

Abstract
A growing number of gene-centric studies have highlighted the emerging significance of lncRNAs in cancer. However, these studies primarily focus on a single cancer type. Therefore, we conducted a pan-cancer analysis of lncRNAs comparing tumor and matched normal expression levels using RNA-Seq data from ∼ 3,000 patients in 8 solid tumor types. While the majority of differentially expressed lncRNAs display tissue-specific expression we discovered 229 lncRNAs with outlier or differential expression across multiple cancers, which we refer to as 'onco-lncRNAs'. Due to their consistent altered expression, we hypothesize that these onco-lncRNAs may have conserved oncogenic and tumor suppressive functions across cancers. To address this, we associated the onco-lncRNAs in biological processes based on their co-expressed protein coding genes. To validate our predictions, we experimentally confirmed cell growth dependence of 2 novel oncogenic lncRNAs, onco-lncRNA-3 and onco-lncRNA-12, and a previously identified lncRNA CCAT1. Overall, we discovered lncRNAs that may have broad oncogenic and tumor suppressor roles that could significantly advance our understanding of cancer lncRNA biology.
Keywords: bioinformatics, Cancer genomics, lncRNA, transcriptome
Introduction
Although many classes of non-coding RNAs have been implicated in cancer, long non-coding RNAs (lncRNAs) are an understudied class of genes with emerging roles in tumor biology. Recent evidence suggests that they are frequently cell-type specific, contribute important functions to numerous systems,1-7 and may interact with known cancer genes such as EZH2.8 Indeed, several well-described examples, such as HOTAIR9 and ANRIL,10 indicate that lncRNAs may be essential players in cancer biology, typically facilitating epigenetic gene repression through chromatin-modifying complexes.11 Moreover, lncRNA expression may confer clinical information about disease outcomes and have utility as diagnostic tests.9,12 Just as other non-coding RNA classes, such as oncomiRs,13 have changed the landscape of cancer research, lncRNAs may similarly play an important role in tumorigenesis. Therefore, comprehensively identifying lncRNAs that are altered in tumors and elucidating their function is a major area of biological and clinical importance.
While lncRNAs have been reported to have tissue-specific expression,5,6 a subset of the more well-characterized lncRNAs appear to be altered across multiple cancer types and display conserved oncogenic roles, such as PVT114 and MEG3.15 However, the majority of cancer lncRNA studies often take a gene-centric approach to explore the clinical and biological significance by investigating a single lncRNA within a specific cancer type. Thus, many oncogenic lncRNAs that are altered across many tumor types have been potentially overlooked. Therefore, we hypothesize that a pan-cancer analysis will reveal (i) lncRNAs previously studied in a single cancer that are actually altered in multiple cancers, (ii) previously unstudied lncRNAs altered in multiple cancers, and (iii) lncRNAs that are altered in only a single cancer type. Once an altered lncRNA has been identified, the next challenge is to elucidate its biological role, as exemplified by the relatively small number of lncRNAs with well-characterized function.16 As in vitro experiments that screen for oncogenic function are labor intensive and time consuming to conduct for numerous lncRNAs, previous studies have used guilt-by-association methods to associate a lncRNA in a pathway or biological process based on the known function of highly co-expressed protein coding genes.1,16-18 When applied across large patient cohorts, this represents an effective option for systematically predicting lncRNA function to guide subsequent functional experiments. Additionally, we identified altered lncRNAs that may be up- or down-regulated due to an amplification or deletion, respectively, and associated lncRNA expression with the mutational status of commonly mutated genes in cancer which could suggest an acquired functional role in tumors. Overall, implicating lncRNAs with previously characterized proteincoding oncogenes and tumor suppressors can place lncRNAs in the context of key biological processes and pathways that will serve as a resource for future studies of lncRNA tumor biology.
Here, we present a systematic pan-cancer analysis of lncRNAs utilizing publicly available tumor and matched normal transcriptome sequencing data across 8 cancer types from The Cancer Genome Atlas (TCGA). Our analysis revealed altered lncRNAs that are specific to a single cancer type, which could serve as putative biomarkers, as well as broadly altered lncRNAs that could serve as key oncogenes and tumor suppressors across multiple cancers. Additionally, to address the challenge of elucidating lncRNA function, we have leveraged the large cohort size to power a guilt-by-association strategy to predict lncRNA functions that are conserved across cancer types. As proof of concept of our functional predictions, we validated the role of 2 uncharacterized lncRNAs, onco-lncRNA-3 and onco-lncRNA-12, and a previously reported lncRNA, CCAT1, in S-phase cell cycle across cancer types. We envision that this work will serve as a roadmap for guiding subsequent studies exploring the oncogenic and tumor suppressive roles of broadly altered lncRNAs.
Results
LncRNA expression across cancers
To assess whether lncRNAs are recurrently altered across multiple cancer types, we conducted a pan-cancer analysis of publicly available RNA-Seq data from 2,878 tumors and 349 matched adjacent normal samples across 8 different cancers that were sequenced as part of TCGA (Table S1): bladder urothelial carcinoma19 (BLCA), breast invasive carcinoma20 (BRCA), colon and rectal adenocarcinoma21 (CRC), head and neck squamous cell carcinoma (HNSC),22 kidney renal cell carcinoma23 (KIRC), lung adenocarcinoma24 (LUAD), lung squamous cell carcinoma25 (LUSC), and uterine corpus endometrial carcinoma26 (UCEC). Although additional cancer types have been sequenced by TCGA we focused on solid tumors with available matched adjacent normal tissue that have been included as part of the TCGA Pan-Cancer analysis27 to facilitate downstream integrative analyses.
We composed a comprehensive transcriptome by merging annotated protein coding genes and lncRNAs from Ensembl,28 UCSC,29 RefSeq,30 and the Human Body Map study.6 To ensure that our analysis focused on transcripts that are reliably expressed, we applied a series of filters (see Methods) that revealed 14,128 coding genes and 1,053 lncRNAs with enriched expression in at least one cancer type (Fig. 1A). Unlike protein coding genes, which often have enriched expression in all 8 cancer types, lncRNAs are not as broadly expressed across multiple cancers, with almost 40% of lncRNAs exhibiting enriched expression in a single cancer type (Fig. 1B). These results are consistent with previous studies that have shown lncRNAs to have more tissue-specific expression patterns than protein coding genes in normal tissues.5,6 The largest proportion of lncRNAs enriched in a single cancer type belonged to KIRC, the fewest to LUAD, and the remaining cancers had a roughly equivalent number of altered lncRNAs (Fig. 1B, inset).
Figure 1.
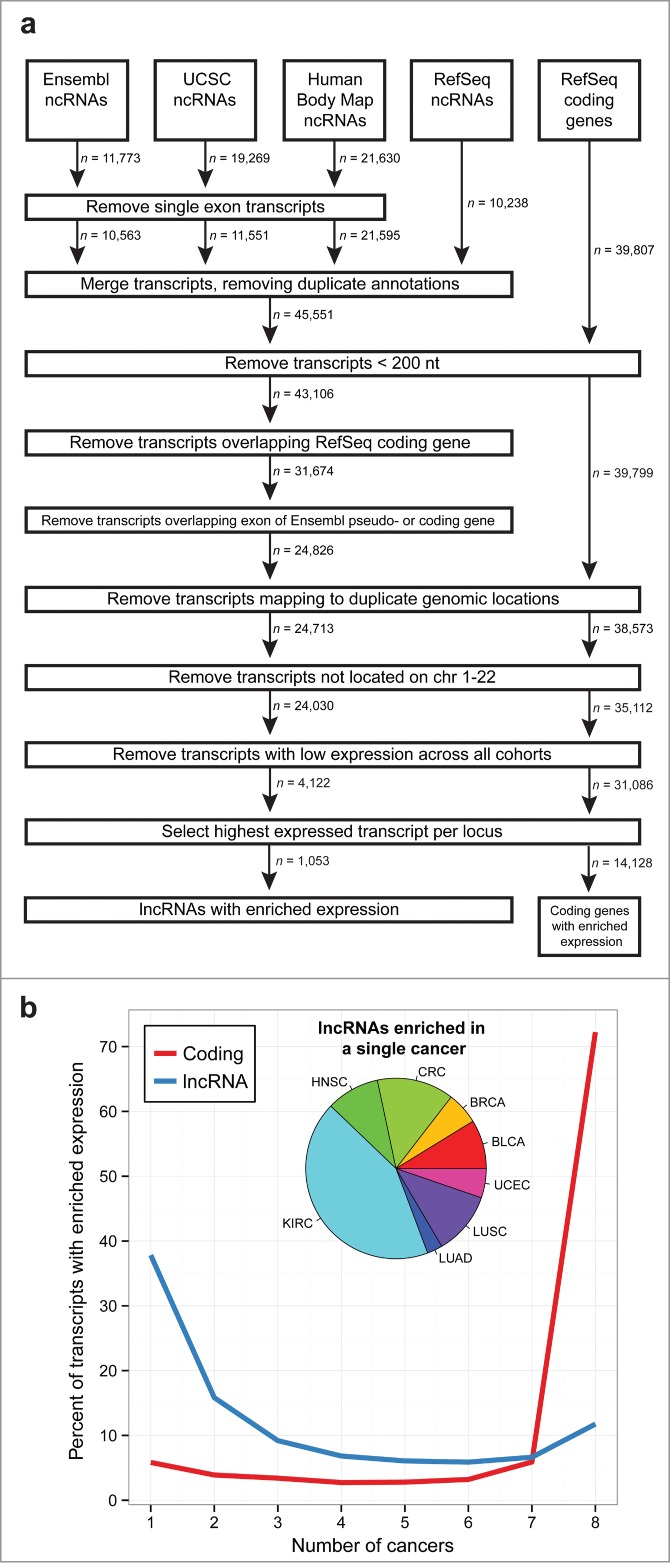
LncRNA expression across 8 cancer types. (A) Schematic of pipeline used to create a comprehensive transcriptome annotation, filter lowly expressed transcripts, and identify altered lncRNAs. (B) Distribution of the number of cancer types that lncRNAs (blue) and protein coding genes (red) are enriched in. While coding genes generally have enriched expression across all cancers, lncRNAs tend to be less broadly enriched across multiple cancers. The inset pie chart shows the distribution of lncRNAs with enriched expression in only a single cancer: bladder urothelial carcinoma (BLCA), breast invasive carcinoma (BRCA), colorectal adenocarcinoma (CRC), head and neck squamous cell carcinoma (HNSC), kidney renal cell carcinoma (KIRC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), and uterine corpus endometrial carcinoma (UCEC).
Differentially expressed LncRNAs
We next investigated which of the lncRNAs and protein coding genes with enriched expression levels were differentially expressed between the paired tumor and adjacent normal tissue samples in each cancer type. On average, we discovered 102 differentially expressed lncRNAs in each cancer type (Fig. 2A, Fig. S1, and Table S2). In contrast, we discovered approximately 1,000 differentially expressed protein coding genes in each cancer type (Fig. S2A and Table S3). Additionally, there is a larger amount of variability in the number of differentially expressed lncRNAs across the 8 cancers compared to coding genes (coefficient of variability: 1.01 vs. 0.38). This increased variability, in addition to the larger number of lncRNAs expressed in a single cancer type, suggests that lncRNAs may be playing a more active role in certain cancers compared to others.
Figure 2.
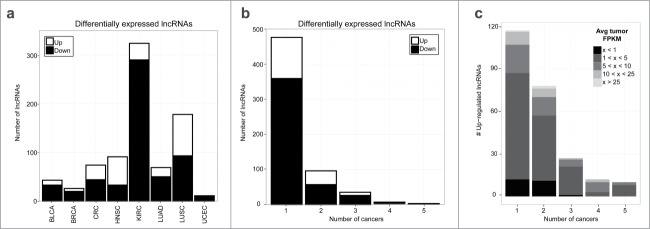
Differentially expressed lncRNAs across 8 cancer types. (A) Number of upregulated (white) and down-regulated (black) lncRNAs that are differentially expressed between paired tumor and adjacent normal samples within each cancer type. (B) Number of cancer types that each lncRNA is differentially expressed in. (C) Average tumor expression (FPKM) for up-regulated lncRNAs. For lncRNAs differentially expressed in multiple cancers, each lncRNA-cancer pair is counted separately.
When looking across all cancers, a majority of the differentially expressed lncRNAs (76%) and protein coding genes (58%) were unique to a single cancer type (Fig. 2B and Fig. S2B). Given the potential utility of lncRNAs that are highly expressed to act as biomarkers, we further evaluated lncRNAs that were differentially expressed in a single cancer type. Interestingly, a subset of the cancer-specific lncRNAs are highly expressed (Fig. 2C, Fig. S3) but have not been previously studied (Table S4 and Fig. S4). For example, TCONS_00011854 is over-expressed in CRC only (Fig. S5) and has higher tumor expression in this cohort than known colorectal cancer biomarkers CCAT131 and CRNDE.32
In addition to discovering cancer-specific lncRNAs, our analysis also revealed a subset of lncRNAs that were differentially expressed across multiple cancer types. Although several protein coding genes were differentially expressed in all 8 cancers, no lncRNAs were differentially expressed in more than 5 cancer types. Often times only a subset of patients show a marked change in gene expression due to a commonality within this subpopulation, which we refer to as 'outliers'. Therefore, to comprehensively discover all lncRNAs altered in multiple cancer types, we also identified lncRNAs with outlier expression profiles (Table S2). After combining the differential expression and outlier results, we identified 229 lncRNAs that were altered in at least 2 cancer types (Table S5). Figure 3A highlights that the altered expression of these lncRNAs are even more widespread than some well-characterized lncRNAs in cancer. We hypothesize that lncRNAs with altered expression in multiple cancer types are likely to have conserved oncogenic or tumor suppressor roles. Therefore, we will refer to these lncRNAs as 'onco-lncRNAs'. Several well-studied lncRNAs are included in our list of onco-lncRNAs, including: CCAT1,31 HULC,33 LCAL1,34 MEG3,15 and UCA1.12 In total 22 onco-lncRNAs have been previously implicated in cancer (Table S6). Additionally, many well-characterized lncRNAs, such as ANRIL and MALAT1, are altered in only a single cancer within our cohort and thus are not considered to be onco-lncRNAs. Figure 3B shows the expression levels of onco-lncRNA-1, which is up-regulated in 5 of the 8 cancers (BLCA, BRCA, LUAD, LUSC, and UCEC) but has only previously been implicated in lung cancer.34 Despite not being significant in the remaining 3 cohorts, it appears that the tumor samples in these cancer types also show a trend of higher tumor expression relative to the normal samples. Additionally, even though our differential expression analysis only included tumors with matched normal tissue, the unpaired tumor samples appear to have expression levels similar to the paired tumor samples. Onco-lncRNA-21, also known as FENDRR, has been implicated in a lethal lung development disorder35 and lung cancer.34 As shown in Figure 3C, in addition to LUAD and LUSC, onco-lncRNA-21 expression levels are also significantly downregulated in BLCA and CRC. Taken together, we have reconfirmed altered expression of lncRNAs previously implicated in cancer interspersed among many uncharacterized lncRNAs that are recurrently altered across multiple cancer types.
Figure 3.

Recurrently altered lncRNAs across 8 cancer types. (A) Heatmap showing well-characterized lncRNAs and onco-lncRNAs that are altered in 4 or more cancer types (red = upregulated, orange = up-regulated and outlier, blue = downregulated, magenta = down-regulated and outlier, gray = outlier). The top panel shows previously characterized lncRNAs and which TCGA cohorts they are altered in. The bottom panel highlights onco-lncRNAs altered in 4 or more cancer types. Boxplots showing expression levels (FPKM) of (B) onco-lncRNA-1 and (C) onco-lncRNA-21 for normal samples (N), tumor samples with a matched normal (T), and unpaired tumor samples (UT) across all 8 cancer types. Red boxes highlight upregulated cancer types and blue boxes highlight downregulated cancer types.
Next, to determine if there are any unique characteristics among the 229 onco-lncRNAs, we compared them with 424 lncRNAs that are differentially expressed in a single cancer and 400 lncRNAs that are not differentially expressed in any cancer type. Onco-lncRNAs have similar numbers of exons (Fig. S6A) and sequence conservation scores (Fig. S6B) compared to both lncRNAs that are altered in a single cancer and lncRNAs that are not altered in cancer. In general, onco-lncRNAs have similar expression levels to lncRNAs altered in a single cancer type, both of which have higher tumor expression levels in upregulated lncRNAs and lower tumor expression levels in down-regulated lncRNAs than unaltered lncRNAs (Fig. S6C and D).
To demonstrate evidence of active promoter regions, we utilized H3K4me3 Chip-seq data from multiple cancer cell lines generated by the University of Washington as part of the ENCODE project.36 Chip-seq coverage within 20kb of the transcript start sites was normalized and averaged across onco-lncRNAs, lncRNAs altered in a single cancer type, and unaltered lncRNAs. All of these groups displayed an enriched histone modification signal near transcriptional start sites (Fig. S7), across the cell line panel, compared with 500 randomly selected sites across the genome. The enrichment signal was stronger in lncRNAs that are not altered in the cancer types studied, which may be due to their consistent, but not differential, expression. The cell lines used for Chip-seq do not encompass all 8 cancer types used for characterizing onco-lncRNAs and therefore may under represent the activity of some onco-lncRNA promoters. However, some of the cancer cell lines were derived from tissues that were not one of the 8 cancer types included as part of our RNA-Seq analysis thereby revealing onco-lncRNA promoter activity, and potential expression, in additional cancer types.
Association with copy number alterations and mutational status
As copy number variation plays an important role in cancer, we next investigated how amplifications and deletions might affect the expression level of lncRNAs. Among the 1,053 expressed lncRNAs, 122 (11.6%) were located within an amplified genomic region of at least one of the cancer types in which it is expressed. Interestingly, the majority of these lncRNAs were not overexpressed in tumor samples; only 13 were up-regulated or outlier lncRNAs. However, of the 13 lncRNAs with altered expression that resided within a copy number amplification, 10 showed significant correlation between copy number and expression levels (Table S7). Among these significant correlations we observed a positive correlation of PVT1 expression with higher copy number in renal cancer (r = 0.183, P < 0.001) which was recently reported as being required for elevated MYC protein levels suggestive of its cancer relevance.37 Additionally, 70 of the downregulated or outlier lncRNAs resided within a copy number deletion, 7 of which showed significant correlation between copy number and expression levels (Table S8). Taken together, this suggests that copy number variation may be causing aberrant expression of a subset of onco-lncRNAs.
Motivated by a recent study that demonstrated the impact of oncogene-activating mutations on lncRNA expression levels,38 we also assessed whether expression levels of lncRNAs altered in at least one cancer are associated with mutational status. To accomplish this, we first identified recurrently mutated coding genes (mutated in at least 5% of tumors, as reported by a TCGA Pan-Cancer analysis27) and lncRNAs that were differentially expressed or an outlier in at least one cancer. Because some genes are highly mutated in certain cancers but not in others, each cancer type was processed separately. For each altered lncRNA and mutated gene pair we tested for a significant difference in expression levels of the lncRNA between samples that are mutated and samples with the wild type allele. We identified 231 (0.9%) significant associations between lncRNA expression levels and mutational status (Table S9). Many lncRNAs showed significant associations with multiple mutations or across multiple cancer types; therefore, this corresponds to 131 unique lncRNAs with a significant association, including 89 onco-lncRNAs (Fig. S8). For example, onco-lncRNA-1, which is upregulated in 5 cancer types, has a significant association with TP53 mutation status in BRCA, LUAD, and UCEC (Fig. 4). A recent study revealed TP53 induced expression of lincRNA-p21, which in turn mediates global repression in the TP53 response.39 Similarly, MEG3 (onco-lncRNA-83) expression was associated with TP53 mutational status and may be regulated via a TP53 binding site within the MEG3 promoter (Table S10). Interestingly, MEG3 has already been found to promote cellular proliferation and induce apoptosis in non-small cell lung carcinoma (NSCLC) by affecting TP53 target gene expression.40 This suggests that the oncogene regulates a lncRNA, MEG3, and that the lncRNA can regulate many downstream targets. Taken together, the association of lncRNA expression with mutational status potentially implicates some of the onco-lncRNAs in specific well-known cancer pathways.
Figure 4.
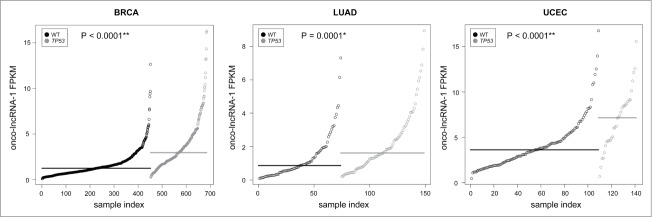
Association between onco-lncRNA-1 expression and TP53 mutational status. Plots showing expression levels (FPKM) of onco-lncRNA-1 for BRCA (left), LUAD (middle), and UCEC (right). Gray points correspond to TP53 mutated samples and black points correspond to TP53 wild type (WT) samples. Samples are ordered on the x-axis by expression within each group. Black and gray horizontal lines display the median expression across each group. P-values for each mutational association are also reported (* FDR< 0.05, ** FDR < 0.01).
Prediction of onco-lncRNA function
We hypothesized that lncRNAs altered across multiple human cancers are likely to be involved in critical oncogenic functions. Therefore, we used a 'guilt-by-association' strategy to predict onco-lncRNA function based on the function of the most highly co-expressed protein coding genes. For each of the 141 onco-lncRNAs that were differentially expressed in the same direction in at least 2 cancer types, the correlation with each protein coding gene was calculated, correlations were combined across all differentially expressed cancer types, and then Gene Set Enrichment Analysis41 (GSEA) was used to identify functional gene sets enriched with the top co-expressed genes (Table S11).
As validation of our co-expression analysis, we explored the correlations between previously characterized lncRNAs and protein-coding genes. As expected, we found that the highest positive correlation (0.724) for the lncRNA CRNDE was with the protein coding gene IRX5. As the CRNDE promoter resides in the same CpG island as that of the adjacent IRX5 gene, methylation of the promoter region results in coordinated expression.32 Similarly, we found that the highest positive correlation (0.77) for the lncRNA FENDRR was with a protein coding gene, FOXF1, transcribed bidirectionally on the opposite strand.35 Additionally, the lncRNA HOTAIR, which resides in a HOXC gene cluster, is known to be co-expressed with HOXC genes, and overlaps HOXC11, displayed its highest positive correlation with multiple HOXC genes including HOXC11 (0.86), HOXC10 (0.72), HOXC13 (0.66), HOXC8 (0.44), and HOXC9 (0.38).42
Next, we clustered the functional gene sets nominated by GSEA, which led to the identification of 3 main clusters (Fig. 5). The top concepts in Cluster 1 involve transcription, cell cycle, DNA replication, and DNA repair. Cluster 2 is driven by concepts related to G protein-coupled receptors (GPCRs), which have been implicated in cancer initiation and progression, largely through activation of AKT/mTOR, MAPK, and Hippo signaling pathways.43 Cluster 3 is largely driven by cell cycle concepts.
Figure 5.

Prediction of onco-lncRNA biological function. (A) Heatmap of functional predictions for lncRNAs differentially expressed in multiple cancers. Onco-lncRNAs are on the x-axis and GSEA functional concepts are on the y-axis. For each onco-lncRNA and functional gene set pair, a GSEA normalized enrichment score (NES) was calculated. Red represents a significant positive association, blue represents a significant negative association, and white represents no significant association. The black boxes highlight 3 distinct clusters of gene sets. (B) Top gene sets, as determined by average absolute NES, contained within each cluster from the heatmap.
Just as many of the protein coding genes that are differentially expressed in multiple cancer types are known to play a central role in cell cycle regulation, such as MYBL2,44 HJURP,45 UBE2C,46 and CDC647 (Table S12), we commonly observed that onco-lncRNAs were enriched with cell cycle gene sets. Furthermore, careful curation of the literature revealed 11 onco-lncRNAs that have been experimentally validated to show a phenotype suggestive of cell cycle upon altering lncRNA expression (Table S13).
As further validation of our functional predictions, we chose to explore the role of Colon Cancer Associated Transcript-1 (CCAT1; onco-lncRNA-40) in cell cycle regulation. Our computational analysis confirmed previous reports that CCAT1 is altered in CRC,31 LUAD,34 and LUSC34 (Fig. 6A). Analysis of CCAT1 co-expressed genes revealed many cell cycle-related gene sets that have high normalized enrichment scores (Fig. 6B). Furthermore, CCAT1 was recently shown to induce cellular proliferation in colorectal cancer by inhibiting G1 arrest.48 Therefore, as a first determination of the potential for CCAT1 to alter cell cycle in lung cancer, and thus demonstrating that the function of CCAT1 is conserved across multiple cancer types, we performed cell growth experiments in 2 lung cancer cell lines previously shown to have high expression of CCAT1.34 Greater than 50% knockdown of CCAT1 in NCI-H322M and NCI-H522 cells using 2 different small interfering RNAs (siRNAs) resulted in decreased cell growth of ∼20% and ∼40%, respectively, as measured by cell counting for 6 d (Fig. 6C–D).
Figure 6.

CCAT1 regulates cell growth in lung cancer. (A) Boxplots showing expression levels (FPKM) of CCAT1 for normal samples (N), tumor samples with a matched normal (T), and unpaired tumor samples (UT) across all 8 cancer types. Red boxes highlight up-regulated cancer types and the yellow box highlights the outlier cancer type. (B) GSEA normalized enrichment scores (NES) for cell cycle related gene sets. (C) Cell growth assay in NCI-H322M cells using CCAT1 siRNAs (* P < 0.05 by a 2-tailed Student's t-test) and qPCR validation of CCAT1 siRNA knockdown. (D) Cell growth assay in NCI-H522 cells using CCAT1 siRNAs (** P < 0.01, # P < 0.001 by a 2-tailed Student's t-test) and qPCR validation of CCAT1 siRNA knockdown. All error bars are mean ± standard error of the mean across n = 3 biological replicates.
In addition to expanding the functional role of a previously characterized lncRNA in additional cancer types, we also sought to explore the role of 2 uncharacterized onco-lncRNAs, onco-lncRNA-3 and onco-lncRNA-12. We found onco-lncRNA-12 to be up-regulated in BRCA, LUAD, and LUSC and have outlier expression in CRC (Fig. 7A). Like many of the onco-lncRNAs, we found that onco-lncRNA-12 also co-expressed with protein coding genes that were enriched for cell cycle-related gene sets (Fig. 7B). As an initial confirmation of the guilt-by-association analysis, we investigated cellular growth as an indication of alteration in cell cycle. Previously, we validated the expression of onco-lcnRNA-12 in a panel of lung cancer cell lines and found it to be upregulated relative to the control cell line BEAS-2B by quantitative PCR.34 Next, we designed 2 siRNAs that achieved greater than 60% knockdown of onco-lncRNA-12 in A549 lung cancer cells and observed a substantial decrease (>25%) in cell growth compared to scrambled control starting at Day 2 and continuing through Day 6 (Fig. 7C). To further support these findings we investigated S-phase of cell cycle by measuring EdU incorporation by flow cytometry analysis. This data confirmed earlier findings showing a 36% and 17% decrease in EdU incorporation in siRNA1 and siRNA2 (P < 0.0002), respectively, compared to the scrambled control knockdown in the A549 lung cell line (Fig. S9). Moreover, onco-lncRNA-12 was also found to be differentially expressed in colon cancer in silico and confirmed by relative quantitative-PCR (qPCR) to be up-regulated in a panel of colon cell lines relative to the control cell line CCD-18Co (Fig. S10A). Measuring EdU incorporation in the colon cell line SW620 recapitulated the findings in the lung cancer cell line. There was also a significant decrease of 24% (p = 0.009) in siRNA1 and 28.9% (p = 0.03) in siRNA2 EdU incorporation compared to 34% EdU incorporation in the scrambled control samples (Fig. S10B). These findings demonstrate lung and colon cancer cell growth dependence and specifically changes in S-phase cell cycle of onco-lncRNA-12.
Figure 7.
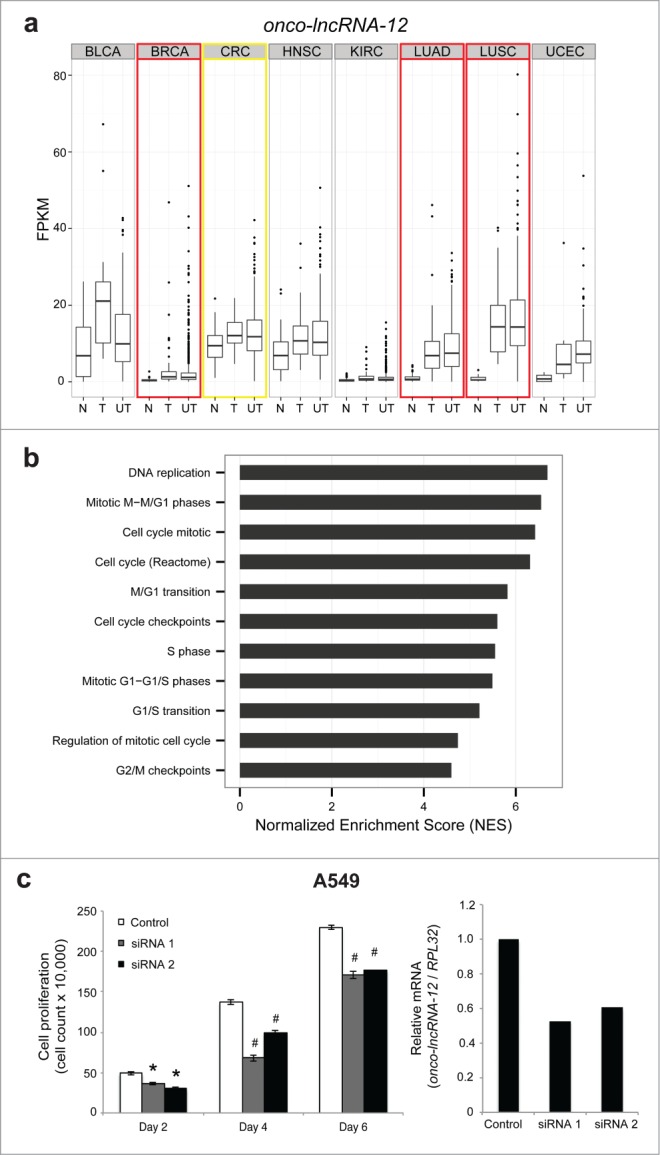
Onco-lncRNA-12 regulates cell growth. (A) Boxplots showing expression levels (FPKM) of onco-lncRNA-12 for normal samples (N), tumor samples with a matched normal (T), and unpaired tumor samples (UT) across all 8 cancer types. Red boxes highlight upregulated cancer types and the yellow box highlights the outlier cancer type. (B) GSEA normalized enrichment scores (NES) for cell cycle related gene sets. (C) Cell growth assay in A549 cells using onco-lncRNA-12 siRNAs (* P < 0.01, # P < 0.005 by a 2-tailed Student's t-test) and qPCR validation of onco-lncRNA-12 siRNA knockdown. All error bars are mean ± standard error of the mean across n = 3 biological replicates.
Lastly, we validated an additional novel onco-lncRNA, onco-lcnRNA-3, as further proof of concept supporting our guilt-by-association analysis. Onco-lcnRNA-3 was previously found to be differentially regulated in LUAD and LUSC as well as differentially expressed in a panel of lung cell lines relative to a control lung cell line by quantitative PCR.34 Moreover, here we found it to be altered across multiple cancer types including BRCA, CRC, HNSC, LUAD, and LUSC. Onco-lncRNA-3 co-expressed with protein coding genes that were enriched for cell cycle-related gene sets. Greater than 50% knockdown of onco-lncRNA-3 in NCI-H322M lung cells with 2 different siRNAs resulted in approximately 15% (p = 0.04) or 12% (p = 0.02), respectively, of EdU incorporation compared to 18% incorporation in scrambled control (Fig. S11). Further investigation of onco-lncRNA-3 in a panel of colon cell lines showed differential expression as measured by quantitative PCR (Fig. S12A). Measuring EdU incorporation in the colon cell line HT-29 highlighted an alteration in S-phase cell cycle with decreased expression of onco-lncRNA-3. Flow cytometry analysis revealed a 7.3% and 5.5% decrease of EdU incorporation for both siRNAs (p = 0.02) compared to 12.3% EdU incorporation for the scrambled control (Fig. 12B).
Taken together, CCAT1, onco-lncRNA-12, and onco-lncRNA-3, highlight the effectiveness of our co-expression analysis to implicate onco-lncRNAs in biological processes such as cell cycle. The data presented here and previous evidence in published literature serve as a proof-of-concept of onco-lncRNAs having conserved phenotypes across cancer types.
Discussion
In this study, we present a pan-cancer lncRNA analysis of ∼3,000 RNA-Seq samples comparing tumor and adjacent normal tissue expression levels. This analysis enabled us to identify “onco-lncRNAs” that are altered across multiple cancer types suggesting a common oncogenic or tumor suppressive function; moreover, this analysis identified lncRNAs that are altered in a single cancer type which may be useful as tissue-specific biomarkers. The potential significance of the onco-lncRNAs is supported by their reliable expression levels, as determined by stringent filters, across hundreds of patients spanning several cancer types, thereby mitigating their likelihood of being transcriptional noise.49 Additionally, given that lncRNAs typically exhibit tissue-specific expression it is even more unexpected to observe lncRNAs that are consistently altered across tumor types.
By conducting a pan-cancer analysis of publicly available data we were able to leverage the large patient cohort size to hone in on a subset of altered lncRNAs that can serve as a valuable resource for the community. However, the advances in transcriptome sequencing over the last few years, during which the data for each cohort was generated, has resulted in some heterogeneity between cancer types such as patient cohort size, number of reads generated, and depth of coverage (Fig. S13–S15). Due to the small variability within a cancer type, compared to across cancer types, we are able to accurately identify altered lncRNAs within each cancer as exemplified by our confirmation of several well-characterized lncRNAs known to play a role in cancer. However, for the cohorts that have fewer reads generated, such as UCEC, we are likely under-representing the quantity of altered lncRNAs due to lower coverage. This in turn may also under-represent the number of onco-lncRNAs or the number of cancer types for which they are actually altered. Despite the lower sequence read coverage in a few of the cancer types, we are still able to detect a subset of altered lncRNAs, suggesting they are more highly expressed and markedly altered. For these reasons, the onco-lncRNAs reported in our study likely represent the most abundant and reliably expressed candidates that warrant further exploration albeit this list will likely expand as additional deeper sequencing is obtained for older, lower coverage cohorts.
To date only a small number of lncRNAs are known to play a role in multiple cancers, such as PVT1 (onco-lncRNA-100) and MEG3 (onco-lncRNA-83). Through our systematic analysis we were able to reveal lncRNAs that may have a significant role in human cancer. First, we discovered known cancer-related lncRNAs to be altered in additional, previously unknown cancers. Examples include: LINC0026150,51 (onco-lncRNA-17), LCAL1 (onco-lncRNA-27),34 BLACAT152 (onco-lncRNA-30), ENST00000547963.153 (onco-lncRNA-32), UCA112 (onco-lncRNA-36), and PCAN-R154 (onco-lncRNA-96). Second, we identified lncRNAs that were previously found to be altered in development with no previous role in human cancer, such as TINCR55 (onco-lncRNA-16) which has been found to play a role in tissue differentiation. Third, we found the majority of onco-lncRNAs have not yet been characterized despite being altered in multiple cancer types. Furthermore, the recent and increasing number of publications implicating a subset of these onco-lncRNAs in cancer further supports their emerging importance and suggests the remaining uncharacterized onco-lncRNAs may also have clinical and biological significance that warrants further exploration.
To date, lncRNA studies often use a gene-centric approach to unveil the clinical significance of a single lncRNA within a specific cancer type. Although such studies may provide promising results by showing a positive association between a lncRNA and a clinical endpoint, there is no guarantee that the best biomarker candidate has been discovered without a comprehensive comparison to lncRNA candidates identified within larger meta-analyses. For instance, gene-centric studies focusing on both CCAT131 and CRNDE32 demonstrate their potential as a putative lncRNA diagnostic biomarkers in colorectal cancer. However, it is unclear how these lncRNAs perform relative to one another as well as additional lncRNAs predicted through a systematic colon cancer transcriptome analysis. In fact, here we report additional colorectal cancer specific lncRNAs displaying higher expression levels than both CCAT1 and CRNDE that may serve as more accurate biomarkers. Taken together, our systematic analysis provides a comprehensive set of lncRNAs for subsequent biomarker evaluation in 8 cancer types.
Despite the discovery of thousands of lncRNAs over the last few years, only a small fraction of these have well defined functional roles. Therefore, in addition to identifying new candidate onco-lncRNAs, we provided an in silico analysis to implicate onco-lncRNAs in key biological processes and pathways that could guide subsequent functional studies. First, given that mutations of well-established oncogenes have been shown to correlate with lncRNA expression, we leveraged matched exome data to discover that 131 lncRNAs associated with mutational status. By focusing on key pan-cancer oncogenes, we have implicated onco-lncRNAs in the context of key oncogenic pathways. Currently, our data supports a model in which the association between lncRNA expression and oncogene mutation status may suggest that a lncRNA resides within the same pathway as the oncogene. It is possible that the lncRNA resides downstream of the oncogene as exemplified by BRAF-regulated lncRNA 1 (BANCR) being recurrently overexpressed due to activation of RAF signaling activation in BRAFV600E-mutant human melanoma.38 Therefore, it is likely that a subset of associations between lncRNA expression and oncogene mutation status may be explained by an oncogene directly activating or repressing transcription of the lncRNA. This is exemplified by the expression of MEG3 (onco-lncRNA-83), which has a TP53 binding site within its promoter, being associated with TP53 mutational status. Our results also build upon the recent finding of signaling pathway activation by an external stimulus such as oxidative stress and cigarette smoke, thereby altering lncRNA expression. For example, smoke and cancer-associated lncRNA-156 was recently shown to act downstream of nuclear factor erythroid 2-related factor (NRF2) and mediate oxidative stress in lung cancer. Similarly we also observed an NRF2 motif upstream of onco-lncRNA-3, which was found to have elevated expression in KEAP1 mutant patients in LUAD. Interestingly KEAP1 is a cytosolic repressor of the NRF2 pathway, which promotes proteasomal degradation via interactions with an ubiquitin ligase. Under oxidative stress KEAP1 is altered such that it can no longer bind to NRF2 resulting in NRF2 accumulation in the nucleus. Recent work has shown that KEAP1 mutant cells protect NRF2 from ubiquitination and degradation, constitutively activating the expression of NRF2 target genes.57 Therefore, it is plausible that there is some interplay between KEAP1 mutant patients and elevated onco-lncRNA-3 expression via NRF2. Taken together, our analysis has provided a foundation for exploring potential mechanisms by which a mutation in an oncogene can potentially lead to altered onco-lncRNA expression. However, further experiments are needed to fully elucidate the relationship between mutational status and lncRNA expression.
We also chose to use a guilt-by-association method to predict lncRNA function by associating a lncRNA with the function of its most highly co-expressed protein coding genes. Guilt-by-association approaches have been successfully used in earlier studies to provide insights into lncRNA function in stem cell pluripotency, adipogenesis, and cancer.39,58,59 However, this approach has a few limitations. First, the relationship between any 2 genes is correlative and therefore does not provide direct evidence of an interaction. As such, some correlated gene pairs may be co-regulated (i.e., bi-directional promoter) but involved in independent processes. Second, the cellular composition of the tumor may vary among patients thereby diluting the correlative signal. Third, many key genes altered in cancer may show more marked changes at the protein level and therefore could be under-represented by an expression-based analysis. Last, while a co-expression analysis considers pairwise relationships, it is likely confounded by more complex interactions involving additional genes and mutations. Despite these limitations, any signal that we are able to observe can offer a starting point for subsequent experimental studies exploring their biological roles. Furthermore, unlike earlier analyses, in our study we extended the guilt-by-association approach to predict lncRNA functions that are conserved across the multiple cancers in which an onco-lncRNA is altered. Just as our methods detected consistently altered protein coding genes that have critical roles in cell cycle regulation, they also revealed a novel subset of cell cycle associated onco-lncRNAs including onco-lncRNA-3 and onco-lncRNA-12, for which we were able to validate their roles in regulating cell growth by altering S-phase cell cycle. Additionally, we chose to study CCAT1 because it was recently shown to regulate cellular proliferation in colorectal cancer.31 Experimental evidence revealed that CCAT1 also regulates cell growth in lung cancer cell lines, thereby demonstrating conserved lncRNA function across multiple cancer types.
Overall, this study has provided a roadmap of critical cancer associated lncRNAs as well as computational strategies for implicating lncRNAs with key cancer genes and biological processes to guide subsequent functional characterization. However, given the broad range of lncRNA functionality there are a variety of mechanisms by which lncRNAs interact with cancer genes. We have observed mutated oncogenes that can activate a pathway to regulate onco-lncRNA expression. Conversely, lncRNAs have been shown to regulate critical cancer genes. This can be exemplified by lncRNAs that modulate chromatin remodeling thereby regulating transcriptional programs that include oncogenes and tumor suppressors.9 Additionally, different classes of lncRNAs can regulate critical cancer genes through a variety of mechanisms. For instance, antisense lncRNAs typically regulate protein-coding genes in close proximity60,61 whereas cytoplasmic competing endogenous RNAs (ceRNAs) will compete with cancer genes harboring similar microRNA binding sites to modulate expression.62 Therefore, given the broad and expanding range of lncRNA functions, our comprehensive study importantly hones in on critical onco-lncRNAs and provides an initial framework for dissecting their emerging regulatory roles with oncogenes and tumor suppressors.
In conclusion, we presented a systematic pan-cancer analysis of lncRNAs using RNA-Seq data across 8 cancer types from TCGA comparing expression levels between tumor and matched adjacent normal tissues. This analysis revealed lncRNAs that are altered in a single cancer type, which could serve as putative biomarkers, as well as 229 onco-lncRNAs that are broadly altered across multiple cancer types and could serve as key oncogenes and tumor suppressors. Our study represents an initial step toward discovering putative oncogenic and tumor suppressor lncRNAs that may play a critical role in cancer and providing potential functional roles that can serve as a resource for future studies exploring their emerging roles in tumorigenesis.
Materials and Methods
TCGA RNA-Seq datasets
The TCGA consortium aligned all RNA-Seq BAM files to hg19 with MapSplice.63 We downloaded the aligned BAM files for the following solid tumor cancers included in the TCGA Pan-Cancer Analysis27: bladder urothelial carcinoma19 (BLCA), breast invasive carcinoma20 (BRCA), colon adenocarcinoma21 (COAD), head and neck squamous cell carcinoma22 (HNSC), kidney renal cell carcinoma23 (KIRC), lung adenocarcinoma24 (LUAD), lung squamous cell carcinoma25 (LUSC), rectum adenocarcinoma21 (READ), and uterine corpus endometrial carcinoma26 (UCEC). When available, RNA-Seq BAM files for matched adjacent normal tissue were also downloaded. Following TCGA practices, the COAD and READ cohorts were merged to form a colorectal cancer (CRC) cohort.21 Sample sizes and read lengths for each cancer type are reported in Supplementary Table 1. UCEC samples and approximately half of the CRC samples were sequenced on Illumina GAIIx while the other samples, including all of the matched tumor and adjacent normal CRC pairs, were sequenced on the Illumina HiSeq platform.
LncRNA annotations
Figure 1A shows the filtering steps used to create a custom annotation file comprising of lncRNAs from multiple sources. First, we downloaded the following gene annotation databases in hg19 coordinates from the UCSC Genome Browser29 on March 24, 2014: RefSeq release 64, Ensembl v75, UCSC Genes build June 2013, and the Human Body Map (known as “lncRNA Transcripts” track in UCSC Genome Browser). The custom annotation file was generated by first removing all protein coding transcripts from Ensembl and UCSC. Next, we removed single-exon transcripts from Ensembl, UCSC, and the Human Body Map because most of these transcripts have not been experimentally validated and transcripts lacking a splice junction could be noise due to potential DNA contamination. However, as some well-characterized single-exon lncRNAs exist, we chose to keep single-exon transcripts from RefSeq. Next, we merged all annotated non-coding transcripts into a single annotation. If a transcript is reported in multiple databases with the exact same exon coordinates, the transcript was included in the merged annotation only once, using one transcript ID (from RefSeq, Ensembl, or UCSC, in that order). All transcripts <200 nt, including RefSeq protein coding transcripts, were removed. To focus on intergenic transcripts, we next removed noncoding transcripts overlapping any exon or intron of a RefSeq protein coding gene or overlapping an exon from an Ensembl transcript annotated as anything other than 'lincRNA', 'antisense', 'retained_intron', or 'processed_transcript'. Due to sequence homology, a minor subset of the reference transcript IDs are not unique to a single genomic location but instead correspond to multiple locations. We removed these transcripts that perfectly map to multiple genomic locations. Finally, due to gender biases of the cohorts and cancers studied, transcripts on chromosomes X and Y were removed, only keeping transcripts mapping to chr 1–22.
Read counts for each transcript were calculated using BEDTools version 2.17.0.64 BedTools handles multi-mapped reads by counting each hit separately. FPKM65 expression values were manually calculated as 109(M / (T *L)) where M is the number of reads mapping to a transcript, T is the total number of mapped reads, and L is the transcript length. For each cancer, we flagged transcripts with low expression (at least 75% of matched tumor and 75% of normal samples had FPKM < 1 or read count < 200). Transcripts with low expression in all cancers were removed. The remaining transcripts were reduced to a set of non-overlapping genes by comparing all overlapping transcripts and keeping the transcript that was expressed in the largest number of cancers. If there was a tie, the transcript with the highest average read count across all expressed cancers was chosen. This filtering revealed 14,128 protein coding genes and 1,053 lncRNAs with enriched expression, which are reported in Supplementary Tables 14 and 15, respectively.
Differential expression analysis
Differential expression analysis was performed on each cancer type separately. Only tumors with a matched adjacent normal were used to identify differentially expressed transcripts. For a given cancer, we only tested transcripts with enriched expression. The remaining transcripts were TMM normalized,66 then edgeR version 3.0.867 was used to test for differential expression between the tumor and normal pairs using a matched pair design with cutoffs of FDR ≤ 10−5 and absolute fold change ≥2.
Outlier analysis
Outlier analysis was performed on each cancer type separately. Read counts for all tumor samples were normalized using the positive quantile transformation.68 For a given cancer, only lncRNAs with maximum FPKM across all tumors ≥25 were tested. P-values were calculated using MIST68 and then corrected for multiple comparisons using the Benjamini & Hockberg FDR correction.69 A significance threshold of FDR < 0.05 was used to determine significant outlier lncRNAs. The same procedure was used to call outliers using the adjacent normal samples and any lncRNAs that were also called outliers in the normal samples were not considered outliers in the tumors.
LncRNA conservation score
phastCons conservation scores70 based on whole genome alignment of 100 vertebrates (vertebrate 100 way) were downloaded from UCSC genome browser.29 The conservation score of a transcript was calculated as the average of the phasCons scores of all nucleotides in the exons.
H3K4me3 histone modification analysis
H3K4me3 Chip-seq aligned BAM and bigWig coverage files were downloaded from UCSC genome browser.29 Bwtool71 was used to calculate the average coverage within 20kb of the transcript start sites of the lncRNAs within each group. Average coverage was then calculated as the total number of reads per million mapped reads.
Copy number analysis
For each cancer type, regions of significant copy number alterations were called using GISTIC72 and the results file 'all_lesions.conf_95.txt' was downloaded for each cancer type from TCGA (www.synapse.org/#!Synapse:syn1713807). Because GISTIC calls were made separately on the COAD and READ cohorts, we chose to divide the CRC cohort into COAD and READ for this analysis. Only tumors with available RNA-Seq data and copy number calls were considered (see Table S1 for sample sizes). Differentially expressed and outlier lncRNAs falling within copy number alterations (CNAs) were determined by intersecting genomic coordinates of upregulated/outlier lncRNAs with amplified regions and down-regulated/outlier lncRNAs with deleted regions (using “wide peak” boundaries). For each lncRNA falling within a CNA, Spearman correlation was calculated between lncRNA FPKM expression values and CNA scores across all tumors. Significance of the correlation was calculated using a permutation test by comparing the true correlation to the distribution of correlations obtained by permuting the order of the CNA scores 1,000 times. P-values < 0.01 were considered significant.
Mutation analysis
For each cancer type, mutation calls were downloaded from TCGA (www.synapse.org/#!Synapse:syn1729383). Attention was restricted to tumor samples with both RNA-Seq and mutation data (see Table S1 for sample sizes). We only considered genes that were reported in an earlier Pan-Cancer study (see Fig. 2)27 and mutated in > 5% of tumors with RNA-Seq data. A Mann-Whitney U test was used to test for significance between mutational status and FPKM expression of each differentially expressed and outlier lncRNA. A one-sided test was used for differentially expressed lncRNAs (testing that mutated samples had higher expression in up-regulated lncRNAs and lower expression in downregulated lncRNAs) and a 2-sided test was used for outlier lncRNAs. For each cancer type, p-values were corrected for multiple comparisons using the Benjamini & Hockberg FDR correction,69 and a significance threshold of FDR < 0.05 was used.
A list of transcription factor binding sites conserved in the human/mouse/rat alignment was downloaded on December 11, 2014 from the UCSC Genome Browser29 (“TFBS Conserved” track). For each onco-lncRNA that is significantly associated with at least one mutation, we identified all transcription factor binding sites within 500 nt upstream of the transcript and on the same strand.
LncRNA functional associations
Prediction of lncRNA function was adapted from a previous study.1 For each cancer type we calculated a co-expression matrix between differentially expressed lncRNAs and all protein coding genes by computing the Spearman correlation across all tumor samples. We restricted attention to onco-lncRNAs that were differentially expressed in at least 2 cancers in the same direction. A meta-correlation was generated by averaging the correlations across all differentially expressed cancers. Functional associations were computed for each lncRNA using Gene Set Enrichment Analysis (GSEA)41 version 2.0.14 by inputting the list of co-expressed genes and testing for associations with the KEGG73 and Reactome74 gene sets. Gene sets with FDR < 0.05 were considered significant. When clustering gene sets using GSEA normalized enrichment scores (NES), negatively associated gene sets were assigned negative NES values and all concepts with FDR > 0.05 were assigned an NES of zero.
Cell culture
Lung cancer cell lines NCI-H322M and A549 were a kind gift from Dr. Brian Van Tine at Washington University. Colon caner cell lines CCD18Co and SW480 were a kind gift from Dr. David Shalloway at Cornell University. Other colon cell lines (HT-29, HT-15, DLD1, SW620, Caco-2 Lovo, HCT-116, and RKO) were a kind gift from Dr. A. Craig Lockhart at Washington University. H322M and A549 cells were grown in RPM1–1640 (Invitrogen, Carsbad, CA) with 10% fetal bovine serum (FBS) (Invitrogen) and 1% penicillin/streptomycin (pen/strep) (Invitrogen) complete media. SW620 cells were grown in DMEM (Invitrogen), 10% FBS, and 1% pen/strep complete media and HT-29 cells were grown in McCoys (Invitrogen), 10% FBS, and 1% pen/strep complete media.
RNA isolation and cDNA synthesis
Total RNA was isolated with the RNeasy Mini Kit (Qiagen, Valencia, CA) with subsequent DNase 1 treatment according to the manufacturer's instructions. cDNA was synthesized from total RNA using High Capacity cDNA Reverse Transcription Kit with random hexamers (Invitrogen).
Quantitative real time PCR
siRNA knockdown was confirmed with quantitative RT-PCR using PowerSyBr Green (Invitrogen) prior to plating for proliferation experiments or day of EdU assay. The comparative CT (ΔΔCT) method was used with values first normalized to the housekeeping gene, RPL32, and then to scrambled control knocdown. All primers were obtained from Integrated DNA Technologies (Coralville, IA). The following primers were used to verify gene expression: CCAT1 Forward (5′-GCCGTGTTAAGCATTGCGAA-3′), CCAT1 Reverse (5′-AGAGTAGTGCCTGGCCTAGA-3′), onco-lncRNA-12 Forward (5′-CGCAAGGACCCTCTGTTAGG-3′), onco-lncRNA-12 Reverse (5′-GAAGGCGGATCGTCTCTCAG-3′), onco-lncRNA-3 Forward (5′-TCCCAATAAACAGGGCAGAC-3′), onco-lncRNA-3 Reserve (5′-CAAGATCACCACACCCCTCT-3′), RPL32 Forward (5′-AGGCATTGACAACAGGGTTC-3′), and RPL32 Reverse (5′-GTTGCACATCAGCAGCACTT-3′). Primer efficiency between 90−110% was determined for each primer candidate.
siRNA knockdown experiments and cellular proliferation assay
Stealth siRNA oligonucleotides were synthesized by Invitrogen. The following siRNA sequences were used for knockdown of CCAT1: CCAT1 siRNA 1 (5′-UGUGGUAGGAAAGAGAAAUGAAUGG-3′), CCAT1 siRNA 2 (5′-GACCACUGCUUUAAAGCCUUUGCAU-3′) or a control (a scrambled-matched %GC oligonucleotide synthesized by Invitrogen). The following siRNA sequences were used for knockdown of onco-lncRNA-12: onco-lncRNA-12 siRNA 1 (5′-CCCAUGUCUGCUGUGCCUUUGUACU-3′) and onco-lncRNA-12 siRNA 2 (5′-CCAGUGUGUGCUGAUGACACAUACA-3′). The following siRNA sequences were used to knockdown onco-lcncRNA-3: onco-lncRNA-3 siRNA 1 (5′-CTCTTCAAGTTGACTGCAGTCCAT-3′) and onco-lncRNA-3 siRNA 2 (5′-TGGCAGCTAAGAATGTGTATCCCA-3′). Cells were transfected with 50 pmol of siRNA and the scrambled control oligo with RNAimax Lipofecatmine (Invitrogen) following manufacturer's instructions. Knockdown efficiency was determined by quantitative PCR at time of plating for assay. After 72 hours, cells were then plated at 200,000 cells/well for cell growth assays. Cells were counted using the Beckman Z1 Coulter Counter at Day 2, 4, and 6. At least 3 biological replicates were performed for each siRNA construct over 2 experiments. S-phase cell cycle was monitored by EdU incorporation following the protocol for the Click-It EdU flow cytometry assay kit Alexa-488 provided by the manufacture (Invitrogen) and cells were analyzed on a BD FACSCan flow cytometer (Franklin Lakes, NJ, USA). Data was analyzed using FlowJo software verson X.07 S (TreeStar, Ashland, OR, USA).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by a LUNGevity Foundation Career Development Award, an American Lung Association Biomedical Research Grant, and a Susan G. Komen CCR Basic/Translational and Clinical Grant, CCR14301279 (to C.A.M.).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al.. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458:223–7; PMID:19182780; http://dx.doi.org/ 10.1038/nature07672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paralkar VR, Mishra T, Luan J, Yao Y, Kossenkov AV, Anderson SM, Dunagin M, Pimkin M, Gore M, Sun D, Konuthula N, Raj A, An X, Mohandas N, Bodine DM, Hardison RC, Weiss MJ.. Lineage and species-specific long noncoding RNAs during erythro-megakaryocytic development. Blood 2014, 123:1927–37; PMID:24497530; http://dx.doi.org/ 10.1182/blood-2013-12-544494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, et al.. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol 2010, 28:503–10; PMID:20436462; http://dx.doi.org/ 10.1038/nbt.1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G, Young G, Lucas AB, Ach R, Bruhn L, et al.. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011, 477:295–300; PMID:21874018; http://dx.doi.org/ 10.1038/nature10398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al.. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res 2012, 22:1775–89; PMID:22955988; http://dx.doi.org/ 10.1101/gr.132159.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL.. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 2011, 25:1915–27; PMID:21890647; http://dx.doi.org/ 10.1101/gad.17446611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hangauer MJ, Vaughn IW, McManus MT.. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 2013, 9:e1003569; PMID:23818866; http://dx.doi.org/ 10.1371/journal.pgen.1003569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prensner JR, Iyer MK, Balbin OA, Dhanasekaran SM, Cao Q, Brenner JC, Laxman B, Asangani IA, Grasso CS, Kominsky HD, et al.. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol 2011, 29:742–9; PMID:21804560; http://dx.doi.org/ 10.1038/nbt.1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai M-C, Hung T, Argani P, Rinn JL, et al.. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464:1071–6; PMID:20393566; http://dx.doi.org/ 10.1038/nature08975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kotake Y, Nakagawa T, Kitagawa K, Suzuki S, Liu N, Kitagawa M, Xiong Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 2011, 30:1956–62; PMID:21151178; http://dx.doi.org/ 10.1038/onc.2010.568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, et al.. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A 2009, 106:11667–72; PMID:19571010; http://dx.doi.org/ 10.1073/pnas.0904715106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X-S, Zhang Z, Wang H-C, Cai J-L, Xu Q-W, Li M-Q, Chen Y-C, Qian X-P, Lu T-J, Yu L-Z, et al.. Rapid identification of UCA1 as a very sensitive and specific unique marker for human bladder carcinoma. Clin Cancer Res 2006, 12:4851–4858; PMID:16914571; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-0134 [DOI] [PubMed] [Google Scholar]
- 13.Hammond SM. RNAi, microRNAs, and human disease. Cancer Chemother Pharmacol 2006, 58 Suppl 1:s63–68; PMID:17093929; http://dx.doi.org/ 10.1007/s00280-006-0318-2 [DOI] [PubMed] [Google Scholar]
- 14.Guan Y, Kuo W-L, Stilwell JL, Takano H, Lapuk AV, Fridlyand J, Mao J-H, Yu M, Miller MA, Santos JL, et al.. Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clin Cancer Res 2007, 13:5745–55; PMID:17908964; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-2882 [DOI] [PubMed] [Google Scholar]
- 15.Zhou Y, Zhang X, Klibanski A.. MEG3 noncoding RNA: a tumor suppressor. J Mol Endocrinol 2012, 48:R45–53; PMID:22393162; http://dx.doi.org/ 10.1530/JME-12-0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma H, Hao Y, Dong X, Gong Q, Chen J, Zhang J, Tian W . Molecular mechanisms and function prediction of long noncoding RNA. Sci World J 2012,2012:e541786; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hung T, Wang Y, Lin MF, Koegel AK, Kotake Y, Grant GD, Horlings HM, Shah N, Umbricht C, Wang P, et al.. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet 2011, 43:621–9; PMID:21642992; http://dx.doi.org/ 10.1038/ng.848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liao Q, Liu C, Yuan X, Kang S, Miao R, Xiao H, Zhao G, Luo H, Bu D, Zhao H, et al.. Large-scale prediction of long non-coding RNA functions in a coding–non-coding gene co-expression network. Nucleic Acids Res 2011, 39:3864–78; PMID:21247874; http://dx.doi.org/ 10.1093/nar/gkq1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The Cancer Genome Atlas Research Network Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014a, 507:315–22; ; http://dx.doi.org/ 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature 2012a, 490:61–70; PMID:23000897; http://dx.doi.org/ 10.1038/nature11412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012b, 487:330–7; PMID:22810696; http://dx.doi.org/ 10.1038/nature11252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The Cancer Genome Atlas Network Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517:576–82; PMID:25631445; http://dx.doi.org/ 10.1038/nature14129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The Cancer Genome Atlas Research Network Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013a, 499:43–9; PMID: 23792563; http://dx.doi.org/ 10.1038/nature12222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.The Cancer Genome Atlas Research Network . Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014b, 511:543–50; PMID: 25079552; http://dx.doi.org/ 10.1038/nature13385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012c, 489:519–25; PMID: 22960745; http://dx.doi.org/ 10.1038/nature11404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.The Cancer Genome Atlas Research Network, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, et al . Integrated genomic characterization of endometrial carcinoma. Nature 2013b, 497:67–73; PMID: 23636398; htpp://dx.doi.org/ 10.1038/nature12113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al.. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502:333–9; PMID:24132290; http://dx.doi.org/ 10.1038/nature12634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al.. Ensembl 2014. Nucleic Acids Res 2014, 42:D749–755; PMID:24316576; http://dx.doi.org/ 10.1093/nar/gkt1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L, Haeussler M, et al.. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res 2015. January 28;43:D670–81; http://dx.doi.org/ 10.1093/nar/gku1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pruitt KD, Brown GR, Hiatt SM, Thibaud-Nissen F, Astashyn A, Ermolaeva O, Farrell CM, Hart J, Landrum MJ, McGarvey KM, et al.. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res 2014, 42, D756–763; PMID:24259432; http://dx.doi.org/ 10.1093/nar/gkt1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nissan A, Stojadinovic A, Mitrani-Rosenbaum S, Halle D, Grinbaum R, Roistacher M, Bochem A, Dayanc BE, Ritter G, Gomceli I, et al.. Colon cancer associated transcript-1: a novel RNA expressed in malignant and pre-malignant human tissues. Int J Cancer J Int Cancer 2012, 130:1598–606; PMID:21547902; http://dx.doi.org/ 10.1002/ijc.26170 [DOI] [PubMed] [Google Scholar]
- 32.Graham LD, Pedersen SK, Brown GS, Ho T, Kassir Z, Moynihan AT, Vizgoft EK, Dunne R, Pimlott L, Young GP, et al.. Colorectal Neoplasia Differentially Expressed (CRNDE), a Novel Gene with Elevated Expression in Colorectal Adenomas and Adenocarcinomas. Genes Cancer 2011, 2:829–40; PMID:22393467; http://dx.doi.org/ 10.1177/1947601911431081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Panzitt K, Tschernatsch MMO, Guelly C, Moustafa T, Stradner M, Strohmaier HM, Buck CR, Denk H, Schroeder R, Trauner M, et al.. Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology 2007, 132:330–42; PMID:17241883; http://dx.doi.org/ 10.1053/j.gastro.2006.08.026 [DOI] [PubMed] [Google Scholar]
- 34.White NM, Cabanski CR, Silva-Fisher JM, Dang HX, Govindan R, Maher CA.. Transcriptome sequencing reveals altered long intergenic non-coding RNAs in lung cancer. Genome Biol 2014, 15:429; PMID:25116943; http://dx.doi.org/ 10.1186/s13059-014-0429-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szafranski P, Dharmadhikari AV, Brosens E, Gurha P, Kolodziejska KE, Zhishuo O, Dittwald P, Majewski T, Mohan KN, Chen B, et al.. Small noncoding differentially methylated copy-number variants, including lncRNA genes, cause a lethal lung developmental disorder. Genome Res 2013, 23:23–33; PMID:23034409; http://dx.doi.org/ 10.1101/gr.141887.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sabo PJ, Hawrylycz M, Wallace JC, Humbert R, Yu M, Shafer A, Kawamoto J, Hall R, Mack J, Dorschner MO, McArthur M, Stamatoyannopoulos JA.. Discovery of functional noncoding elements by digital analysis of chromatin structure. Proc Natl Acad Sci U S A. 2004, 101, 16837–42; PMID:15550541; http://dx.doi.org/ 10.1073/pnas.0407387101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tseng YY, Moriarity BS, Gong W, Akiyama R, Tiwari A, Kawakami H, Ronning P, Reuland B, Guenther K, Beadnel,l TC, Essig J, Otto GM, O'Sullivan MG, Largaespada DA, Schwertfeger KL, Marahrens Y, Kawakami Y, Bagchi A . PVT1 dependence in cancer with MYC copy-number increase. Nature 2014, 512:82–6; PMID:25043044; http://dx.doi.org/ 10.1038/nature13311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flockhart RJ, Webster DE, Qu K, Mascarenhas N, Kovalski J, Kretz M, Khavari PA.. BRAFV600E remodels the melanocyte transcriptome and induces BANCR to regulate melanoma cell migration. Genome Res 2012, 22:1006–14; PMID:22581800; http://dx.doi.org/ 10.1101/gr.140061.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, Attard,i LD, Regev A, Lander ES, Jacks T, Rinn JL.. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142:409–19; PMID:20673990; http://dx.doi.org/ 10.1016/j.cell.2010.06.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu KH, Li W, Liu XH, Sun M, Zhang ML, Wu WQ, Xie WP, Hou YY.. Long non-coding RNA MEG3 inhibits NSCLC cells proliferation and induces apoptosis by affecting p53 expression. BMC Cancer 2013, 13:461; PMID:24098911; http://dx.doi.org/ 10.1186/1471-2407-13-461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005, 102:15545–50; PMID:16199517; http://dx.doi.org/ 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schorderet P, Duboule D.. Structural and functional differences in the long non-coding RNA hotair in mouse and human. PLoS Genet 2011, 7:e1002071; PMID:21637793; http://dx.doi.org/ 10.1371/journal.pgen.1002071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Hayre M, Degese MS, Gutkind JS.. Novel insights into G protein and G protein-coupled receptor signaling in cancer. Curr Opin Cell Biol 2014, 27:126–35; PMID:24508914; http://dx.doi.org/ 10.1016/j.ceb.2014.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Joaquin M, Watson RJ.. Cell cycle regulation by the B-Myb transcription factor. Cell Mol Life Sci CMLS 2003, 60:2389–401; http://dx.doi.org/ 10.1007/s00018-003-3037-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dunleavy EM, Roche D, Tagami H, Lacoste N, Ray-Gallet D, Nakamura Y, Daigo Y, Nakatani Y, Almouzni-Pettinotti G.. HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell 2009, 137:485–97; PMID:19410545; http://dx.doi.org/ 10.1016/j.cell.2009.02.040 [DOI] [PubMed] [Google Scholar]
- 46.Hao Z, Zhang H, Cowell J.. Ubiquitin-conjugating enzyme UBE2C: molecular biology, role in tumorigenesis, and potential as a biomarker. Tumour Biol J Int Soc Oncodevelopmental Biol Med 2012, 33:723–30; http://dx.doi.org/ 10.1007/s13277-011-0291-1 [DOI] [PubMed] [Google Scholar]
- 47.Borlado LR, Méndez J.. CDC6: from DNA replication to cell cycle checkpoints and oncogenesis. Carcinogenesis 2008, 29:237–43; PMID:18048387; http://dx.doi.org/ 10.1093/carcin/bgm268 [DOI] [PubMed] [Google Scholar]
- 48.Kim T, Cui R, Jeon Y-J, Lee J-H, Lee JH, Sim H, Park JK, Fadda P, Tili E, Nakanishi H, et al.. Long-range interaction and correlation between MYC enhancer and oncogenic long noncoding RNA CARLo-5. Proc Natl Acad Sci U S A 2014, 111:4173–8; PMID:24594601; http://dx.doi.org/ 10.1073/pnas.1400350111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ponjavic J, Ponting CP, Lunter G.. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res 2007, 17:556–65; PMID:17387145; http://dx.doi.org/ 10.1101/gr.6036807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cao W-J, Wu H-L, He B-S, Zhang Y-S, Zhang Z-Y.. Analysis of long non-coding RNA expression profiles in gastric cancer. World J Gastroenterol 2013, 19:3658–64; PMID:23801869; http://dx.doi.org/ 10.3748/wjg.v19.i23.3658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin Z-Y, Chuang W-L.. Genes responsible for the characteristics of primary cultured invasive phenotype hepatocellular carcinoma cells. Biomed Pharmacother 2012, 66:454–8; PMID:22681909; http://dx.doi.org/ 10.1016/j.biopha.2012.04.001 [DOI] [PubMed] [Google Scholar]
- 52.He W, Cai Q, Sun F, Zhong G, Wang P, Liu H, Luo J, Yu H, Huang J, Lin T.. linc-UBC1 physically associates with polycomb repressive complex 2 (PRC2) and acts as a negative prognostic factor for lymph node metastasis and survival in bladder cancer. Biochim. Biophys. Acta 2013, 1832:1528–37; PMID:23688781; http://dx.doi.org/ 10.1016/j.bbadis.2013.05.010 [DOI] [PubMed] [Google Scholar]
- 53.Li J, Chen Z, Tian L, Zhou C, He MY, Gao Y, Wang S, Zhou F, Shi S, Feng X, et al.. LncRNA profile study reveals a three-lncRNA signature associated with the survival of patients with oesophageal squamous cell carcinoma. Gut 2014, 63:1700–10; PMID:24522499; http://dx.doi.org/ 10.1136/gutjnl-2013-305806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Du Z, Fei T, Verhaak RGW, Su Z, Zhang Y, Brown M, Chen Y, Liu XS.. Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nat Struct Mol Biol 2013, 20:908–13; PMID:23728290; http://dx.doi.org/ 10.1038/nsmb.2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kretz M, Siprashvili Z, Chu C, Webster DE, Zehnder A, Qu K, Lee CS, Flockhart RJ, Groff AF, Chow J, Johnston D, Kim GE, Spitale RC, Flynn RA, Zheng GX, Aiyer S, Raj A, Rinn JL, Chang HY, Khavari PA.. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 2013, 493:231–5; PMID:23201690; http://dx.doi.org/ 10.1038/nature11661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thai P, Statt S, Chen CH, Liang E, Campbell C, Wu R.. Characterization of a novel long noncoding RNA, SCAL1, induced by cigarette smoke and elevated in lung cancer cell lines. Am J Respir Cell Mol Biol 2013, 49:204–11; PMID:23672216; http://dx.doi.org/ 10.1165/rcmb.2013-0159RC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, Shyr C, Wakabayashi N, Kensler TW, Wasserman WW, Biswal S.. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res 2010, 38:5718–34; PMID:20460467; http://dx.doi.org/ 10.1093/nar/gkq212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Loewer S, Cabili MN, Guttman M, Loh YH, Thomas K, Park IH, Garber M, Curran M, Onder T, Agarwal S, Manos PD, Datta S, Lander ES, Schlaeger TM, Daley GQ, Rinn JL.. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet 2010, 42:1113–7; PMID:21057500; http://dx.doi.org/ 10.1038/ng.710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao XY, Li S, Wang GX, Yu Q, Lin JD.. A long noncoding RNA transcriptional regulatory circuit drives thermogenic adipocyte differentiation. Mol Cell 2014, 55:372–82; PMID:25002143; http://dx.doi.org/ 10.1016/j.molcel.2014.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xue Y, Ma G, Zhang Z, Hua Q, Chu H, Tong N, Yuan L, Qin C, Yin C, Zhang Z, Wang M.. A novel antisense long noncoding RNA regulates the expression of MDC1 in bladder cancer. Oncotarget 2015, 6:484–93; PMID:25514464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takayama K, Horie-Inoue K, Katayama S, Suzuki T, Tsutsumi S, Ikeda K, Urano T, Fujimura T, Takagi K, Takahashi S, Homma Y, Ouchi Y, Aburatani H, Hayashizaki Y, Inoue S.. Androgen-responsive long noncoding RNA CTBP1-AS promotes prostate cancer. EMBO J 2013, 32:1665–80; PMID:23644382; http://dx.doi.org/ 10.1038/emboj.2013.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.de Giorgio A, Krel L J, Harding V, Stebbing J, Castellano L.. Emerging roles of competing endogenous RNAs in cancer: insights from the regulation of PTEN. Mol Cell Biol 2013, 33:3976–82; PMID:23918803; http://dx.doi.org/ 10.1128/MCB.00683-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, et al.. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res 2010, 38:e178; PMID:20802226; http://dx.doi.org/ 10.1093/nar/gkq622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Quinlan AR, Hall IM.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26:841–2; PMID:20110278; http://dx.doi.org/ 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L.. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010, 28:511–5; PMID:20436464; http://dx.doi.org/ 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Robinson MD, Oshlack A.. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 2010, 11:R25; PMID:20196867; http://dx.doi.org/ 10.1186/gb-2010-11-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robinson MD, McCarthy DJ, Smyth GK.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26:139–40; PMID:19910308; http://dx.doi.org/ 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pawlikowska I, Wu G, Edmonson M, Liu Z, Gruber T, Zhang J, Pounds S.. The most informative spacing test effectively discovers biologically relevant outliers or multiple modes in expression. Bioinformatics 2014, 30:1400–8; PMID:24458951; http://dx.doi.org/ 10.1093/bioinformatics/btu039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Benjamini Y, Hochberg Y.. Controlling the False Discovery Rate- A Practical and Powerful Approach to Multiple Testing. J R Stat Soc Ser B 1995, 57:289–300. [Google Scholar]
- 70.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, Weinstock GM, Wilson RK, Gibbs RA, Kent WJ, Miller W, Haussler D.. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 2005, 15:1034–50; PMID:16024819; http://dx.doi.org/ 10.1101/gr.3715005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pohl A, Beato M.. bwtool: a tool for bigWig files. Bioinformatics 2014, 30:1618–9; PMID:24489365; http://dx.doi.org/ 10.1093/bioinformatics/btu056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G.. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 2011, 12:R41; PMID:21527027; http://dx.doi.org/ 10.1186/gb-2011-12-4-r41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M.. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 2014, 42:D199–205; PMID:24214961; http://dx.doi.org/ 10.1093/nar/gkt1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Croft D, Mundo AF, Haw R, Milacic M, Weiser J, Wu G, Caudy M, Garapati P, Gillespie M, Kamdar MR, et al.. The Reactome pathway knowledgebase. Nucleic Acids Res 2014, 42:D472–477; PMID:24243840; http://dx.doi.org/ 10.1093/nar/gkt1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.