

Highlights
-
•
Succinyl-CoA binding to ALAS is facilitated by the CoA moiety of the molecule.
-
•
The and values of ALAS are significantly different.
-
•
A 23-fold increase in the value was observed with the K221V variant.
-
•
The increased of K221V is not due to a weakened succinyl-CoA binding affinity.
-
•
The K221V substitution reduced the rate of quinonoid intermediate formation.
Abbreviations: ALA, 5-aminolevulinic acid; ALAS2, erythroid specific isoform of 5-aminolevulinate synthase; mALAS2, erythroid specific isoform of murine 5-aminolevulinate synthase; PLP, pyridoxal 5′-phosphate; SDS–PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; XLPP, X-linked protoporphyria; XLSA, X-linked sideroblastic anemia
Keywords: 5-Aminolevulinate synthase, Pyridoxal phosphate, Heme, Enzymatic mechanism, Sideroblastic anemia, Porphyria
Abstract
5-Aminolevulinate synthase (ALAS) catalyzes the initial step of mammalian heme biosynthesis, the condensation between glycine and succinyl-CoA to produce CoA, CO2, and 5-aminolevulinate. The crystal structure of Rhodobacter capsulatus ALAS indicates that the adenosyl moiety of succinyl-CoA is positioned in a mainly hydrophobic pocket, where the ribose group forms a putative hydrogen bond with Lys156. Loss-of-function mutations in the analogous lysine of human erythroid ALAS (ALAS2) cause X-linked sideroblastic anemia. To characterize the contribution of this residue toward catalysis, the equivalent lysine in murine ALAS2 was substituted with valine, eliminating the possibility of a hydrogen bond. The K221V substitution produced a 23-fold increase in the and a 97% decrease in . This reduction in the specificity constant does not stem from lower affinity toward succinyl-CoA, since the of K221V is lower than that of wild-type ALAS. For both enzymes, the value is significantly different from the . That K221V has stronger binding affinity for succinyl-CoA was further deduced from substrate protection studies, as K221V achieved maximal protection at lower succinyl-CoA concentration than wild-type ALAS. Moreover, it is the CoA, rather than the succinyl moiety, that facilitates binding of succinyl-CoA to wild-type ALAS, as evident from identical and values. Transient kinetic analyses of the K221V-catalyzed reaction revealed that the mutation reduced the rates of quinonoid intermediate II formation and decay. Altogether, the results imply that the adenosyl-binding site Lys221 contributes to binding and orientation of succinyl-CoA for effective catalysis.
1. Introduction
5-Aminolevulinate (ALA) represents the first committed precursor of heme biosynthesis that is produced by all heme-synthesizing organisms [1]. In non-plant eukaryotes and certain bacteria, ALA [with coenzymeA (CoA) and CO2 as byproducts] is synthesized from glycine and succinyl-CoA, during a condensation reaction catalyzed by the pyridoxal 5′-phosphate (PLP)-dependent enzyme, 5-aminolevulinate synthase (ALAS) [1,2]. Disturbances in the rate of ALA biosynthesis due to loss-of-function and gain-of-function mutations in the erythroid-specific ALAS gene (ALAS2) result in the erythropoietic disorders X-linked sideroblastic anemia (XLSA) and X-linked protoporphyria (XLPP), respectively [3–5].
The complex, multi-intermediate reaction cycle of ALAS, which has been a subject of extensive experimental studies [6–13], is outlined in Scheme 1. In the absence of substrates, PLP is covalently attached to an active site lysine (Lys313 of murine ALAS2 [abbreviated as mALAS2]) forming what is commonly referred as an internal aldimine (Scheme 1, I) [14,15]. Transaldimination occurs upon the entry of glycine, during which the internal aldimine is displaced and an external aldimine adduct is formed between PLP and the amino acid substrate (Scheme 1, II) [6,8]. Subsequent removal of the pro-R proton of glycine generates an initial, resonance stabilized, quinonoid intermediate (Scheme 1, III) that acts as a nucleophile in the condensation with succinyl-CoA [11–13]. During this process, CoA is released, and decarboxylation of the resulting 2-amino-3-ketoadipate intermediate (Scheme 1, V), yields a second quinonoid intermediate [referred to as quinonoid intermediate II (Scheme 1, VII)], which is proposed to be in a rapid equilibrium with an enol (Scheme 1, VI) [6–8,12]. The release of ALA from the active site, guided by conformational transitions, represents the last, and rate-limiting, step of the reaction [6–8].
Scheme 1.

Proposed chemical mechanism of mALAS2. (I) Internal aldimine complex, (II) glycine-external aldimine, (III) quinonoid intermediate I, (IV) glycine–succinyl-CoA condensation intermediate, (V) 2-amino-3-ketoadipate intermediate, (VI) enol intermediate, (VII) quinonoid intermediate II, (VIII) ALA-external aldimine. This scheme was originally published in Ref. [12] in the Journal of Biological Chemistry. © The American Society for Biochemistry and Molecular Biology.
ALAS is a homodimeric enzyme whose individual active sites are located at the dimeric interface and are composed of amino acids from each subunit [16,17]. The internal aldimine adduct resides deep in the active site interior, where glycine also binds upon its entry [17]. Crystallographic data indicate that association of succinyl-CoA with Rhodobacter capsulatus ALAS entails interactions with amino acids from the N-terminal, catalytic, and C-terminal domains of one monomer and the catalytic domain from the opposite monomer [17]. These extensive interactions ensure optimal positioning between the carbonyl group of the succinyl-moiety and the Cα of glycine, which is necessary for effective condensation of the substrates during catalysis. The adenosyl group of succinyl-CoA is important for initial recognition and is positioned within a mainly hydrophobic pocket, located on the active site surface [17]. Lys156 (R. capsulatus ALAS numbering; Lys221 in mALAS2) outlines the surface of this pocket, and its side chain associates with the ribose group through a putative hydrogen bond (Fig. 1) [17]. Mutations in the analogous lysine of human ALAS2 have been reported in cases of pyridoxine-responsive XLSA, implicating that this amino acid has an important physiological role in catalysis [18]. However, in the absence of a crystallographic structure of the reported XLSA variant, [i.e., K299Q [18] (human ALAS numbering; Lys221 in mature mALAS2)] and detailed kinetic studies, a potential hydrogen bond between the Gln side chain and the ribose group cannot be ruled out and the effects of the mutation in the positioning of the succinyl-CoA substrate within the active site and catalysis are difficult to predict. The pantetheine moiety of succinyl-CoA occupies a narrow channel that connects the surface of the active site to the interior, while the positioning of the succinyl moiety is coordinated by amino acids from the glycine-rich stretch and flexible active site loop [17]. This latter interaction stabilizes the closed conformation of ALAS by reducing the segmental motions of the active site loop. Furthermore, it is clear from the crystal structure of ALAS [17] that the association of succinyl-CoA with the enzyme profoundly minimizes the exposure of the active site interior to the solvent, partly due to stabilization of the closed conformation and occupancy of the narrow channel by the pantetheine moiety. However, at present, it is not known how catalysis might be influenced by reducing the exposure of the active site to the solvent.
Fig. 1.
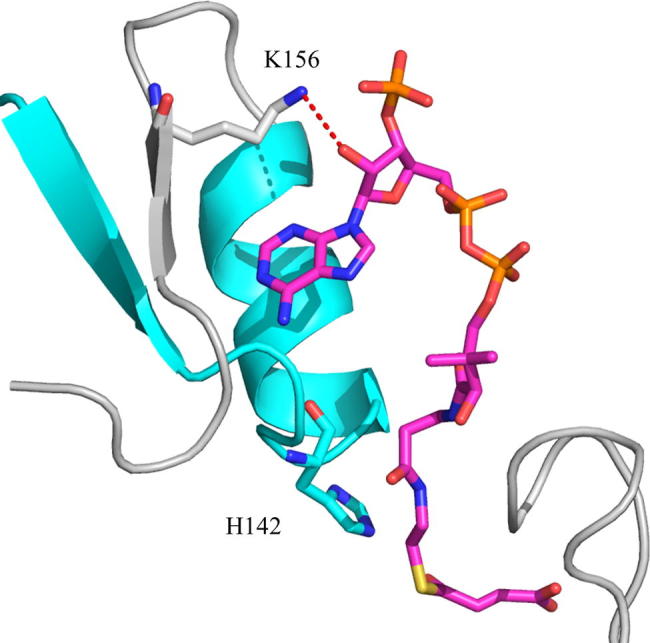
Lys156 (gray) of R. capsulatus ALAS coordinates the positioning of the adenosyl group of succinyl-CoA (magenta). On opposite sides of Lys156 are β-strand 134–138 and α-helix 143–150 (cyan), which are connected by a linker that contains the catalytic His142. R. capsulatus ALAS Lys156 and His142 correspond to Lys221 and His207 in mALAS2. Image created with Pymol using PDB: 2BWO.
The elucidation of the crystal structure of ALAS in complex with succinyl-CoA was followed by several studies that investigated the catalytic role of active site amino acids that coordinate the positioning of the succinyl-moiety [11,12,19], while largely ignoring those that associate with the adenosyl group, even though substitutions in this region can cause XLSA [18]. In this study, we characterized the catalytic role of Lys221 from the adenosyl-binding pocket of mALAS2 by disrupting the putative hydrogen bond that its side chain forms with the ribose moiety through substitution with the hydrophobic amino acid valine. Our study reveals that K221V increases by 23-fold, and that this increase does not originate from weaker binding affinity, but instead from effects of the mutation on individual catalytic rates, including that of quinonoid intermediate II formation and decay. Moreover, significant differences between and were detected, implicating that the latter constant should not be used as a measure of binding affinity. Furthermore, we discerned that binding of succinyl-CoA to wild-type mALAS2 is primarily facilitated by the CoA moiety, and that Lys221 contributes toward the recognition of CoA.
2. Materials and methods
2.1. Reagents
The following reagents were obtained from Fisher Scientific: glycine, potassium phosphate monobasic, potassium phosphate dibasic, HEPES, and glycerol. A bicinchoninic acid assay kit was from Thermo Scientific. Pyridoxal 5′-phosphate, bovine serum albumin (BSA) standards, DEAE resin, ACA-44 ultrogel, and sodium cyanoborohydride were from Sigma–Aldrich.
2.2. Construction and isolation of the K221V variant
The K221V variant was obtained by screening a library of mALAS2 variants for ALAS activity, as described previously for the T148A variant [12].
2.3. Overproduction, purification, and determination of protein concentrations
Wild-type mALAS2 and the K221V variant were overproduced and purified as described elsewhere [20]. SDS–PAGE analyses corroborated that the eluted proteins were purified to homogeneity. The protein concentration used for steady-state kinetic measurements was quantified using the bicinchoninic acid (BCA) assay, according to a protocol provided by the manufacturer (Thermo Scientific™ Pierce™) that entails incubation of the proteins of interest and BSA standards for 30 min at 37 °C upon the addition of the working reagent. The concentration of the enzymes used for transient kinetic measurements was determined spectroscopically [21].
2.4. Steady-state kinetics
A continuous spectrophotometric assay [22] was used to determine the steady-state kinetic activities of wild-type mALAS2 and K221V enzyme variant. ALAS activity was measured at pH 7.5 and 37 °C using a Shimadzu UV-2401 PC spectrophotometer. The final concentration of enzyme was 2 μM (wild-type mALAS2 or K221V), while the concentration of substrates ranged from (0.25–10)Km. All reactions were performed in a buffer composed of 20 mM HEPES, pH 7.5, and 10% glycerol. Secondary plots of apparent Vmax as a function of substrate concentration were constructed and the steady-state kinetic parameters were obtained from fitting the data to the Michaelis–Menten equation using non-linear regression analyses (SigmaPlot software).
2.5. Equilibrium dissociation constants for succinyl-CoA and CoA
The equilibrium dissociation constants (Kd) for succinyl-CoA and CoA were determined at 37 °C and pH 7.5 by measuring the quenching of intrinsic protein fluorescence at 340 nm (λex = 280 nm) upon titration of the enzyme (wild-type mALAS2 or K221V) with increasing concentration of ligand (succinyl-CoA or CoA). Titrations were performed by adding small aliquots of increasing concentrations of succinyl-CoA or CoA (from a 24 mM stock) to a reaction containing 1 μM enzyme (wild-type mALAS2 or K221V) in a buffer composed of 20 mM HEPES, pH 7.5, and 10% glycerol while stirring (the buffer pH did not change upon addition of ligands). Following incubation period of 1 min at 37 °C, spectra were collected using a Shimadzu RF5301 PC spectrofluorophotometer. Ligand addition did not significantly alter the final volume of the reaction (∼4% change in the final volume). The fraction of ligand bound (F.B) to the enzyme was obtained by normalizing the data using Eq. (1),
| (1) |
where x represents the maximal fluorescence emission at specific ligand concentration, xi is the emission in the absence of ligands, and xf is the emission at the highest concentration of ligand when the enzyme was fully saturated. The equilibrium dissociation constant, Kd, was determined by fitting the data to Eq. (2), using SigmaPlot software for the non-linear regression analyses
| (2) |
2.6. Stopped-flow absorption spectroscopy experiments
A KinTek stopped-flow apparatus (model SF-2001) was used to monitor transient changes in absorbance at a single wavelength (either 420 or 510 nm) upon rapid mixing of enzyme and reactants. Prior to the kinetic measurements, the purified, concentrated proteins were dialyzed into a 50 mM phosphate, pH 7.5, buffer with 10% glycerol (v/v) and 40 μM PLP. All reactions were performed at 18 °C in a buffer composed of 50 mM phosphate, pH 7.5, (37 mM K2HPO4/13 mM KH2PO4) and 10% glycerol (v/v). Reactions under single turnover conditions, with enzyme molar concentration in excess over succinyl-CoA, were monitored by following the changes in absorbance at 510 nm. One of the syringes contained 120 μM enzyme (either wild-type mALAS2 or K221V) and 200 mM glycine in the above buffer, while the other syringe contained 40 μM succinyl-CoA in the same buffer. The formation of the external aldimine, under multiple turnover conditions, was monitored at 420 nm; one of the syringes contained 60 μM enzyme (wild-type mALAS2 or K221V) and buffer, and the other syringe 200 mM glycine in the same buffer. Reductions of the internal aldimine with sodium cyanoborohydride were monitored at 420 nm. One of the syringes contained 40 μM enzyme (wild-type mALAS2 or K221V) in phosphate buffer, with succinyl-CoA concentrations ranging from 0 to 2.5 mM, while 5 mM sodium cyanoborohydride in the same phosphate buffer was present in the other syringe. The concentration of reactants in the observation chamber of all stopped-flow experiments reported in this study is one-half of that present in the syringes. The progress curves of the reactions were described using an equation with either one or three exponentials:
| (3) |
where At is the absorbance at time t, a is the amplitude of each phase, k is the observed rate constant for each phase and c is the final absorbance. For the sodium cyanoborohydride substrate protection experiments, in order to determine the concentration of succinyl-CoA at which one-half of the maximal protection is achieved, the reduction rates were normalized using Eq. (1), where x represents the rate at a given succinyl-CoA concentration, xi is the rate in the absence of succinyl-CoA, and xf is the rate at the highest concentration of substrate when a plateau was reached. The data were fitted to Eq. (4) using non-linear regression analysis (Sigma-Plot) to obtain the constant (K′) that signifies the succinyl-CoA concentration when the protection against reduction is 1/2the maximal
| (4) |
3. Results
3.1. Screening and selection of the K221V variant
A combinatorial active-site saturation test (CAST) [23] library of mALAS2 variants, in which random mutations were introduced simultaneously in the adenosyl-binding site, specifically at positions 214 and 221, was screened for functional ALAS variants using the ALA auxotrophic strain of HemA− Escherichia coli HU227 cells [24]. Permissible substitutions at position 221, which is occupied by Lys in wild-type mALAS2, included Ala, Arg, Asn, His, Ile, Leu, Ser, Thr and Val (data not shown). Cys, Phe, Pro and Tyr were also tolerated substitutions provided that wild-type mALAS2 I214 was simultaneously mutated (data not shown). Since our objective in examining the catalytic role of K221 was to eliminate the putative hydrogen bond between its side chain and the ribose group of the succinyl-CoA adenylate, we focused on the characterization of K221V in relation to wild-type mALAS2.
3.2. Steady-state kinetics
The steady-state kinetic parameters and specificity constants for wild-type mALAS2 and the K221V variant, determined at pH 7.5 and 37 °C, are reported in Table 1. In comparison to the wild-type enzyme, the kcat value for the K221V variant was moderately reduced. However, relative to the wild-type enzyme, the K221V mutation resulted in a 23-fold increase in the value and 97% reduction in . Interestingly, even though Lys221 does not participate in the binding of glycine, a 6-fold increase in the was also detected for the K221V variant.
Table 1.
Steady-state kinetic parameters and specificity constants.
| Enzyme | kcat (s−1) | (mM) | (μM) | (s−1 M−1) | (s−1 M−1) |
|---|---|---|---|---|---|
| wt mALAS2 | 0.25 ± 0.004 | 8 ± 0.7 | 1.0 ± 0.1 | 31.2 ± 3.0 | 2.5 × 105 ± 0.3 × 105 |
| K221V | 0.17 ± 0.002 | 50 ± 13 | 23 ± 2 | 3.4 ± 0.7 | 0.07 × 105 ± 0.007 × 105 |
3.3. Equilibrium dissociation constants (Kd) for succinyl-CoA and CoA
The Kd values for succinyl-CoA and CoA were obtained by monitoring the quenching of the intrinsic protein fluorescence (pH 7.5 and 37 °C) that resulted upon titration of the enzyme (wild-type mALAS2 or K221V) with increasing concentration of ligand (Fig. 2 and Table 2). Our data reveal that the binding of succinyl-CoA to the wild-type enzyme is primarily facilitated by the CoA, and not the succinyl moiety, since the Kd values for succinyl-CoA and CoA are comparable within error (Table 2). Interestingly, in comparison to the wild-type enzyme, the K221V mutation lowered the by ∼25%, indicating stronger affinity of the variant toward succinyl-CoA. However, the binding of CoA to the variant enzyme is weakened by the mutation, as evident from the increased . This implies an important role for the succinyl-moiety in the binding of succinyl-CoA to the active site of the K221V enzyme. Importantly, for either enzyme, there are significant differences between the and [Tables 1 and 2; (see Section 4)].
Fig. 2.

Binding isotherms resulting from the titration of wild-type mALAS2 (A) and K221V (B) with increasing concentrations of succinyl-CoA, and from the titration of wild-type mALAS2 (C) and K221V (D) with increasing concentrations of CoA. All reactions were done in duplicates.
Table 2.
Equilibrium dissociation constants for succinyl-CoA and CoA.
| Enzyme | (μM) | (μM) |
|---|---|---|
| wt mALAS2 | 297 ± 7 | 302 ± 12 |
| K221V | 223 ± 8 | 431 ± 17 |
3.4. Single turnover reactions: quinonoid intermediate II formation
The pre-steady-state rates for quinonoid intermediate II formation [λmax = 510 nm (Scheme 1, VII)] resulting from the reaction between the enzyme (wild-type or K221V)-glycine complex and succinyl-CoA were determined during a single turnover, with the enzyme molar concentration in excess over succinyl-CoA. The reactions were conducted at pH 7.5 and 18 °C, rather than 37 °C, because at the higher temperature, it was challenging to characterize the individual kinetic phases associated with the transient formation and decay of the quinonoid intermediate II. As presented in Fig. 3, the reactions for either the wild-type or variant enzyme are best described by an equation with three exponentials. Previous studies with mALAS2 led to the assignment of the three kinetic phases to: (k1) formation of the quinonoid intermediate II upon decarboxylation of the 2-amino-3-ketoadipate intermediate [Scheme 1, V], (k2) decay of the quinonoid intermediate II due to protonation of the enol [Scheme 1, VI], and (k3) release of ALA from the active site, guided by conformational transitions [7]. Even though Lys221 is located on the surface of the active site [17], and, consequently, does not directly act as an acid-base catalyst during the formation or decay of the quinonoid intermediate II, its substitution with a valine resulted in a decrease in the rates (Table 3). Specifically, relative to the wild-type reaction, the K221V variant catalyzes formation of the quinonoid intermediate II with a rate that is decreased by 29% (k1 = 2.7 ± 0.02 s−1 vs. k1 = 3.8 ± 0.03 s−1), while decreases of 39% and 21% were detected for k2 and k3, respectively (Table 3).
Fig. 3.
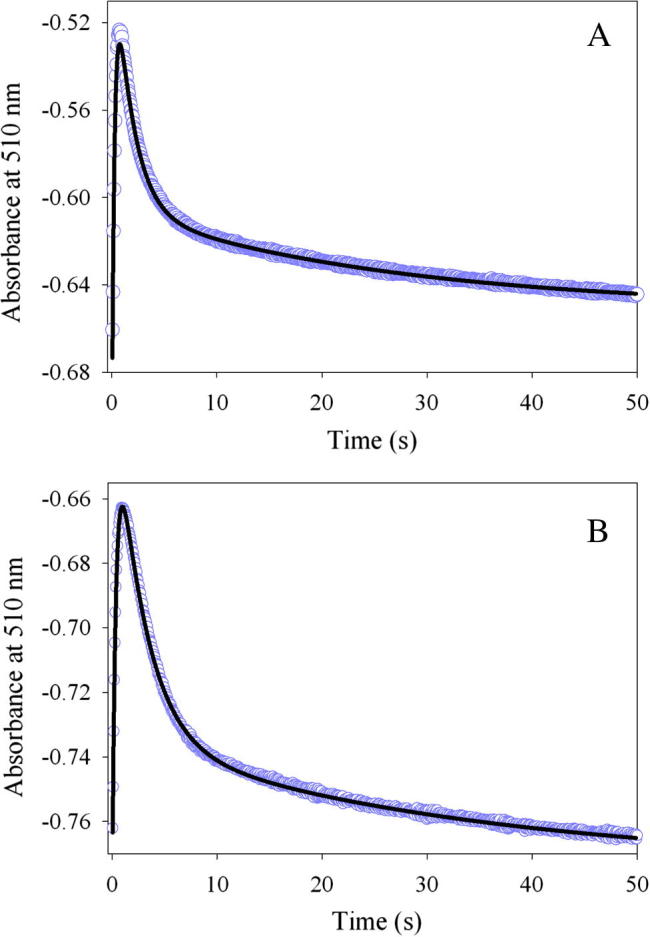
Progress curves for the single turnover reactions between (A) wild-type mALAS2-glycine complex or (B) K221V-glycine complex and succinyl-CoA monitored at 510 nm. The kinetic traces (data points in purple), collected at pH 7.5 and 18 °C, were fitted to a three-exponential equation, and the fitted lines are shown in black.
Table 3.
Observed rate constants for the reaction between the enzyme–glycine complex and succinyl-CoA.
| Enzyme | k1a (s−1) | k2b (s−1) | k3c (s−1) |
|---|---|---|---|
| wt mALAS2 | 3.8 ± 0.03 | 0.59 ± 0.005 | 0.038 ± 0.001 |
| K221V | 2.7 ± 0.02 | 0.36 ± 0.003 | 0.030 ± 0.001 |
Rate for quinonoid intermediate II formation.
Rate for first step of quinonoid intermediate II decay.
Rate for second step of quinonoid intermediate II decay.
3.5. Multiple turnover reactions: glycine-associated external aldimine formation
The reaction between the enzyme (wild-type mALAS2 or K221V) and glycine was examined by monitoring the changes in absorbance at 420 nm (at pH 7.5 and 18 °C) associated with the formation of the external aldimine complex (Scheme 1, II). Because of the high Km for glycine (Table 1), we had to resort to examining the reaction under multiple, rather than single, turnover conditions. Monophasic processes best describe the kinetic traces produced by the reactions of glycine with either the wild-type or variant enzymes (Fig. 4). Importantly, the K221V mutation has no pronounced effect on this transaldimination reaction, yielding an observed rate constant, k1 = 0.09 ± 0.001 s−1, which is nearly indistinguishable from the rate for the wild-type reaction (k1 = 0.08 ± 0.001 s−1).
Fig. 4.
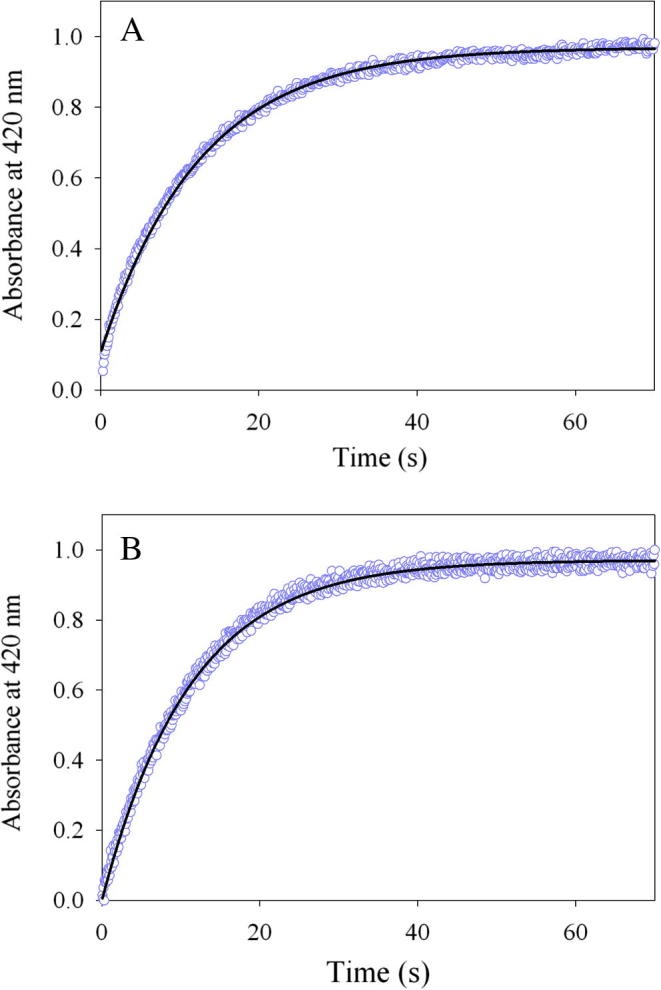
Progress curves for the multi-turnover reactions between (A) wild-type mALAS2 or (B) K221V and glycine monitored at 420 nm. The kinetic traces (data points in purple), collected at pH 7.5 and 18 °C, are best described by an equation with one exponential, and the fitted lines are shown in black.
3.6. Substrate protection studies: reduction with sodium cyanoborohydride
Since succinyl-CoA binds on the enzymatic surface and minimizes the exposure of the active site to the solvent, while the internal aldimine adduct (PLP-Lys313; mALAS2 numbering [Scheme 1, I]) is positioned deep within the active site interior [17], the rate of reduction of the internal aldimine with imine-specific reducing agents will depend on the strength of association of succinyl-CoA with ALAS. Thus, we performed substrate protection studies with succinyl-CoA by monitoring the transient decrease in absorbance at 420 nm upon reaction with sodium cyanoborohydride (a selective imine reducing agent under physiological conditions) [25]. The kinetic traces that resulted from reduction of the wild-type mALAS2 or K221V internal aldimine adducts, either in the presence or absence of succinyl-CoA, are best-described by a single-exponential equation (Fig. 5A and B). Upon increase in the concentration of succinyl-CoA, for either enzyme, we detected a decrease in the rate of internal aldimine reduction until a plateau was reached, and by fitting the normalized rates to an equation for a hyperbola (Fig. 5C and D), we obtained the concentration of succinyl-CoA at which one-half of the maximal protection against reduction is observed (K′). The data reveal that with K221V, maximal protection is reached at a lower succinyl-CoA concentration (K′ = 136 ± 11 μM) relative to wild-type mALAS2 (K′ = 261 ± 75 μM).
Fig. 5.

Selected kinetic traces for the sodium cyanoborohydride reductions of the (A) wild type mALAS2 and (B) K221V internal aldimine adducts monitored at 420 nm. Monophasic processes best described the kinetic traces in the absence (purple) or presence of 1.25 mM succinyl-CoA (gray) and the fitted lines are shown in black. The rates were normalized as described in Section 2, and K′ = 261 ± 75 μM and K′ = 136 ± 11 μM were obtained by fitting the normalized rates for wild-type mALAS2 (C) and the K221V enzyme (D) to Eq. (4), respectively. For the succinyl-CoA concentrations reported in (C) and (D), each symbol represents the average of three reaction rates and the error bars associated with the determined maximal protection values are obscured by the symbols.
4. Discussion
The elucidation of the crystal structure of R. capsulatus ALAS in complex with succinyl-CoA contributed to identification of the amino acids implicated in coordination of this substrate within the enzymatic active site [17]. Thus far, however, kinetic and mechanistic studies have been directed toward understanding the catalytic role of active site amino acids that coordinate the positioning of the succinyl-moiety of succinyl-CoA [11,12,19], while the catalytic importance of residues that are involved in the recognition of the adenosyl moiety has never been appraised and characterized. In this study, we performed detailed steady and pre-steady state kinetic analyses in order to understand the role of Lys221 of mALAS2 in catalysis. This amino acid, which is located on the surface of the mainly hydrophobic, adenosyl-binding pocket of ALAS, forms a hydrogen bond with the ribose group of succinyl-CoA (Fig. 1), and its physiological significance for effective catalysis is evident from reports that substitutions at this position lead to the erythropoietic disorder XLSA [18].
When the hydrogen bond between the side chain of Lys221 and the adenosyl moiety of succinyl-CoA was disrupted by introducing a valine, we observed that the mutation had only a moderate effect on the catalytic turnover of mALAS2, resulting in a 32% reduction in the kcat value (Table 1). While researchers have identified numerous XLSA-associated mutations in the human ALAS2 (hALAS2) gene [3], apart from a recent study reporting Vmax values for two XLSA hALAS2 variants, in which the mutations perturbed protein–protein interactions without impacting the catalytic activity [26], information is lacking on how the majority of XLSA mutations affect the catalytic turnover (kcat) of hALAS2. Even when the catalytic activities of the XLSA variants were estimated [18,27–30], their measurements were limited to initial reaction velocities and, often, involved the use of cell lysates or ALAS fusion proteins [18,27–30]. Therefore, at present, it is not known the extent of decrease or the lower limit in the kcat value of ALAS2 associated with the manifestation of XLSA. Despite the mild effects of the K221V mutation on the catalytic turnover of mALAS2, the K221V mutation has a profound effect on the specificity constant for succinyl-CoA, with a 97% decrease in the value of the K221V variant relative to that of the wild-type enzyme (Table 1). This reduction in the specificity constant, which is due to a 23-fold increase in , does not stem from weakened affinity of the variant toward succinyl-CoA, since contrary to expectations, the value for the K221V variant is actually lower than the one for the wild-type enzyme, implicating tighter binding (Table 2).
Even for a simple catalytic mechanism that consists of a single reaction intermediate, the Michaelis constant, Km, which is indicative of the substrate concentration when v = 1/2 Vmax, is a complex function consisting of multiple rates [31], and thus, it should not be misinterpreted with the equilibrium dissociation constant (Kd), because the two will only be identical when k−1 is significantly greater than kcat. In the case of mALAS2, comparison between and reveals significant differences between the two constants. Specifically, for the wild-type enzyme, the dissociation constant is 300-fold greater than the Michaelis constant, while for the K221V variant, the ratio between these two constants is reduced to 10-fold. Therefore, our results imply that the increase in does not originate from weakened affinity of the K221V variant toward succinyl-CoA, but rather from effects of the mutation on specific catalytic rate(s). Given the role of Lys221 in coordinating the positioning of succinyl-CoA [17], it is possible that the mutation affects the rate of condensation between the substrates, probably by impairing the attainment of a catalytically competent orientation between glycine and succinyl-CoA. However, presently, there is no assay that would allow us to measure the condensation rates and examine the validity of this hypothesis.
One of the reaction rates that we do know to be perturbed by the K221V mutation is that of quinonoid intermediate II catalysis (Table 3). Our single turnover transient kinetic studies revealed that the K221V variant catalyzes the formation and decay of the quinonoid intermediate II with rates that are reduced by about one-third relative to the wild-type reaction. This finding is rather intriguing, particularly since Lys221 is located on the enzymatic surface, far from any catalytic center [17], and is not directly involved in acid-base catalysis. The most probable explanation for this detrimental effect is that the mutation results in subtle structural perturbations of the active site, leading to an increase in the energetic barrier required for quinonoid intermediate II catalysis. The analogous lysine (Lys156) in R. capsulatus ALAS occupies a strand (β 156–159; R. capsulatus ALAS numbering) in the catalytic domain that is part of a mostly parallel, seven stranded β-sheet, covered on either side by nine helices [17]. On opposite sides of Lys156 are β-strand 134–138 and α-helix 143–150 (Fig. 1), which are connected by a linker that contains His142—the amino acid postulated to act as an acid catalyst during the formation of the quinonoid intermediate II [7]. Thus, it is possible that structural perturbations introduced by the K221V mutation indirectly affect the optimal positioning of the catalytic histidine.
Our data also indicate that the binding of succinyl-CoA to the active site of mALAS2 is primarily facilitated by the CoA, rather than the succinyl moiety, as evident from the identical Kd values for the two ligands (Table 2). Moreover, from the increase in that was detected for the K221V variant (Table 2), it can be deduced that Lys221 is important for initial recognition of the CoA moiety. Intriguingly, even though the K221V variant displays weaker binding toward CoA, its affinity for succinyl-CoA is actually higher than that of wild-type mALAS2 (Table 2), implicating that through stronger interactions with the succinyl moiety, the active site of the variant appears to compensate for its reduction in the affinity toward CoA.
The crystal structure of R. capsulatus ALAS [17] indicates that association of succinyl-CoA with the enzyme leads to significant reduction in the exposure of the active site to the solvent. This minimized exposure is due to stabilization of the closed conformation and occupancy of the active site channel by the pantetheine moiety. In order to validate the crystallographic predictions experimentally, we performed substrate protection studies with succinyl-CoA by monitoring the reduction of the internal aldimine, which is located in the active site interior [17], and the results revealed that succinyl-CoA binding does indeed minimize the exposure of the active site interior to the solvent (Fig. 5). These studies also indicate that with K221V, maximal level of protection is achieved at lower concentrations of succinyl-CoA relative to wild-type mALAS2, which is consistent with the finding that the variant has a lower than the wild-type enzyme.
In summary, the data presented in this study reveal that the K221V mutation reduces the specificity constant for succinyl-CoA, and that the increase in does not originate from weaker binding affinity, but rather from impairments in individual catalytic rates, including that of quinonoid intermediate II catalysis. Furthermore, it is possible that in vivo, substitutions at position Lys221 might have more pronounced effect on ALA biosynthesis, particularly if this lysine participates in protein–protein interactions with the β-subunit of ATP-specific succinyl-CoA synthetase (SUCLA2), the protein that is believed to channel succinyl-CoA to the active site of ALAS [32]. In fact, impairment in the ability to interact with the β-subunit of ATP-specific succinyl-CoA synthetase was detected in several ALAS2 variants associated with XLSA [26,32]. Thus, future endeavors should be directed toward detailed understanding of the structural regions of ALAS that are necessary for these protein–protein interaction.
Author contribution statement
B.M.S. and G.C.F. conceived and designed the project; B.M.S. acquired the data; B.M.S. and G.C.F. analyzed and interpreted the data; B.M.S. and G.C.F. wrote the paper.
Acknowledgements
This work was supported by grants from the American Heart Association (#10GRNT4300073) and the National Institutes of Health (#GM080270).
References
- 1.Fratz E.J., Stojanovski B.M., Ferreira G.C. Toward heme: 5-aminolevulinate synthase and initiation of porphyrin synthesis. In: Ferreira G.C., Kadish K.M., Smith K.M., Guilard R., editors. The Handbook of Porphyrin Science. World Scientific Publishing Co.; Singapore: 2013. pp. 1–78. [Google Scholar]
- 2.Hunter G.A., Ferreira G.C. Molecular enzymology of 5-aminolevulinate synthase, the gatekeeper of heme biosynthesis. Biochim. Biophys. Acta. 2011;1814:1467–1473. doi: 10.1016/j.bbapap.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bottomley S.S., Fleming M.D. Sideroblastic anemias: molecular basis, pathophysiology, and clinical aspects. In: Ferreira G.C., Kadish K.M., Smith K.M., Guilard R., editors. The Handbook of Porphyrin Science. World Scientific Publishing Co.; Singapore: 2013. pp. 44–87. [Google Scholar]
- 4.Whatley S.D., Ducamp S., Gouya L., Grandchamp B., Beaumont C., Badminton M.N., Elder G.H., Holme S.A., Anstey A.V., Parker M., Corrigall A.V., Meissner P.N., Hift R.J., Marsden J.T., Ma Y., Mieli-Vergani G., Deybach J.C., Puy H. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am. J. Hum. Genet. 2008;83:408–414. doi: 10.1016/j.ajhg.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balwani M., Doheny D., Bishop D.F., Nazarenko I., Yasuda M., Dailey H.A., Anderson K.E., Bissell D.M., Bloomer J., Bonkovsky H.L., Phillips J.D., Liu L., Desnick R.J. Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and X-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-linked protoporphyria. Mol. Med. 2013;19:26–35. doi: 10.2119/molmed.2012.00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hunter G.A., Ferreira G.C. Pre-steady-state reaction of 5-aminolevulinate synthase. Evidence for a rate-determining product release. J. Biol. Chem. 1999;274:12222–12228. doi: 10.1074/jbc.274.18.12222. [DOI] [PubMed] [Google Scholar]
- 7.Hunter G.A., Zhang J., Ferreira G.C. Transient kinetic studies support refinements to the chemical and kinetic mechanisms of aminolevulinate synthase. J. Biol. Chem. 2007;282:23025–23035. doi: 10.1074/jbc.M609330200. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J., Ferreira G.C. Transient state kinetic investigation of 5-aminolevulinate synthase reaction mechanism. J. Biol. Chem. 2002;277:44660–44669. doi: 10.1074/jbc.M203584200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akhtar M., Jordan P.M. The mechanism of action of δ-aminolaevulinate synthetase and the synthesis of stereospecifically tritiated glycine. Chem. Commun. 1968:1691–1692. [Google Scholar]
- 10.Abboud M.M., Jordan P.M., Akhtar M. Biosynthesis of 5-aminolevulinic acid: involvement of a retention–inversion mechanism. J. Chem. Soc. Chem. Commun. 1974:643–644. [Google Scholar]
- 11.Kaufholz A.L., Hunter G.A., Ferreira G.C., Lendrihas T., Hering V., Layer G., Jahn M., Jahn D. Aminolevulinic acid synthase of Rhodobacter capsulatus: high-resolution kinetic investigation of the structural basis for substrate binding and catalysis. Biochem. J. 2013;451:205–216. doi: 10.1042/BJ20121041. [DOI] [PubMed] [Google Scholar]
- 12.Stojanovski B.M., Hunter G.A., Jahn M., Jahn D., Ferreira G.C. Unstable reaction intermediates and hysteresis during the catalytic cycle of 5-aminolevulinate synthase: implications from using pseudo and alternate substrates and a promiscuous enzyme variant. J. Biol. Chem. 2014;289:22915–22925. doi: 10.1074/jbc.M114.574731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nandi D.L. Studies on δ-aminolevulinic acid synthase of Rhodopseudomonas spheroides. Reversibility of the reaction, kinetic, spectral, and other studies related to the mechanism of action. J. Biol. Chem. 1978;253:8872–8877. [PubMed] [Google Scholar]
- 14.Ferreira G.C., Neame P.J., Dailey H.A. Heme biosynthesis in mammalian systems: evidence of a Schiff base linkage between the pyridoxal 5′-phosphate cofactor and a lysine residue in 5-aminolevulinate synthase. Protein Sci. 1993;2:1959–1965. doi: 10.1002/pro.5560021117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferreira G.C., Vajapey U., Hafez O., Hunter G.A., Barber M.J. Aminolevulinate synthase: lysine 313 is not essential for binding the pyridoxal phosphate cofactor but is essential for catalysis. Protein Sci. 1995;4:1001–1006. doi: 10.1002/pro.5560040520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan D., Ferreira G.C. Active site of 5-aminolevulinate synthase resides at the subunit interface. Evidence from in vivo heterodimer formation. Biochemistry. 1996;35:8934–8941. doi: 10.1021/bi952918m. [DOI] [PubMed] [Google Scholar]
- 17.Astner I., Schulze J.O., van den Heuvel J., Jahn D., Schubert W.D., Heinz D.W. Crystal structure of 5-aminolevulinate synthase, the first enzyme of heme biosynthesis, and its link to XLSA in humans. EMBO J. 2005;24:3166–3177. doi: 10.1038/sj.emboj.7600792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cotter P.D., May A., Fitzsimons E.J., Houston T., Woodcock B.E., Al-Sabah A.I., Wong L., Bishop D.F. Late-onset X-linked sideroblastic anemia. Missense mutations in the erythroid delta-aminolevulinate synthase (ALAS2) gene in two pyridoxine-responsive patients initially diagnosed with acquired refractory anemia and ringed sideroblasts. J. Clin. Invest. 1995;96:2090–2096. doi: 10.1172/JCI118258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lendrihas T., Zhang J., Hunter G.A., Ferreira G.C. Arg-85 and Thr-430 in murine 5-aminolevulinate synthase coordinate acyl-CoA-binding and contribute to substrate specificity. Protein Sci. 2009;18:1847–1859. doi: 10.1002/pro.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferreira G.C., Dailey H.A. Expression of mammalian 5-aminolevulinate synthase in Escherichia coli. Overproduction, purification, and characterization. J. Biol. Chem. 1993;268:584–590. [PubMed] [Google Scholar]
- 21.Stojanovski B.M., Breydo L., Hunter G.A., Uversky V.N., Ferreira G.C. Catalytically active alkaline molten globular enzyme: effect of pH and temperature on the structural integrity of 5-aminolevulinate synthase. Biochim. Biophys. Acta. 2014;1844:2145–2154. doi: 10.1016/j.bbapap.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hunter G.A., Ferreira G.C. A continuous spectrophotometric assay for 5-aminolevulinate synthase that utilizes substrate cycling. Anal. Biochem. 1995;226:221–224. doi: 10.1006/abio.1995.1217. [DOI] [PubMed] [Google Scholar]
- 23.Reetz M.T., Carballeira J.D., Peyralans J., Hobenreich H., Maichele A., Vogel A. Expanding the substrate scope of enzymes: combining mutations obtained by CASTing. Chemistry. 2006;12:6031–6038. doi: 10.1002/chem.200600459. [DOI] [PubMed] [Google Scholar]
- 24.Umanoff H., Russell C.S., Cosloy S.D. Availability of porphobilinogen controls appearance of porphobilinogen deaminase activity in Escherichia coli K-12. J. Bacteriol. 1988;170:4969–4971. doi: 10.1128/jb.170.10.4969-4971.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lane C.F. Sodium cyanoborohydride – a highly selective reducing agent for organic functional groups. Synthesis. 1975;3:135–146. [Google Scholar]
- 26.Bishop D.F., Tchaikovskii V., Hoffbrand A.V., Fraser M.E., Margolis S. X-linked sideroblastic anemia due to carboxyl-terminal ALAS2 mutations that cause loss of binding to the beta-subunit of succinyl-CoA synthetase (SUCLA2) J. Biol. Chem. 2012;287:28943–28955. doi: 10.1074/jbc.M111.306423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Furuyama K., Fujita H., Nagai T., Yomogida K., Munakata H., Kondo M., Kimura A., Kuramoto A., Hayashi N., Yamamoto M. Pyridoxine refractory X-linked sideroblastic anemia caused by a point mutation in the erythroid 5-aminolevulinate synthase gene. Blood. 1997;90:822–830. [PubMed] [Google Scholar]
- 28.Harigae H., Furuyama K., Kimura A., Neriishi K., Tahara N., Kondo M., Hayashi N., Yamamoto M., Sassa S., Sasaki T. A novel mutation of the erythroid-specific delta-aminolaevulinate synthase gene in a patient with X-linked sideroblastic anaemia. Br. J. Haematol. 1999;106:175–177. doi: 10.1046/j.1365-2141.1999.01479.x. [DOI] [PubMed] [Google Scholar]
- 29.Kadirvel S., Furuyama K., Harigae H., Kaneko K., Tamai Y., Ishida Y., Shibahara S. The carboxyl-terminal region of erythroid-specific 5-aminolevulinate synthase acts as an intrinsic modifier for its catalytic activity and protein stability. Exp. Hematol. 2012;40(477–486):e471. doi: 10.1016/j.exphem.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 30.Cox T.C., Bottomley S.S., Wiley J.S., Bawden M.J., Matthews C.S., May B.K. X-linked pyridoxine-responsive sideroblastic anemia due to a Thr388-to-Ser substitution in erythroid 5-aminolevulinate synthase. N. Engl. J. Med. 1994;330:675–679. doi: 10.1056/NEJM199403103301004. [DOI] [PubMed] [Google Scholar]
- 31.Johnson K.A. The Enzymes. Academic Press; New York: 1992. Transient-state kinetic analysis of enzyme reaction pathways; pp. 1–61. [Google Scholar]
- 32.Furuyama K., Sassa S. Interaction between succinyl CoA synthetase and the heme-biosynthetic enzyme ALAS-E is disrupted in sideroblastic anemia. J. Clin. Invest. 2000;105:757–764. doi: 10.1172/JCI6816. [DOI] [PMC free article] [PubMed] [Google Scholar]