

Abstract
OBJECTIVES:
The survival rate of thalassemia patients has not been conclusively established, and the factors associated with survival remain unclear. This study aimed to determine the survival rate of thalassemia among patients in southern Iran and to identify the factors associated with mortality from thalassemia.
METHODS:
This retrospective cohort study was conducted based on a retrospective review of the medical records of 911 beta-thalassemia patients in 2014. Data analysis was conducted using the Kaplan-Meier method and Cox regression analysis.
RESULTS:
Overall, 212 patients (23.3%) died, and 26.8% had thalassemia intermedia. The 20-year, 40-year, and 60-year survival rates were 85%, 63%, and 54%, respectively. Both crude and adjusted analyses found that education, marital status, ferritin levels, and comorbidities were related to mortality.
CONCLUSIONS:
Sociodemographic and hematological factors were found to be significantly associated with the survival rate of thalassemia. Addressing these factors may help healthcare providers and physicians to provide the best possible care and to improve the survival rate.
Keywords: Survival, Thalassemia, Kaplan-Meier estimate, Iran
INTRODUCTION
Inherited hemoglobin disorders, such as thalassemia, are now considered among the most important public health challenges worldwide [1]. Recent surveys have estimated that 5.2% of the world population are carriers of such disorders, and over 330,000 affected infants are born annually, 80% of whom are born in developing countries [2]. Moreover, an estimated number of 18,000 deaths occurred due to thalassemia in 2010 [3].
In order to improve the overall health of the population and to reduce the burden of thalassemia on families, a thalassemia program was implemented in 1997 in the Iranian primary healthcare system [4]. As a result, the number of newborns with beta-thalassemia has decreased, and the heavy financial burden of the disease has been mitigated by the relatively low-cost measures of premarital screening and genetic counseling [5]. However, some barriers, such as cultural factors, have hindered the implementation of a well-organized prenatal diagnostic system [6].
Despite significant improvements over the last decades in treatment and the quality of life of patients [7], as well as decreases in mortality [8,9], many new cases of beta-thalassemia continue to emerge every year [10]. Several studies have investigated potential prognostic factors affecting the survival rate of beta-thalassemia patients, including birth cohort [11], sex [8], comorbidities [12], ferritin and hemoglobin levels [13], and the type of transfused blood [13].
Nonetheless, significant contributions remain to be made in order to better understand the prognostic factors affecting the long-term survival of thalassemia patients, particularly in areas with a high prevalence of the disorder. Without reliable information on the prognostic factors that affect the survival rate, it is difficult to perform effective interventions to improve the life expectancy of thalassemia patients. Thus, our study aimed to determine the survival rate of treated thalassemia patients in southern Iran and to identify factors that are related to the survival rate.
MATERIALS AND METHODS
This retrospective cohort study was conducted by enrolling 911 beta-thalassemia patients in 2014 in Shiraz, the largest city in southern Iran. The flowchart of the study is shown in Figure 1.
Figure 1.
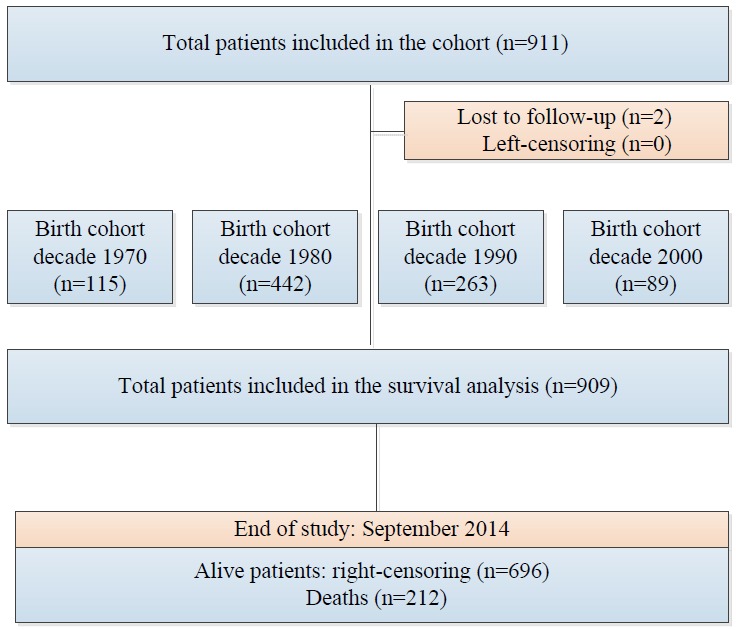
Flowchart of this retrospective study.
A program of thalassemia prevention was established in 1997 in Iran [4]. Due to some cultural barriers, new thalassemia patients are still born annually [14]. However, the disease is detected at the time of birth. Thalassemia patients are tracked prospectively using referral hospitals, and a paper-based database was created in 2004. There was no left-censoring in this study. The data of all patients, using the census method, were cross-sectionally extracted from patients’ medical records based on a predetermined checklist including age (calendar age at the time of enrollment in the study), sex, education level, marital status (single or married), consanguinity, date of birth, date of death (if applicable), blood group, ferritin and hemoglobin levels, blood transfusions, type of iron chelation drugs (desferrioxamine, deferiprone, exjade, desferal, or esferal), type of beta-thalassemia (major or intermedia), and comorbidities (heart failure, type 2 diabetes, hypogonadism, thyroidism, parathyroidism, osteoporosis, HIV/AIDS, or hepatitis B or C). Ferritin and hemoglobin levels were measured every six months and once a month, respectively. The average of the last four measurements (except for two patients) was used to represent the levels of ferritin and hemoglobin for the purpose our analysis.
This study was approved by the Fasa University of Medical Sciences (no. 94038).
The survival time for each patient, in terms of person-years, was defined as the interval between the date of birth and the date of death or September 2014 (the end of the study). Cumulative survival rates were estimated using the Kaplan-Meier method. The log-rank test was used to compare the survival curves across the subgroups. A univariate (crude analysis) and multivariate (adjusted analysis) Cox proportional hazards model was also used to identify risk factors and protective factors relating to mortality. Some prognostic factors, such as marital status and comorbidities, were not present at the onset of disease. As a result, age may have been a potential confounder, which was adjusted for in our analysis. For this purpose, our sample was divided into four birth cohorts (1970s, 1980s, 1990s, and 2000 to the present) and the post-2000 birth cohort was considered to be the reference group. Accordingly, in both the crude and the multivariate analyses, the birth cohort variable was adjusted for in order to identify the effect of marital status and comorbidities on the survival rate. The Akaike information criterion was used to compare the crude and adjusted models [15].
Since different types of iron chelation drugs were used by the patients, we adjusted for the effect of the types of the drugs in both the univariate and multivariate models. All statistical analyses were performed at the p<0.05 significance level using Stata version 11 (StataCorp, College Station, TX, USA).
RESULTS
A total of 911 beta-thalassemia patients were included, with 20,662.7 person-years of follow-up. The maximum follow-up time was 66.1 years. Overall, 212 (23.3%) patients died, two of whom had been lost to follow-up. The total mortality rate over the study period was 1.02 per 100 person-years. Of the patients, 50.8% were female and 11.0% were married. Overall, 40.2% of patients had not completed their formal education (no diploma). Comorbidities were observed in 27.3% of the patients, and 26.8% had thalassemia intermedia. The additional demographic characteristics of the subjects are presented in Table 1. The 20-year, 40-year, and 60-year survival rates of the patients were 85%, 63%, and 54%, respectively.
Table 1.
Descriptive statistics of the thalassemia patients included in this study
| n (%) | |
|---|---|
| Sex | |
| Male | 448 (49.2) |
| Female | 463 (50.8) |
| Education | |
| No diploma 1 | 322 (40.2) |
| Diploma | 478 (59.8) |
| Consanguinity | |
| No | 408 (49.6) |
| Yes | 415 (50.4) |
| Marital status | |
| Single | 738 (89) |
| Married | 91 (11) |
| Level of hemoglobin (g/dL) | |
| <9 | 420 (46.2) |
| ≥9 | 489 (53.8) |
| Level of ferritin (ng/mL) | |
| <2,500 | 543 (60) |
| ≥2,500 | 362 (40) |
| Comorbidities | |
| No | 603 (72.7) |
| Yes | 227 (27.3) |
| Type of thalassemia | |
| Intermedia | 244 (26.8) |
| Major | 667 (73.2) |
The patients who had not completed high school.
The results of the crude and adjusted analyses, using the Cox proportional hazard model, are shown in Table 2. In our crude analysis, sex, education, consanguinity, marital status, ferritin and hemoglobin levels, the type of beta-thalassemia, and comorbidities were found to be related to mortality. After adjusting for the birth cohort and other factors, the mortality risk of married subjects was found to be 69% lower than that of single subjects (hazard ratio [HR], 0.31; 95% confidence interval [CI], 0.12 to 0.82). Education level was also associated with survival, and more highly educated (diploma) patients had a lower risk of mortality than those with a lower level of education (no diploma; HR, 0.40; 95% CI, 0.26 to 0.62). Patients with comorbidities had a fourfold higher risk of mortality (HR, 3.7; 95% CI, 2.37 to 5.75), and survival was also associated with ferritin levels. Beta-thalassemia major patients did not have a significantly elevated risk of mortality in comparison to patients with thalassemia intermedia (HR, 1.54; 95% CI, 0.94 to 2.53).
Table 2.
Crude and adjusted analysis of the prognostic factors affecting survival time among thalassemia patients using the Cox proportional hazard model
| Crude |
p-value | Adjusted1 |
p-value | |||
|---|---|---|---|---|---|---|
| HR | 95% Cl | HR | 95% Cl | |||
| Sex | ||||||
| Male | 1.00 | - | - | 1.00 | - | - |
| Female | 0.65 | 0.45, 0.92 | 0.02 | 1.01 | 0.66, 1.52 | 0.98 |
| Education | ||||||
| No diploma2 | 1.00 | - | - | 1.00 | - | - |
| Diploma | 0.40 | 0.27, 0.59 | <0.001 | 0.40 | 0.26, 0.62 | <0.001 |
| Consanguinity | ||||||
| No | 1.00 | - | - | 1.00 | - | - |
| Yes | 1.63 | 1.15, 2.31 | 0.006 | 1.18 | 0.78, 1.78 | 0.31 |
| Marital status3 | ||||||
| Single | 1.00 | - | - | 1.00 | - | - |
| Married | 0.25 | 0.11,0.57 | 0.001 | 0.31 | 0.12, 0.82 | 0.02 |
| Level of hemoglobin (g/dL) | ||||||
| <9 | 1.00 | - | - | 1.00 | - | - |
| ≥9 | 0.62 | 0.44, 0.88 | 0.008 | 0.67 | 0.44, 1.01 | 0.05 |
| Level of ferritin (ng/mL) | ||||||
| <2,500 | 1.00 | - | - | 1.00 | - | - |
| ≥2,500 | 4.79 | 3.15, 7.27 | <0.001 | 4.86 | 2.96, 7.99 | <0.001 |
| Comorbidities3 | ||||||
| No | 1.00 | - | - | 1.00 | - | - |
| Yes | 3.96 | 2.70, 5.80 | <0.001 | 3.70 | 2.37, 5.75 | <0.001 |
| Type of thalassemia | ||||||
| lntermedia | 1.00 | - | - | 1.00 | - | - |
| Major | 1.78 | 1.18-2.70 | 0.007 | 1.54 | 0.94, 2.53 | 0.09 |
| Decade of birth | ||||||
| 2000 to present | 1.00 | - | - | 1.00 | - | - |
| 1990-1999 | 0.28 | 0.06, 1.29 | 0.10 | 0.12 | 0.02, 0.62 | 0.01 |
| 1980-1989 | 0.08 | 0.02, 0.36 | 0.001 | 0.03 | 0.01, 0.17 | <0.001 |
| 1979 and earlier | 0.02 | 0.003, 0.09 | < 0.001 | 0.01 | 0.001, 0.04 | <0.001 |
The type of iron chelation drug used by the patients was adjusted for in both the crude and adjusted models. The survival time was defined as the interval between the date of birth and the date of death or September 2014 (the end of the study).
HR, hazard ratio; CI, confidence interval.
Adjusted for birth cohort and all other variables in the table.
The patients who had not completed high school. The Akaike information criterion showed that the model including a birth cohort variable (adjusted model) fit better than the unadjusted model.
Odds ratio adjusted for birth cohort.
The Kaplan-Meier survival curves for thalassemia patients according to sex, consanguinity status, comorbidities, and type of thalassemia are presented in Figure 2.
Figure 2.

Kaplan-Meier survival curves for the thalassemia patients enrolled in this study stratified according to (A) sex (p=0.022), (B) consanguinity status (p=0.006), (C) comorbidities (p=0.001), and (D) type of thalassemia (p=0.004).
DISCUSSION
A total of 54% of the thalassemia patients survived the study period, meaning that the overall median survival time could not be calculated. However, the 20-year, 40-year, and 60-year survival rates of the patients were 85%, 63%, and 54%, respectively. Solid evidence indicates that the survival rate of thalassemia patients has been significantly improved. A cohort study conducted by Yavarian et al. [16] in 2006 in southern Iran showed that only 50% of beta-thalassemia major patients survived after age 30. Another study conducted in the southeast of Iran showed that the 25-year survival of beta-thalassemia major patients was 59.3% [13].
The crude analysis found that female gender was a protective factor against mortality, but the multivariate analysis did not confirm this association, suggesting that sex was a confounder variable. One possible reason could be noted for this important observation; namely, females were more likely to adhere to the recommended chelation therapy. This explanation is corroborated by the fact that the mean levels of serum ferritin were significantly higher in males than in females (2,557.3 ng/mL vs. 2,343.9 ng/mL; p<0.05, respectively). Other studies have addressed this issue. Borgna-Pignatti et al. [9] found that females had a higher survival rate than males, and similarly suggested that this association may have been due to better compliance on the part of females. Telfer et al. [8] suggested that further investigation of the different survival rates between females and males is necessary. Another study found no significant sex-based difference in the survival rate [13].
Our study strongly indicated that lower ferritin concentrations predicted longer survival, which is consistent with previous evidence [9,13,16]. Recent studies have demonstrated a better prognosis for survival in transfused patients whose ferritin concentrations remained below 2,500 ng/mL [9,13]. It has been found that chronic diseases are associated with a reduced survival rate. Our results were consistent with those of other studies [13,17]. Thalassemia is usually accompanied by other diseases. For example, 54.7% of Italian thalassemia patients were found to have hypogonadism [9], as well of 32.5% of thalassemia patients in Cyprus [18]. Accordingly, the early diagnosis and prevention of comorbidities is necessary to improve the quality of life in these patients. We also found that mean serum levels of ferritin were higher in patients with at least one comorbidity. This information may be especially useful in developing countries where monitoring serum ferritin levels is the only available tool for assessing iron overload.
We found consanguinity to have a significant association with mortality in the crude analysis, but this association was not significant in the adjusted analysis. Consanguinity may be a possible confounder regarding the survival rate of thalassemia. An earlier study likewise did not find a significant association between the survival time of beta-thalassemia and consanguinity [17]. Of the subjects in our study, 50.6% were the products of first-cousin or second-cousin marriages. This rate is similar to those reported in other studies conducted in Iran [19,20]. Although the thalassemia prevention program has improved the general knowledge and attitudes of high-risk couples [21], a high rate of consanguinity still appears to be present in the community.
Our finding that patients with lower levels of hemoglobin had a lower survival rate is consistent with that of a previous study carried out in another part of Iran [13]. In both crude and adjusted analyses, education level and marital status were also found to be associated with mortality. Highly educated and married subjects had a lower mortality risk. We found no previous study that would allow this finding to be contextualized.
One of our other findings was that thalassemia major patients had a lower survival rate than thalassemia intermedia patients. The thalassemia major patients differed from the thalassemia intermedia patients in a number of respects: namely, they had somewhat higher levels of ferritin and comorbidities. However, contrary to our expectations, the findings of our study showed that patients with thalassemia intermedia had a lower quality of life than those with thalassemia major [22].
A potential limitation of this study should be noted. We conducted a retrospective cohort study and estimated the survival rate using the data recorded. However, prospective studies with reliable sources of data employ the best design for estimating survival rates. Therefore, the quality and precision of our estimated rates primarily depend on the quality of the data present in the medical records. This could be a source of information bias. Another limitation is that a number of the thalassemia patients died before the period of our study, which may have affected the generalizability of the survival rates reported in this study.
Sociodemographic and hematological factors were significantly associated with the survival rate of beta-thalassemia. Addressing these factors may help healthcare providers and physicians to provide the best possible care and to improve the survival rate.
Acknowledgments
We would like to thank the Vice-Chancellor of Health of the Shiraz University of Medical Sciences for technical support of this study.
Footnotes
The authors have no conflicts of interest to declare for this study.
REFERENCES
- 1.Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79:704–712. [PMC free article] [PubMed] [Google Scholar]
- 2.Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86:480–487. doi: 10.2471/BLT.06.036673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samavat A, Modell B. Iranian national thalassaemia screening programme. BMJ. 2004;329:1134–1137. doi: 10.1136/bmj.329.7475.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khorasani G, Kosaryan M, Vahidshahi K, Shakeri S, Nasehi MM. Results of the national program for prevention of beta-thalassemia major in the Iranian Province of Mazandaran. Hemoglobin. 2008;32:263–271. doi: 10.1080/03630260802004269. [DOI] [PubMed] [Google Scholar]
- 6.Haghpanah S, Johari S, Parand S, Bordbar MR, Karimi M. Family planning practices in families with children affected by β-thalassemia major in Southern Iran. Hemoglobin. 2013;37:74–79. doi: 10.3109/03630269.2012.745419. [DOI] [PubMed] [Google Scholar]
- 7.Gollo G, Savioli G, Balocco M, Venturino C, Boeri E, Costantini M, et al. Changes in the quality of life of people with thalassemia major between 2001 and 2009. Patient Prefer Adherence. 2013;7:231–236. doi: 10.2147/PPA.S42133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Telfer P, Coen PG, Christou S, Hadjigavriel M, Kolnakou A, Pangalou E, et al. Survival of medically treated thalassemia patients in Cyprus. Trends and risk factors over the period 1980-2004. Haematologica. 2006;91:1187–1192. [PubMed] [Google Scholar]
- 9.Borgna-Pignatti C, Rugolotto S, De Stefano P, Zhao H, Cappellini MD, Del Vecchio GC, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica. 2004;89:1187–1193. [PubMed] [Google Scholar]
- 10.Dehshal MH, Ahmadvand A, Darestani SY, Manshadi M, Abolghasemi H. Secular trends in the national and provincial births of new thalassemia cases in Iran from 2001 to 2006. Hemoglobin. 2013;37:124–137. doi: 10.3109/03630269.2013.772062. [DOI] [PubMed] [Google Scholar]
- 11.Kosaryan M, Vahidshahi K, Karami H, Forootan MA, Ahangari M. Survival of thalassemic patients referred to the Boo Ali Sina Teaching Hospital, Sari, Iran. Hemoglobin. 2007;31:453–462. doi: 10.1080/03630260701641294. [DOI] [PubMed] [Google Scholar]
- 12.Borgna-Pignatti C, Cappellini MD, De Stefano P, Del Vecchio GC, Forni GL, Gamberini MR, et al. Survival and complications in thalassemia. Ann N Y Acad Sci. 2005;1054:40–47. doi: 10.1196/annals.1345.006. [DOI] [PubMed] [Google Scholar]
- 13.Roudbari M, Soltani-Rad M, Roudbari S. The survival analysis of beta thalassemia major patients in South East of Iran. Saudi Med J. 2008;29:1031–1035. [PubMed] [Google Scholar]
- 14.Moradi G, Ghaderi E. Chronic disease program in Iran: Thalassemia control program. Chronic Dis J. 2013;1:98–106. [Google Scholar]
- 15.Hein MJ, Schubauer-Berigan MK, Deddens JA. Evaluating bias from birth-cohort effects in the age-based cox proportional hazards model. Epidemiology. 2011;22:249–256. doi: 10.1097/EDE.0b013e3182093912. [DOI] [PubMed] [Google Scholar]
- 16.Yavarian M, Farsheedfar GR, Karimi M, Almoazzez M, Harteveld CL, Giordano PC. Survival analysis of transfusion dependent β-thalassemia major patients. J Res Health Sci. 2006;6:8–13. [Google Scholar]
- 17.Zamani R, Khazaei S, Rezaeian S. Survival analysis and its associated factors of Beta thalassemia major in hamadan province. Iran J Med Sci. 2015;40:233–239. [PMC free article] [PubMed] [Google Scholar]
- 18.Toumba M, Sergis A, Kanaris C, Skordis N. Endocrine complications in patients with Thalassaemia Major. Pediatr Endocrinol Rev. 2007;5:642–648. [PubMed] [Google Scholar]
- 19.Asadi-Pooya AA, Doroudchi M. Thalassemia major and consanguinity in Shiraz city, Iran. Turk J Haematol. 2004;21:127–130. [PubMed] [Google Scholar]
- 20.Miri-Moghaddam E, Zadeh-Vakili A, Rouhani Z, Naderi M, Eshghi P, Khazaei Feizabad A. Molecular basis and prenatal diagnosis of β-thalassemia among Balouch population in Iran. Prenat Diagn. 2011;31:788–791. doi: 10.1002/pd.2767. [DOI] [PubMed] [Google Scholar]
- 21.Kosaryan M, Vahidshahi K, Siami R, Nazari M, Karami H, Ehteshami S. Knowledge, attitude, and practice of reproductive behavior in Iranian minor thalassemia couples. Saudi Med J. 2009;30:835–839. [PubMed] [Google Scholar]
- 22.Safizadeh H, Farahmandinia Z, Nejad SS, Pourdamghan N, Araste M. Quality of life in patients with thalassemia major and intermedia in kerman-iran (I.R.) Mediterr J Hematol Infect Dis. 2012;4:e2015031. doi: 10.4084/MJHID.2012.058. [DOI] [PMC free article] [PubMed] [Google Scholar]