polaribacter. The genome sequences of phages P12002L and P12002S represent the first addition of Polaribacter phage genomes to public sequence databases.
Virus information
Classification and features
P12002L and P12002S are lytic phages that infect the bacterial strain Polaribacter sp. IMCC12002. A coastal seawater sample collected off Incheon Harbor, located on the west coast of South Korea, was serially diluted with autoclaved seawater and spread onto marine agar plates (Difco). Strain IMCC12002 was established from a colony grown on a plate after two weeks of incubation at 20 °C in the dark by three rounds of plate streaking. Phylogenetic analyses based on 16S rRNA gene sequences placed strain IMCC12002 within the genus Polaribacter of the family Flavobacteriaceae. Sequence similarities of the 16S rRNA gene between IMCC12002 and type strains of the genus Polaribacter, calculated using EzTaxon-e server [11], ranged from 95.0 % to 98.4 %, with a maximum value found for Polaribacter irgensii 23-PT [12]. Strain IMCC12002 was routinely grown in mR2A media.
Phages P12002L and P12002S were isolated from a surface seawater sample collected from the same station where the host strain IMCC12002 was isolated previously. An enrichment culture was performed to increase the concentration of phages putatively infecting strain IMCC12002 prior to plaque assay. Broth culture of IMCC12002 (20 ml) in the exponential phase was mixed with 380 ml of a 0.2-μm filtered seawater sample and 100 ml of 5× mR2A broth, and was further incubated at 20 °C for a week. During incubation, 10 ml of culture broth was withdrawn three times (at 2-day intervals) and vortexed for 5 min after the addition of 2 ml of chloroform. The chloroform was separated by centrifugation and the aqueous phase was recovered and stored at 4 °C for subsequent use in plaque assays. Plaque assays were performed using the double agar layer method. Bottom and top agar layers were prepared by the addition of Bacto Agar (BD Difco) to mR2A broth, at concentrations of 1.5 % and 0.7 % (w/v), respectively. Exponentially growing IMCC12002 (0.5 ml) and the chloroform-treated enrichment culture (0.5 ml; diluted if necessary) were added to 6 ml of molten (50 °C) top agar and poured onto bottom agar plates. After solidification of the top agar layer, plates were incubated at 20 °C for a week. Two plaques were selected from the assay plates, then were further purified through three cycles of picking, elution, dilution, and plaque assay, and were established and designated phages P12002L and P12002S. The two phages formed clear plaques on the host bacterial lawn after 1–2 days of incubation at 20 °C. The diameters of plaques were 1–2 mm for both phages, although the two phages seemed to show a difference in plaque size on the original assay plates.
Morphological and genomic characterization were performed for classification of the two phages, P12002L and P12002S, using viral particles amplified and purified as described below (see Genome sequencing information). For morphological characterization, purified phage particles were adsorbed onto 200-mesh formvar and carbon-coated copper grids (Electron Microscopy Sciences), stained with uranyl acetate solution (2 %, w/v), and then examined by transmission electron microscope (CM200, Phillips). Both phages had isometric heads of about 55 nm in diameter and long non-contractile tails of about 150 nm in length (Fig. 1). For genomic characterization, nucleic acids were extracted from phage particles and treated with restriction enzymes. The genomic material of both phages were digested by enzymes such as Nde I and Hha I, showing that the two phages possessed dsDNA as their genomic material. Taken together, these results indicated the affiliation of these phages with the family Siphoviridae [13].
Fig. 1.
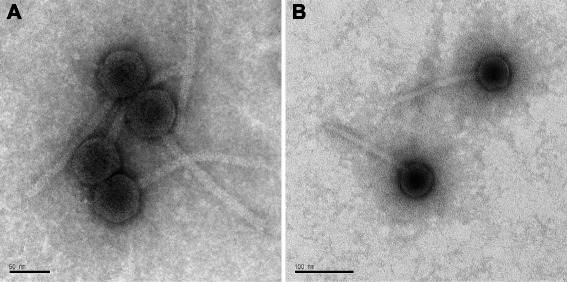
Transmission electron micrographs of Polaribacter phages P12002L (a) and P12002S (b). Scale bar, 50 nm in (a) and 100 nm in (b)
Genes encoding TerL were predicted from the genomes of both phages and were used for inference of the phylogenetic positions of the phages. Although mosaicism is known to be rather prevalent in phage genomes [14] and the family Siphoviridae is underexplored with regard to marker (signature) genes [15], the terminase large subunit gene has been widely used as a marker gene in phage studies [16, 17]. TerL amino acid sequences of the two phages were identical and only distantly related to those of other phages representing the diverse genera of Siphoviridae [18] (Fig. 2). The most similar TerL proteins among prokaryotic isolates were found mainly in the genomes of Bacteroides, most of which were isolated from animals including humans. Among phage isolates included in the nr database of GenBank, the highest similarities were recorded for Cellulophaga phage phi10:1 [8] and Flavobacterium phage 11b [19], both of which were isolated from marine environments and that are not yet assigned to any known genera of Siphoviridae. Taken together, TerL-based phylogeny suggested that P12002L and P12002S, the siphoviruses isolated in this study, occupied a phylogenetic position distinct from previously established genera of the family Siphoviridae.
Fig. 2.
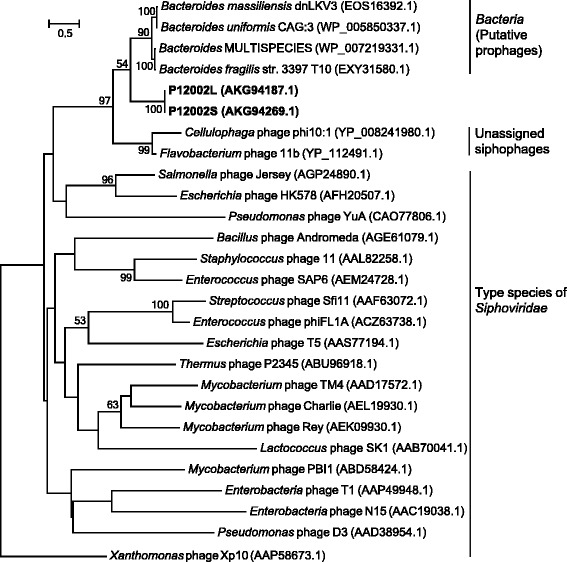
Maximum-likelihood phylogenetic tree based on TerL protein sequences indicating the position of P12002L and P12002S (shown in bold). All sequences, except for P12002L and P12002S, were obtained from GenBank. Sequences were aligned by MUSCLE [36]. Tree building was performed by RAxML [37], using the “-f a” method, PROTGAMMAJTT model, and MRE-based bootstopping criterion. Bootstrap replicate searches were performed 400 times, and bootstrap values (>50 %) are shown at branch nodes. Xanthomonas phage Xp10 was set as the root after tree building. Bar, 0.5 substitutions per amino acid position
Information on the isolation, classification, and general features of the two phages are presented in Table 1.
Table 1.
Classification and general features of phages P12002L and P12002S according to the MIGS recommendations [39]
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain: unassigned (ds DNA viruses) | ||
| Phylum: unassigned | |||
| Class: unassigned | |||
| Order Caudovirales | TAS [13] | ||
| Family Siphoviridae | TAS [13] | ||
| Genus: unassigned | |||
| Species: unassigned | |||
| Strains: P12002L, P12002S | |||
| Particle shape | Isometric capsid with a long non-contractile tail | IDA | |
| MIGS-6 | Habitat | Marine water column | IDA |
| MIGS-15 | Biotic relationship | Intracellular parasite of Polaribacter strain IMCC12002 | IDA |
| MIGS-14 | Pathogenicity | Non-pathogenic | NAS |
| MIGS-4 | Geographic location | The Yellow Sea, Incheon, South Korea | IDA |
| MIGS-5 | Sample collection | March 16, 2010 | IDA |
| MIGS-4.1 | Latitude | 37° 29′ 51′′ N | IDA |
| MIGS-4.2 | Longitude | 126° 38′ 26′′ E | IDA |
| MIGS-4.3 | Depth | 0.3 m | IDA |
| MIGS-4.4 | Altitude | - | - |
aEvidence codes - IDA Inferred from Direct Assay, TAS Traceable Author Statement, NAS Non-traceable Author Statement. These evidence codes are from the Gene Ontology project [40]
Genome sequencing information
Genome project history
Genome sequencing of the two phages, P12002L and P12002S, was performed as a part of a research project that aimed to increase the number of marine phage genomes in public databases, which is expected to lay the foundation for a more thorough understanding of the (meta)genomic diversity in marine environments, as exemplified by studies on pelagiphages and SAR116 phages [20–22]. Previously, we reported the genome sequences of several other marine phages infecting diverse host bacteria [23–25], which have been utilized for marine virus studies [26, 27].
Genome sequencing of P12002L and P12002S was performed using an Illumina HiSeq platform using a 100-bp paired-end library generated from DNA samples extracted from concentrated and purified phage particles. Assembly by SOAPdenovo [28], followed by gap closing by PCR, resulted in a single circular contig for each phage. Gene calling and annotation were carried out mainly using the RAST server [29]. The complete genome sequences and annotation information of both phages were submitted to GenBank, with accession numbers KR136259 (P12002L) and KR136260 (P12002S). Information on these genome sequencing projects is available through the Genomes OnLine Database under project numbers Gp0115710 (P12002L) and Gp0115711 (P12002S), and a summary of these projects is shown in Table 2.
Table 2.
Project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Finished |
| Number of contigs | 1 | |
| MIGS-28 | Libraries used | One paired-end Illumina library (per each phage) |
| MIGS-29 | Sequencing platforms | Illumina HiSeq |
| MIGS-31.2 | Fold coverage | P12002L, ~29,000×; P12002S, ~33,000× |
| MIGS-30 | Assemblers | SOAPdenovo |
| MIGS-32 | Gene calling method | RAST gene caller |
| GenBank ID | P12002L, KR136259; P12002S, KR136260 | |
| GenBank Date of Release | May 16, 2015 | |
| GOLD ID | P12002L, Gp0115710; P12002S, Gp0115711 | |
| MIGS-13 | Source material identifiera | P12002L, P12002S |
| Project relevance | Diversity of marine bacteriophage |
aViruses have not been deposited yet
Growth conditions and genomic DNA preparation
Phages P12002L and P12002S, isolated as described above, were subsequently amplified in liquid culture using bacterial strain IMCC12002 as a host. IMCC12002 was grown at 20 °C with shaking at 100 rpm in 200 ml of mR2A broth until the late lag or early exponential phase before concentrated phage particles were inoculated. Host bacteria were clearly lysed within 24 h of phage inoculation. After lysis, extracellular nucleic acids were digested by the addition of DNase I (Sigma) and RNase A (Qiagen) (each 1 μg ml−1), followed by incubation at 37 °C for 1 h. Subsequently, sodium chloride was added at a final concentration of 0.5 M and lysed cultures were stored in iced water for 1 h before removal of cellular debris by centrifugation (12,000 × g, 30 min). PEG 8000 (Sigma) was added to recovered supernatants (10 %, w/v) and the mixture was stored at 4 °C overnight. Phage particles were pelleted by centrifugation (12,000 × g, 30 min). After discarding supernatants, pellets were resuspended in SM buffer. PEG was removed by treatment with an equal volume of chloroform, followed by phase separation by centrifugation (2,500 × g, 1 h). Phage particles in the aqueous phase were further purified by equilibrium gradient ultracentrifugation (SW 55 Ti Rotor, 40,000 rpm, 24 h, 4 °C) after addition of CsCl (Sigma; 0.75 g per ml of phage concentrates). Phage bands formed in ultracentrifuge tubes were withdrawn with syringes. After removal of CsCl by repeated ultrafiltration, phage samples were used for DNA extraction with a DNeasy Blood & Tissue Kit (Qiagen).
Genome sequencing and assembly
Extracted DNA samples were used for generation of shotgun libraries (one library for each phage), which were sequenced (2× 100 bp) by Macrogen, Inc. (Korea) using an Illumina HiSeq platform. In total, about 7.1 and 8.3 million pairs of reads were obtained for P12002L and P12002S, respectively. Raw reads were assembled by SOAPdenovo after being partitioned into subsets composed of 50,000–200,000 paired reads, with a k-mer value of 85. Partition seemed to be necessary since the assembly of non-partitioned whole sequence data resulted in a greater number of shorter contigs when compared to the assembly results obtained from partitioned subsets. For both phages, several partitioned subsets of different sizes were assembled separately, which produced nearly the same single contig for all subsets. Primers were designed from the ends of contigs with an outward orientation and used in PCR, with the genomic DNA of phages used as templates. The sequences of PCR products were determined by Sanger sequencing and were used to close the circular contigs for both phages, eventually resulting in a single contig for each phage. Coverages, roughly estimated from the sequencing statistics and final contig sizes, were about 29,000 and 33,000 for P12002L and P12002S, respectively (Table 2).
Genome annotation
Gene calling and annotation of the complete phage genomes were performed by the RAST server with FIGfam version of release 70. Gene caller was set to ‘RAST’, and ‘Fix frameshifts’ and ‘Backfill gaps’ were selected. Functional annotation of protein coding genes was improved by RPS-BLAST searches against the Conserved Domain Database (CDD), available through the web interface [30], and HMMER searches against UniProtKB [31], the latter of which was also used to predict signal peptides and transmembrane helices by the Phobius program [32]. BLASTp searches against NCBI nr database were also performed. To calculate COG statistics from RPS-BLAST results, two files downloaded from the NCBI FTP server were used: “cognames2003–2014.tab” and “fun2003–2014.tab” [33]. Coding densities of the genomes were calculated using the “Create Intergenic Features” function of Artemis [34], based on Genbank files generated by the RAST server.
Genome properties
The complete genomes of two phages were assembled into single circular contigs of 48,689 bp (P12002L) and 49,847 bp (P12002S) in length. Considering that the genomes of tailed dsDNA phages are known to be linear, this circular assembly suggests that the genomes were circularly permuted or terminally redundant [35]. The G + C contents of the two genomes were 28.9 %. The number of protein coding genes predicted in the two phage genomes were 82 (P12002L) and 86 (P12002S). No RNA genes or pseudogenes were predicted. Among the protein coding genes, only 16 and 14 genes in P12002L and P12002S, respectively, were assigned putative functions, while the other genes were annotated as hypothetical proteins. The annotation summaries for the two genomes are presented in Table 3 and detailed annotation information is shown in Additional file 1: Tables S1 and S2. Distributions of the protein functions among COG functional categories are shown in Table 4.
Table 3.
Genome statistics
| Attribute | P12002L | P12002S | ||
|---|---|---|---|---|
| Value | % of Totala | Value | % of Totala | |
| Genome size (bp) | 48,689 | 100.00 | 49,847 | 100.00 |
| DNA coding (bp) | 43,666 | 89.68 | 44,538 | 89.35 |
| DNA G + C (bp) | 14,088 | 28.93 | 14,422 | 28.93 |
| DNA scaffolds | 1 | 100.00 | 1 | 100.00 |
| Total genes | 82 | 100.00 | 86 | 100.00 |
| Protein coding genes | 82 | 100.00 | 86 | 100.00 |
| RNA genes | 0 | 0.00 | 0 | 0.00 |
| Pseudo genes | 0 | 0.00 | 0 | 0.00 |
| Genes in internal clusters | 0 | 0.00 | 0 | 0.00 |
| Genes with function prediction | 16 | 19.51 | 14 | 16.28 |
| Genes assigned to COGs | 9 | 10.98 | 9 | 10.47 |
| Genes with Pfam domains | 18 | 21.95 | 15 | 17.44 |
| Genes with signal peptides | 1 | 1.22 | 0 | 0.00 |
| Genes with transmembrane helices | 4 | 4.89 | 8 | 9.30 |
| CRISPR repeats | 0 | 0.00 | 0 | 0.00 |
aThe total is based on the size of the genome in base pairs or the total number of protein-coding genes in the annotated genome
Table 4.
Number of genes associated with general COG functional categories
| Code | P12002L | P12002S | Description | ||
|---|---|---|---|---|---|
| Value | % age | Value | % age | ||
| J | 0 | 0 | 0 | 0 | Translation, ribosomal structure and biogenesis |
| A | 0 | 0 | 0 | 0 | RNA processing and modification |
| K | 0 | 0 | 0 | 0 | Transcription |
| L | 5 | 6.10 | 6 | 6.98 | Replication, recombination and repair |
| B | 0 | 0 | 0 | 0 | Chromatin structure and dynamics |
| D | 1 | 1.22 | 1 | 1.16 | Cell cycle control, cell division, chromosome partitioning |
| V | 1 | 1.22 | 2 | 2.33 | Defense mechanisms |
| T | 0 | 0 | 0 | 0 | Signal transduction mechanisms |
| M | 0 | 0 | 0 | 0 | Cell wall/membrane biogenesis |
| N | 0 | 0 | 0 | 0 | Cell motility |
| U | 0 | 0 | 0 | 0 | Intracellular trafficking and secretion |
| O | 0 | 0 | 0 | 0 | Posttranslational modification, protein turnover, chaperones |
| C | 0 | 0 | 0 | 0 | Energy production and conversion |
| G | 0 | 0 | 0 | 0 | Carbohydrate transport and metabolism |
| E | 0 | 0 | 0 | 0 | Amino acid transport and metabolism |
| F | 0 | 0 | 0 | 0 | Nucleotide transport and metabolism |
| H | 0 | 0 | 0 | 0 | Coenzyme transport and metabolism |
| I | 0 | 0 | 0 | 0 | Lipid transport and metabolism |
| P | 0 | 0 | 0 | 0 | Inorganic ion transport and metabolism |
| Q | 0 | 0 | 0 | 0 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 4 | 4.88 | 3 | 3.49 | General function prediction only |
| S | 0 | 0 | 1 | 1.16 | Function unknown |
| X | 1 | 1.22 | 1 | 1.16 | Mobilome: prophages, transposons |
| - | 73 | 89.02 | 77 | 89.53 | Not in COGs |
The two phage genomes showed a modular structure, were syntenic over the whole genomes, and shared many genes, including terminase, carboxypeptidase, tail length tape measure protein, methyltransferase, replication initiation protein DnaA, and endonucleases (Fig. 3).
Fig. 3.
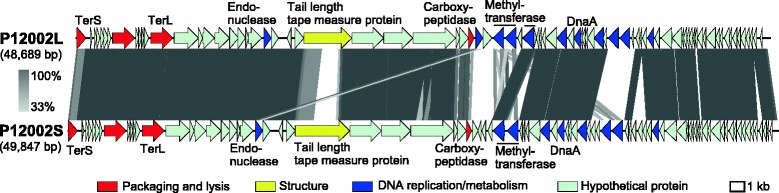
Genome maps of P12002L and P12002S. Protein coding genes are represented by arrows and their functional categories are coded in color as follows: Red, Packaging and lysis; Yellow, Structure; Blue, DNA metabolism/replication. Similarites between the two genomes were calculated based on tBLASTx and indicated in grey according to the scale at the left. This figure was drawn by Easyfig [38]
Conclusion
P12002L and P12002S, two lytic siphophages isolated from coastal seawater of Korea, infect Polaribacter sp. strain IMCC12002, and therefore represent the first phages of the genus Polaribacter, a taxonomic group found widely in the marine water column. Complete genome sequences of the two phages were obtained by Illumina sequencing, and were annotated by the RAST server and searches against various databases. The two phages showed synteny over the whole length (≈50 kb) of their genomes and shared many genes. A phylogenetic analysis of TerL protein sequences suggested that the two phages might constitute a novel genus-level group of Siphoviridae.
Acknowledgements
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (NRF-2013R1A1A2063660), and also by a grant from the Marine Biotechnology Program (PJT200620, Genome Analysis of Marine Organisms and Development of Functional Applications), funded by the Ministry of Oceans and Fisheries, Korea.
Abbreviation
- TerL
Terminase large subunit
- mR2A
R2A broth dissolved in diluted aged seawater (seawater:distilled water = 4:1)
Additional file
Detailed annotation information of the two phage genomes. (PDF 80 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HJ isolated the two phages, in addition to the host strain IMCC12002, and performed laboratory work related to genome sequencing. IK analyzed the genome sequences and drafted the manuscript. IK and J-CC together organized this study and wrote the manuscript. All authors read and approved the final manuscript.
References
- 1.Danovaro R, Corinaldesi C, Dell’Anno A, Fuhrman JA, Middelburg JJ, Noble RT, et al. Marine viruses and global climate change. FEMS Microbiol Rev. 2011;35:993–1034. doi: 10.1111/j.1574-6976.2010.00258.x. [DOI] [PubMed] [Google Scholar]
- 2.Hurwitz BL, Sullivan MB. The Pacific Ocean Virome (POV): A marine viral metagenomic dataset and associated protein clusters for quantitative viral ecology. PLoS One. 2013;8:e57355. doi: 10.1371/journal.pone.0057355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brum JR, Ignacio-Espinoza JC, Roux S, Doulcier G, Acinas SG, Alberti A, et al. Patterns and ecological drivers of ocean viral communities. Science. 2015;348:1261498. doi: 10.1126/science.1261498. [DOI] [PubMed] [Google Scholar]
- 4.Breitbart M. Marine viruses: Truth or dare. Ann Rev Mar Sci. 2012;4:425–48. doi: 10.1146/annurev-marine-120709-142805. [DOI] [PubMed] [Google Scholar]
- 5.Klindworth A, Mann AJ, Huang S, Wichels A, Quast C, Waldmann J, et al. Diversity and activity of marine bacterioplankton during a diatom bloom in the North Sea assessed by total RNA and pyrotag sequencing. Mar Genomics. 2014;18:185–92. doi: 10.1016/j.margen.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Williams TJ, Wilkins D, Long E, Evans F, DeMaere MZ, Raftery MJ, et al. The role of planktonic Flavobacteria in processing algal organic matter in coastal East Antarctica revealed using metagenomics and metaproteomics. Environ Microbiol. 2013;15:1302–17. doi: 10.1111/1462-2920.12017. [DOI] [PubMed] [Google Scholar]
- 7.Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM, et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science. 2012;336:608–11. doi: 10.1126/science.1218344. [DOI] [PubMed] [Google Scholar]
- 8.Holmfeldt K, Solonenko N, Shah M, Corrier K, Riemann L, VerBerkmoes NC, et al. Twelve previously unknown phage genera are ubiquitous in global oceans. Proc Natl Acad Sci U S A. 2013;110:12798–803. doi: 10.1073/pnas.1305956110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirchman DL. The ecology of Cytophaga–Flavobacteria in aquatic environments. FEMS Microbiol Ecol. 2002;39:91–100. doi: 10.1111/j.1574-6941.2002.tb00910.x. [DOI] [PubMed] [Google Scholar]
- 10.Yilmaz P, Iversen MH, Hankeln W, Kottmann R, Quast C, Glöckner FO. Ecological structuring of bacterial and archaeal taxa in surface ocean waters. FEMS Microbiol Ecol. 2012;81:373–85. doi: 10.1111/j.1574-6941.2012.01357.x. [DOI] [PubMed] [Google Scholar]
- 11.Kim O-S, Cho Y-J, Lee K, Yoon S-H, Kim M, Na H, et al. Introducing EzTaxon-e: a prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species. Int J Syst Evol Microbiol. 2012;62:716–21. doi: 10.1099/ijs.0.038075-0. [DOI] [PubMed] [Google Scholar]
- 12.Gosink JJ, Woese CR, Staley JT. Polaribacter gen. nov., with three new species, P. irgensii sp. nov., P. franzmannii sp. nov. and P. filamentus sp. nov., gas vacuolate polar marine bacteria of the Cytophaga-Flavobacterium-Bacteroides group and reclassification of ‘Flectobacillus glomeratus’ as Polaribacter glomeratus comb. nov. Int J Syst Evol Microbiol. 1998;48:223–35. doi: 10.1099/00207713-48-1-223. [DOI] [PubMed] [Google Scholar]
- 13.King AM, Adams MJ, Carstens EB, Lefkowitz EJ. Virus taxonomy: Ninth report of the International Committee on Taxonomy of Viruses. San Diego: Elsevier; 2012. [Google Scholar]
- 14.Hatfull GF, Hendrix RW. Bacteriophages and their genomes. Curr Opin Virol. 2011;1:298–303. doi: 10.1016/j.coviro.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adriaenssens EM, Cowan DA. Using signature genes as tools to assess environmental viral ecology and diversity. Appl Environ Microbiol. 2014;80:4470–80. doi: 10.1128/AEM.00878-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Casjens SR, Gilcrease EB, Winn-Stapley DA, Schicklmaier P, Schmieger H, Pedulla ML, et al. The generalized transducing Salmonella bacteriophage ES18: Complete genome sequence and DNA packaging strategy. J Bacteriol. 2005;187:1091–104. doi: 10.1128/JB.187.3.1091-1104.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang S, Wang K, Jiao N, Chen F. Genome sequences of siphoviruses infecting marine Synechococcus unveil a diverse cyanophage group and extensive phage–host genetic exchanges. Environ Microbiol. 2012;14:540–58. doi: 10.1111/j.1462-2920.2011.02667.x. [DOI] [PubMed] [Google Scholar]
- 18.Adriaenssens EM, Edwards R, Nash JHE, Mahadevan P, Seto D, Ackermann H-W, et al. Integration of genomic and proteomic analyses in the classification of the Siphoviridae family. Virology. 2015;477:144–54. doi: 10.1016/j.virol.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 19.Borriss M, Lombardot T, Glöckner F, Becher D, Albrecht D, Schweder T. Genome and proteome characterization of the psychrophilic Flavobacterium bacteriophage 11b. Extremophiles. 2007;11:95–104. doi: 10.1007/s00792-006-0014-5. [DOI] [PubMed] [Google Scholar]
- 20.Zhao Y, Temperton B, Thrash JC, Schwalbach MS, Vergin KL, Landry ZC, et al. Abundant SAR11 viruses in the ocean. Nature. 2013;494:357–60. doi: 10.1038/nature11921. [DOI] [PubMed] [Google Scholar]
- 21.Kang I, Oh H-M, Kang D, Cho J-C. Genome of a SAR116 bacteriophage shows the prevalence of this phage type in the oceans. Proc Natl Acad Sci U S A. 2013;110:12343–8. doi: 10.1073/pnas.1219930110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang I, Cho J-C. Depth-specific distribution of the SAR116 phages revealed by virome binning. J Microbiol Biotechn. 2014;24:592–6. doi: 10.4014/jmb.1312.12062. [DOI] [PubMed] [Google Scholar]
- 23.Kang I, Jang H, Cho J-C. Complete genome sequences of two Persicivirga bacteriophages, P12024S and P12024L. J Virol. 2012;86:8907–8. doi: 10.1128/JVI.01327-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang I, Jang H, Oh H-M, Cho J-C. Complete genome sequence of Marinomonas bacteriophage P12026. J Virol. 2012;86:8909–10. doi: 10.1128/JVI.01328-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kang I, Jang H, Oh H-M, Cho J-C. Complete genome sequence of Celeribacter bacteriophage P12053L. J Virol. 2012;86:8339–40. doi: 10.1128/JVI.01153-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chow C-ET, Winget DM, White RA, III, Hallam SJ, Suttle CA. Combining genomic sequencing methods to explore viral diversity and reveal potential virus-host interactions. Front Microbiol. 2015;6:265. doi: 10.3389/fmicb.2015.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakowski EG, Munsell EV, Hyatt M, Kress W, Williamson SJ, Nasko DJ, et al. Ribonucleotide reductases reveal novel viral diversity and predict biological and ecological features of unknown marine viruses. Proc Natl Acad Sci U S A. 2014;111:15786–91. doi: 10.1073/pnas.1401322111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–72. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aziz R, Bartels D, Best A, DeJongh M, Disz T, Edwards R, et al. The RAST server: Rapid Annotations using Subsystems Technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015;43:D222–D6. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, et al. HMMER web server: 2015 update. Nucleic Acids Res. 2015;43:W30–W8. doi: 10.1093/nar/gkv397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Käll L, Krogh A, Sonnhammer ELL. A combined transmembrane topology and signal peptide prediction method. J Mol Biol. 2004;338:1027–36. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 33.Galperin MY, Makarova KS, Wolf YI, Koonin EV. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015;43:D261–D9. doi: 10.1093/nar/gku1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream M-A, et al. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–5. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 35.Baudoux AC, Hendrix RW, Lander GC, Bailly X, Podell S, Paillard C, et al. Genomic and functional analysis of Vibrio phage SIO-2 reveals novel insights into ecology and evolution of marine siphoviruses. Environ Microbiol. 2012;14:2071–86. doi: 10.1111/j.1462-2920.2011.02685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sullivan MJ, Petty NK, Beatson SA. Easyfig: a genome comparison visualizer. Bioinformatics. 2011;27:1009–10. doi: 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotech. 2008;26:541–7. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: Tool for the unification of biology. Nat genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]