

Abstract
Inflammatory bowel disease (IBD) is the result of a combination of environmental, genetic and immunologic factors that trigger an uncontrolled immune response within the intestine, which results in inflammation among genetically predisposed individuals. Several studies have reported that the prevalence of classic cardiovascular risk factors is lower among subjects with IBD than in the general population, including obesity, dyslipidaemia, diabetes and hypertension. Therefore, given the risk profile of IBD subjects, the expected cardiovascular morbidity and mortality should be lower in these patients than in the general population. However, this is not the case because the standardized mortality ratio is not reduced and the risk of coronary heart disease is increased in patients with IBD. It is reasonable to hypothesize that other factors not considered in the classical stratification of cardiovascular risk may be involved in these subjects. Therefore, IBD may be a useful model with which to evaluate the effects of chronic low-grade inflammation in the development of cardiovascular diseases. Arterial stiffness is both a marker of subclinical target organ damage and a cardiovascular risk factor. In diseases characterized by chronic systemic inflammation, there is evidence that the inflammation affects arterial properties and induces both endothelial dysfunction and arterial stiffening. It has been reported that decreasing inflammation via anti tumor necrosis factor alpha therapy decreases arterial stiffness and restores endothelial function in patients with chronic inflammatory disorders. Consistent with these results, several recent studies have been conducted to determine whether arterial properties are altered among patients with IBD. In this review, we discuss the evidence pertaining to arterial structure and function and present the available data regarding arterial stiffness and endothelial function in patients with IBD.
Keywords: Arterial stiffness, Ulcerative colitis, Pulse wave velocity, Crohn’s disease, Inflammation, Tumour necrosis factor alpha
Core tip: The prevalence of classic cardiovascular risk factors, including obesity, dyslipidaemia, diabetes and hypertension, is lower among patients with inflammatory bowel disease (IBD) than in the general population. However, the risk of coronary heart disease is increased in IBD patients. Chronic inflammation may explain the difference between expected and observed cardiovascular risk. Arterial stiffness, a marker of subclinical target organ damage and a cardiovascular risk factor, is increased in chronic inflammatory disorders. In this review, we discuss the evidence pertaining to arterial structure and function and present the available data regarding arterial stiffness and endothelial function in patients with IBD.
INTRODUCTION
The idea that “man is as old as his arteries” was postulated by William Osler more than a century ago[1]. This axiom, initially used only in the setting of atherosclerosis, has also been used in the setting of increased arterial stiffness. Many studies, including a recent meta-analysis, have reported that aortic stiffness predicts an individual’s risk of developing cardiovascular disease independently of the classic risk factors[2,3]. Moreover, arterial stiffness and endothelial function have been identified as markers of subclinical target organ damage[4]. As target organ damage predicts cardiovascular death independently of the classic cardiovascular risk factors, it has been suggested that identifying organ damage, particularly among individuals at moderate risk for cardiovascular disease (CVD), may be useful[4]. Among these patients, the presence of increased arterial stiffness is sufficient to reclassify their risk of CVD from moderate to high[4].
A relationship between arterial stiffness and several markers of inflammation has been described in healthy subjects and hypertensive individuals[5,6], as well as in patients with chronic inflammatory disorders[7-10], in whom arterial stiffening occurs independently of atherosclerosis and is related to disease duration[8]. Chronic inflammation has also been linked to endothelial dysfunction[7].
Inflammatory bowel disease (IBD) is a chronic inflammatory condition that results from a combination of environmental, genetic and immunologic factors that trigger an uncontrolled immune response within the intestine in genetically predisposed individuals[11]. The dysfunction of the intestinal immune system and cross-reactivity against host epithelial cells have both been implicated as the primary mechanisms by which said inflammation occurs[12]. Therefore, among patients with IBD, it is reasonable that the chronic low-grade inflammation and the acute inflammation that occur during relapses of the disease may affect arterial properties. Several groups have studied both endothelial function and arterial stiffness in subjects with IBD. In this review, we briefly describe the physiology of the arterial system and the available data regarding both arterial stiffness and endothelial function in the setting of IBD.
PHYSIOLOGY OF THE ARTERIAL SYSTEM
The human arterial system is designed to receive pulsatile blood from the left ventricle and distribute it as a steady flow through the peripheral capillaries. Two distinct functions of the arterial tree may be schematized as follows: the ability (1) to deliver blood from the left ventricle (LV) to the capillaries of organs and tissues (conduit function); and (2) to dampen the blood flow and pressure oscillations generated by the heart, ensuring peripheral organ perfusion at both a steady flow rate and pressure (cushioning function)[13].
The efficiency of the conduit function is a consequence of both arterial diameter and the low resistance offered by large arteries to flow (in the supine position, mean blood pressure drops between the ascending aorta and the arteries in the forearm and leg by no more than 2-4 mmHg).
The thoracic aorta and its principal branches are rich in elastic fibres (elastic arteries). In the abdominal aorta and smaller arteries, the numbers of elastic fibres progressively decrease and are replaced by muscular fibres (muscular arteries). The presence of elastic fibres within the walls of the large arteries enables them to dampen blood pressure fluctuations, confining flow pulsations to the larger arteries, particularly the proximal aorta, and storing the stroke volume during systole. Under physiologic conditions, approximately half of the stroke volume is forwarded directly to the peripheral tissues, whereas the remaining 50% of the stroke volume is momentarily stored within the aorta and the large elastic arteries stretching the arterial walls (Figure 1). Approximately 10% of the energy produced by the heart is stored within the arterial wall by increasing the distension of the arteries. During diastole, the energy imbricated within the arterial wall is discharged, and the stored blood is forwarded to the peripheral tissues, ensuring continuous flow and contributing to the maintenance of sufficient diastolic blood pressure.
Figure 1.
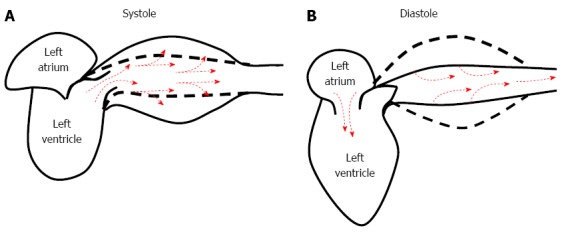
Role of arterial compliance in the damping of blood flow and the pressure oscillations generated by the heart. A: During systole, a portion of the stroke volume is forwarded directly to the peripheral tissues; approximately 50% of the stroke volume is momentarily stored within the aorta and stretches the arterial walls; B: During diastole, the energy imbricated within the arterial wall is discharged, and the stored blood is forwarded into the peripheral tissues, ensuring continuous flow and contributing to the maintenance of sufficient diastolic blood pressure.
With ageing, repetitive pulsations (approximately 35 million/year) cause fatigue and fracture the elastin lamellae of the elastic arteries. Ageing is also associated with a number of molecular changes in the load-bearing media of the elastic arteries, as follows: the orderly arrangement of elastic fibres and laminae is gradually lost over time, and thinning, splitting, fraying and fragmentation are observed. The degeneration of the elastic fibres is associated with an increase in collagenous material and these changes are often accompanied by calcium deposition and degenerated elastic fibres[14,15]. These processes result in both the stiffening and the dilatation of the large arteries and the early return of the reflected pressure waves to the heart. In the setting of increased arterial stiffness, the aorta and the elastic arteries cannot be stretched during systole. Consequently, the entire stroke volume flows through the arterial system and peripheral tissues only during systole, increasing systolic blood pressure and decreasing diastolic blood pressure.
ARTERIAL STIFFNESS AND REFLECTED WAVES
The integration of the conduit and cushioning functions results in pressure wave propagation and reflection (Figure 2). At each level of the arterial tree, the arterial pulse may be divided into two components, a forward or incident pressure wave, which originates at the level of the left ventricle, and a backward pressure wave, the sum of the reflected waves that originates primarily at the level of the high-resistance arterioles. The forward pressure wave generated within the aorta is propagated to arteries throughout the body. The progressive and physiological increase in arterial stiffness from the proximal aorta to the peripheral muscular arteries, together with the changes in aortic geometry, local arterial branching and luminal narrowing, produces an impedance mismatch and causes partial reflections of the forward pressure waves. The reflected pressure waves travel back to the central aorta and participate in changes in the amplitude of both the systolic blood pressure and the pulse pressure along the arterial tree. The stiffer the elastic and muscular arteries, the faster the forward and backward pulse waves within the arterial tree, and the earlier the return of the backward wave to the ascending aorta. Consequently, at the level of the proximal aorta, the backward wave interacts with the forward wave during diastole in subjects with elastic arteries (i.e., in youth) and during systole in subjects with increased arterial stiffness (i.e., either in the elderly or in the setting of pathological conditions). This causes both a greater peak in aortic pressure during systole and a larger decline during diastole.
Figure 2.
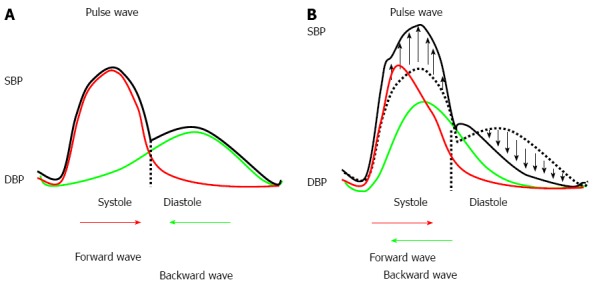
Arterial stiffness and reflection waves. A: Pulse wave in subjects with normal arterial stiffness; B: Pulse waves in subjects with increased arterial stiffness. DBP: Diastolic blood pressure; SBP: Systolic blood pressure.
HOW TO MEASURE ARTERIAL STIFFNESS IN CLINICAL PRACTICE
Several techniques and devices have been validated to measure arterial stiffness in clinical practice[2]. A direct reflection of arterial stiffness, pulse wave velocity (PWV) represents the gold standard for assessing regional arterial stiffness in daily practice[2]. PWV is the speed at which the pressure wave generated by cardiac ejection is propagated through the arterial tree. PWV is usually measured noninvasively from pressure waveforms obtained transcutaneously at the level of the right common carotid artery and the right femoral artery (carotid-femoral PWV), and the time delay (transit time) measured between the feet of the two waveforms, where the foot of the wave is defined at the end of diastole, when the steep rise of the wavefront begins. The distance covered by the waves is assimilated to the surface distance between the two recording sites[2]. PWV is classically calculated by dividing the distance travelled (in metres) by the time delay (in seconds) between the arrival of the pulse wave at the level of two different measuring sites (Figure 3). Carotid-femoral PWV is equivalent to the stiffness of the aorta. An increased carotid-femoral PWV is considered both a marker of target organ damage and a cardiovascular risk factor[16]. PWV may also be measured at the level of the peripheral muscular arteries (i.e., the brachial artery, carotid-radial PWV); however, the role of muscular artery stiffness in the prediction of cardiovascular risk remains a matter of debate.
Figure 3.
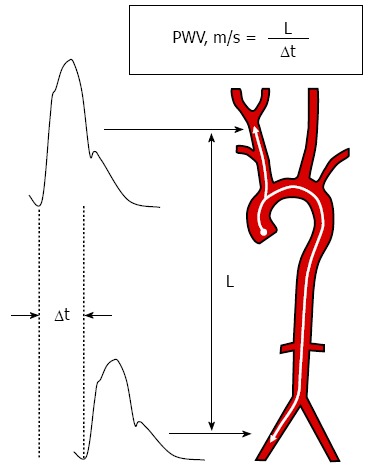
Reference technique utilized to measure carotid-femoral pulse wave velocity. PWV: Pulse wave velocity; L: The distance between the two measurement sites; Δt: The time lag between the pulse waves acquired at the proximal (carotid) and distal (femoral) sites.
PWV should not be confused with blood velocity, as the former is related to the transmission of energy through the arterial wall and varies between 4 and 12 m/s with both age and pressure, whereas the latter is related to the displacement of mass through an incompressible blood column and varies in order of cm/s.
ARTERIAL STIFFNESS AND CARDIOVASCULAR EVENTS, THE PATHOPHYSIOLOGICAL BASIS
In the setting of arterial stiffness, the early return of the reflected waves is primarily responsible for the rise in central systolic blood pressure, the drop in diastolic blood pressure and the associated increase in pulse pressure. The increased central systolic blood pressure is responsible for the augmented systolic work (left ventricular load), left ventricular oxygen requirements and the resultant risk of left ventricular hypertrophy[17]. The physiologic return of the reflected waves at the level of the proximal aorta during diastole is important in maintaining adequate perfusion in the myocardial microvasculature. Therefore, the decrease in central diastolic blood pressure caused by the increased arterial stiffness is responsible for the decreased coronary artery perfusion pressure observed during diastole, as well as the increased risk of myocardial infarction. Moreover, in the setting of left ventricular hypertrophy, the perfusion of the myocardial microvasculature is decreased because the hypertrophied heart contracts and relaxes more slowly, and the duration of systole is subsequently increased, whereas the duration of diastole is decreased. The elevated central pulse pressure that drives cerebral blood flow is responsible for the increased risk of stroke among patients with increased arterial stiffness.
ARTERIAL STIFFNESS AND INFLAMMATION
Several factors have been implicated in the pathophysiology of arterial stiffening. An emerging causal factor is the presence of systemic inflammation. This relationship has been described in various chronic inflammatory disease states, including systemic vasculitis[9], systemic lupus erythematosus[8], rheumatoid arthritis[8] and HIV[18]. Even acute, mild and transient inflammatory stimuli have been associated with the deterioration of the elastic properties of the large arteries[19]. It should be noted that in chronic inflammatory disorders, arterial stiffening may occur independently of atherosclerosis and has been linked to the duration of the inflammatory disease in question[8].
ARTERIAL STRUCTURE AND FUNCTION IN THE SETTING OF IBD
Only a limited number of studies have evaluated endothelial function in subjects with IBD. In a study published in 2007, microvascular dysfunction was linked to the loss of nitric oxide generation by the microvascular endothelium[20]. More recently, endothelial dysfunction was also reported in arterial districts far from the intestinal tract. In 2009 a study described low flow-mediated dilatation and shear stress reactive hyperaemia[21]. These results were confirmed by independent research groups that studied both adult patients[22] and paediatric patients[23]. The number of circulating endothelial precursor cells, markers of endothelial function, was significantly reduced in patients with Crohn’s disease (CD), as well as patients with ulcerative colitis (UC), compared with healthy controls, whereas the number of apoptotic endothelial precursor cells was higher in both patients with CD and patients with UC[22]. It was also observed that endothelial function improves following the administration of tumour necrosis factor-alpha (TNF-α) antagonists[24].
Only a limited number of studies have evaluated arterial stiffness in the setting of IBD (Table 1). The first study that measured the PWV in IBD was published by our group in 2012[25]. The stiffness of the elastic arteries (carotid-femoral PWV) and the muscular arteries (carotid-radial PWV) were both increased in subjects with IBD; no significant difference in PWV was noted between the patients with UC and those with CD. These results were subsequently duplicated by independent research groups[26-29]. A correlation between disease duration, a surrogate marker of the chronic inflammatory burden, and arterial stiffness was also reported by our group[25,29] and confirmed by an independent research group[27]. A relationship between arterial stiffness and several markers of inflammation (higher disease activity and more extensive involvement) was also reported[26]. Interestingly, in each of the studies that described increased arterial stiffness in the setting of IBD, only a few subjects (0%-19%) were treated with anti TNF-α therapy. By contrast, in two studies published by the same group[30,31], a higher percentage of patients, approximately 50%, were treated with anti TNF-α therapy; arterial stiffness was found to be only slightly increased in the setting of IBD. These findings were consistent with those of recent reports demonstrating that arterial stiffness[30] and endothelial function[24] both improved following the administration of anti TNF-α therapy among subjects with IBD, findings suggestive of a pivotal role for this cytokine in the pathogenesis of arterial dysfunction.
Table 1.
Studies that have measured carotid-femoral pulse wave velocity in subjects with inflammatory bowel disease
| Ref. | Anti TNF-α therapy (%) |
Subjects, n |
Pulse wave velocity (m/s) |
||||
|
IBD |
Controls |
IBD |
Controls | ||||
| CD | UC | CD | UC | ||||
| Zanoli et al[25] (2012) | 13 | 16 | 16 | 32 | 6.5 ± 1.5 | 6.8 ± 1.3 | 6.0 ± 0.8a |
| Akdogan et al[26] (2013) | 5 | 0 | 37 | 30 | - | 8.9 ± 3.0 | 7.2 ± 1.7c |
| Theocharidou et al[31] (2013) | 44 | 43 | 23 | 44 | 6.8 ± 1.3 | 6.3 ± 1.1 | 6.1 ± 0.9 |
| Zanoli et al[29] (2014) | 19 | 34 | 40 | 80 | 8.0 ± 1.6 | 7.8 ± 1.7 | 7.0 ± 1.1bcd |
| Korkmaz et al[27] (2014) | 2 | 18 | 84 | 74 | 6.4 ± 1.2 | 6.6 ± 1.2 | 5.9 ± 1.2a |
| Theocharidou et al[31] (2014) | 46 | 29 | 15 | 44 | 7 ± 1.2 | 6.3 ± 1.2d | 6.4 ± 0.9d |
| Aytaç et al[28] (2015) | 0 | 25 | 30 | 25 | 9.6 ± 1.4 | 9.3 ± 1.3 | 7.6 ± 0.3ef |
Data are presented as percentages (%), counts or mean ± SD.
P < 0.05 vs the whole group of subjects with IBD;
P < 0.001 vs the whole group of subjects with IBD;
P < 0.05 vs subjects with ulcerative colitis;
P < 0.001 vs subjects with Crohn’s disease;
P < 0.05 vs subjects with ulcerative colitis;
P < 0.001 vs subjects with Crohn’s disease. CD: Crohn’s disease; UC: Ulcerative colitis; IBD: Inflammatory bowel disease.
HOW DOES INFLAMMATION AFFECT ARTERIAL ELASTIC PROPERTIES?
Inflammation may stiffen the large arteries via several mechanisms (Figure 4). First, in diseases characterized by chronic inflammation, including IBD, endothelial dysfunction has been reported by multiple groups[7,20-23]. It has been suggested that endothelium-derived factors such as nitric oxide and endothelin-1 may influence the development of arterial stiffness[32,33]. The presence of endothelial dysfunction may result in functional arterial stiffening and the concomitant reduction of nitric oxide bioavailability and the increased activity of an opposing mediator, endothelin-1 (Figure 4A). This mechanism may also be responsible for the reversible increase in the stiffness observed among subjects suffering from acute inflammation.
Figure 4.
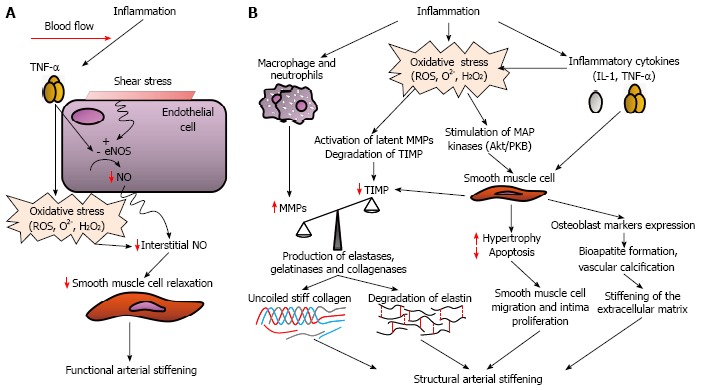
Potential mechanisms by which inflammation can induce functional (A) and structural (B) arterial stiffening. eNOS: Endothelial nitric oxide synthase; H2O2: Hydrogen peroxide; IL-1: Interleukin-1; MMPs: Matrix metalloproteinases; NO: Nitric oxide; O2-: Superoxide; ROS: Reactive oxygen species; TIMP: Tissue inhibitor of matrix metalloproteinases; TNF-α: Tumor necrosis factor alpha.
Endothelial dysfunction may also be associated with both the hyperplasia of vascular smooth muscle cells and the increased synthesis of collagen[34], resulting in structural arterial stiffening (Figure 4B). Moreover, the increased levels of circulating inflammatory mediators (i.e., interleukin-1 and TNF-α) promote white blood cell infiltration into blood vessels and changes in vascular smooth muscle phenotypes, which may release matrix metalloproteinases. The increased fragmentation of elastin molecules may be mediated by the activation of both matrix metalloproteinases and serine proteinases[35,36]. In addition to elastin degradation, matrix metalloproteinases also have collagenolytic activity, which results in the generation of uncoiled and stiffer collagen[35]. The increased matrix metalloproteinase activity may also be mediated by the presence of oxidative stress, which may activate latent matrix metalloproteinases and degrade tissue inhibitors of matrix metalloproteinases, as well as the increased activity of cell adhesion molecules[37,38]. In the setting of chronic inflammation, vascular smooth muscle cells also express osteoblast markers, take up phosphate and produce bioapatite, resulting in medial calcification and reduced vessel elasticity[39], and also produce C-reactive protein (CRP). CRP has an active role in promoting vascular inflammation and reducing endothelial function. Perivascular and vasa vasorum inflammation may result in vessel ischaemia, particularly in the setting of thrombo-occlusion, which may also promote both matrix remodelling and arterial stiffening.
INCREASED CARDIOVASCULAR RISK IN SUBJECTS WITH A LOW PREVALENCE OF CLASSIC CARDIOVASCULAR RISK FACTORS, THE IBD PARADOX
By definition, the higher the prevalence of classic cardiovascular risk factors, the higher the risk of cardiovascular events. However, upon the review of the literature, this axiom does not appear to apply to subjects with IBD. Several studies have reported that the prevalence of classic cardiovascular risk factors is lower among subjects with IBD than in the general population[40-43]. In particular, body mass index and lipid levels are lower in patients with IBD[40-43]. These patients also have lower rates of diabetes, obesity and hypertension[43]. Therefore, given the risk profile of IBD subjects, the expected cardiovascular morbidity and mortality would be expected be lower in these patients than in the general population. However, although the standardized mortality ratio was not reduced[44], the risk of coronary heart disease was reportedly increased in patients with IBD[43,45]. It is reasonable to hypothesize that other factors not considered in the classical stratification of cardiovascular risk may be involved in these subjects. Therefore, IBD may be a useful model with which to evaluate the effects of chronic low-grade inflammation in the development of cardiovascular diseases. In contrast to other clinical models of chronic inflammation in which the prevalence of classic cardiovascular risk factors is comparable with those of the general population, among subjects with IBD, the low cardiovascular risk associated with the low prevalence of classic cardiovascular risk factors may partially offset the cardiovascular burden associated with chronic inflammation[46]. An improved understanding of these concomitant but opposing effects, which are currently not considered in the cardiovascular risk stratification of patients with IBD, may result in the development of specific programs of intervention aimed at reducing cardiovascular risk. As is the case with other diseases characterized by chronic inflammation[7], increased arterial stiffness may represent a link between chronic low-grade inflammation and increased cardiovascular risk among patients with IBD. Additional studies are necessary to determine whether reduced arterial stiffness and improved endothelial function (with anti TNF-α therapy) decreases the risk of cardiovascular events among subjects with IBD.
In conclusion, there is evidence indicating that in the setting of IBD, as with other chronic inflammatory disorders, endothelial function is reduced, and arterial stiffness is increased. Treatment with anti TNF-α therapy appears to be associated with improvements in both endothelial function and arterial stiffness. Additional studies are necessary to determine whether the improvements in arterial stiffness and endothelial function are associated with a decreased risk of cardiovascular events in subjects with IBD.
Footnotes
Conflict-of-interest statement: None of the authors have any conflicts of interest to report.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: March 31, 2015
First decision: May 18, 2015
Article in press: September 2, 2015
P- Reviewer: Desai DC, Hokamavan A, Langenberg DR S- Editor: Yu J L- Editor: A E- Editor: Wang CH
References
- 1.Osler W. The Principles and Practice of Medicine. 3rd ed. New York, NY: Appleton; 1898. [Google Scholar]
- 2.Laurent S, Cockcroft J, Van Bortel L, Boutouyrie P, Giannattasio C, Hayoz D, Pannier B, Vlachopoulos C, Wilkinson I, Struijker-Boudier H. Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur Heart J. 2006;27:2588–2605. doi: 10.1093/eurheartj/ehl254. [DOI] [PubMed] [Google Scholar]
- 3.Ben-Shlomo Y, Spears M, Boustred C, May M, Anderson SG, Benjamin EJ, Boutouyrie P, Cameron J, Chen CH, Cruickshank JK, et al. Aortic pulse wave velocity improves cardiovascular event prediction: an individual participant meta-analysis of prospective observational data from 17,635 subjects. J Am Coll Cardiol. 2014;63:636–646. doi: 10.1016/j.jacc.2013.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.ESH/ESC Task Force for the Management of Arterial Hypertension. 2013 Practice guidelines for the management of arterial hypertension of the European Society of Hypertension (ESH) and the European Society of Cardiology (ESC): ESH/ESC Task Force for the Management of Arterial Hypertension. J Hypertens. 2013;31:1925–1938. doi: 10.1097/HJH.0b013e328364ca4c. [DOI] [PubMed] [Google Scholar]
- 5.Pietri P, Vyssoulis G, Vlachopoulos C, Zervoudaki A, Gialernios T, Aznaouridis K, Stefanadis C. Relationship between low-grade inflammation and arterial stiffness in patients with essential hypertension. J Hypertens. 2006;24:2231–2238. doi: 10.1097/01.hjh.0000249701.49854.21. [DOI] [PubMed] [Google Scholar]
- 6.Yasmin CM, Wallace S, Mackenzie IS, Cockcroft JR, Wilkinson IB. C-reactive protein is associated with arterial stiffness in apparently healthy individuals. Arterioscler Thromb Vasc Biol. 2004;24:969–974. doi: 10.1161/01.ATV.zhq0504.0173. [DOI] [PubMed] [Google Scholar]
- 7.Mäki-Petäjä KM, Hall FC, Booth AD, Wallace SM, Yasmin PW, Harish S, Furlong A, McEniery CM, Brown J, Wilkinson IB. Rheumatoid arthritis is associated with increased aortic pulse-wave velocity, which is reduced by anti-tumor necrosis factor-alpha therapy. Circulation. 2006;114:1185–1192. doi: 10.1161/CIRCULATIONAHA.105.601641. [DOI] [PubMed] [Google Scholar]
- 8.Roman MJ, Devereux RB, Schwartz JE, Lockshin MD, Paget SA, Davis A, Crow MK, Sammaritano L, Levine DM, Shankar BA, et al. Arterial stiffness in chronic inflammatory diseases. Hypertension. 2005;46:194–199. doi: 10.1161/01.HYP.0000168055.89955.db. [DOI] [PubMed] [Google Scholar]
- 9.Booth AD, Wallace S, McEniery CM, Yasmin J, Jayne DR, Wilkinson IB. Inflammation and arterial stiffness in systemic vasculitis: a model of vascular inflammation. Arthritis Rheum. 2004;50:581–588. doi: 10.1002/art.20002. [DOI] [PubMed] [Google Scholar]
- 10.Schillaci G, De Socio GV, Pucci G, Mannarino MR, Helou J, Pirro M, Mannarino E. Aortic stiffness in untreated adult patients with human immunodeficiency virus infection. Hypertension. 2008;52:308–313. doi: 10.1161/HYPERTENSIONAHA.108.114660. [DOI] [PubMed] [Google Scholar]
- 11.Karlinger K, Györke T, Makö E, Mester A, Tarján Z. The epidemiology and the pathogenesis of inflammatory bowel disease. Eur J Radiol. 2000;35:154–167. doi: 10.1016/s0720-048x(00)00238-2. [DOI] [PubMed] [Google Scholar]
- 12.Yu Y, Sitaraman S, Gewirtz AT. Intestinal epithelial cell regulation of mucosal inflammation. Immunol Res. 2004;29:55–68. doi: 10.1385/IR:29:1-3:055. [DOI] [PubMed] [Google Scholar]
- 13.Nichols WW, O’Rourke MF. McDonald’s Blood Flow in Arteries: Theoretical, Experimental and Clinical Principles. 5th ed. London: Hodder Arnold; 2005. [Google Scholar]
- 14.Khoshdel AR, Thakkinstian A, Carney SL, Attia J. Estimation of an age-specific reference interval for pulse wave velocity: a meta-analysis. J Hypertens. 2006;24:1231–1237. doi: 10.1097/01.hjh.0000234098.85497.31. [DOI] [PubMed] [Google Scholar]
- 15.Laurent S, Boutouyrie P, Lacolley P. Structural and genetic bases of arterial stiffness. Hypertension. 2005;45:1050–1055. doi: 10.1161/01.HYP.0000164580.39991.3d. [DOI] [PubMed] [Google Scholar]
- 16.Papa A, Santoliquido A, Danese S, Covino M, Di Campli C, Urgesi R, Grillo A, Guglielmo S, Tondi P, Guidi L, et al. Increased carotid intima-media thickness in patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2005;22:839–846. doi: 10.1111/j.1365-2036.2005.02657.x. [DOI] [PubMed] [Google Scholar]
- 17.Katz AM. Cardiomyopathy of overload. A major determinant of prognosis in congestive heart failure. N Engl J Med. 1990;322:100–110. doi: 10.1056/NEJM199001113220206. [DOI] [PubMed] [Google Scholar]
- 18.Seaberg EC, Benning L, Sharrett AR, Lazar JM, Hodis HN, Mack WJ, Siedner MJ, Phair JP, Kingsley LA, Kaplan RC. Association between human immunodeficiency virus infection and stiffness of the common carotid artery. Stroke. 2010;41:2163–2170. doi: 10.1161/STROKEAHA.110.583856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vlachopoulos C, Dima I, Aznaouridis K, Vasiliadou C, Ioakeimidis N, Aggeli C, Toutouza M, Stefanadis C. Acute systemic inflammation increases arterial stiffness and decreases wave reflections in healthy individuals. Circulation. 2005;112:2193–2200. doi: 10.1161/CIRCULATIONAHA.105.535435. [DOI] [PubMed] [Google Scholar]
- 20.Horowitz S, Binion DG, Nelson VM, Kanaa Y, Javadi P, Lazarova Z, Andrekopoulos C, Kalyanaraman B, Otterson MF, Rafiee P. Increased arginase activity and endothelial dysfunction in human inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1323–G1336. doi: 10.1152/ajpgi.00499.2006. [DOI] [PubMed] [Google Scholar]
- 21.Roifman I, Sun YC, Fedwick JP, Panaccione R, Buret AG, Liu H, Rostom A, Anderson TJ, Beck PL. Evidence of endothelial dysfunction in patients with inflammatory bowel disease. Clin Gastroenterol Hepatol. 2009;7:175–182. doi: 10.1016/j.cgh.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 22.Garolla A, D’Incà R, Checchin D, Biagioli A, De Toni L, Nicoletti V, Scarpa M, Bolzonello E, Sturniolo GC, Foresta C. Reduced endothelial progenitor cell number and function in inflammatory bowel disease: a possible link to the pathogenesis. Am J Gastroenterol. 2009;104:2500–2507. doi: 10.1038/ajg.2009.332. [DOI] [PubMed] [Google Scholar]
- 23.Aloi M, Tromba L, Di Nardo G, Dilillo A, Del Giudice E, Marocchi E, Viola F, Civitelli F, Berni A, Cucchiara S. Premature subclinical atherosclerosis in pediatric inflammatory bowel disease. J Pediatr. 2012;161:589–94.e1. doi: 10.1016/j.jpeds.2012.03.043. [DOI] [PubMed] [Google Scholar]
- 24.Schinzari F, Armuzzi A, De Pascalis B, Mores N, Tesauro M, Melina D, Cardillo C. Tumor necrosis factor-alpha antagonism improves endothelial dysfunction in patients with Crohn’s disease. Clin Pharmacol Ther. 2008;83:70–76. doi: 10.1038/sj.clpt.6100229. [DOI] [PubMed] [Google Scholar]
- 25.Zanoli L, Cannavò M, Rastelli S, Di Pino L, Monte I, Di Gangi M, Boutouyrie P, Inserra G, Laurent S, Castellino P. Arterial stiffness is increased in patients with inflammatory bowel disease. J Hypertens. 2012;30:1775–1781. doi: 10.1097/HJH.0b013e3283568abd. [DOI] [PubMed] [Google Scholar]
- 26.Akdoğan RA, Durakoğlugil ME, Kocaman SA, Çiçek Y, Durakoğlugil T, Ergül E, Rakıcı H. Increased pulse wave velocity and carotid intima-media thickness in patients with ulcerative colitis. Dig Dis Sci. 2013;58:2293–2300. doi: 10.1007/s10620-013-2634-9. [DOI] [PubMed] [Google Scholar]
- 27.Korkmaz H, Sahin F, Ipekci SH, Temel T, Kebapcilar L. Increased pulse wave velocity and relationship with inflammation, insulin, and insulin resistance in inflammatory bowel disease. Eur J Gastroenterol Hepatol. 2014;26:725–732. doi: 10.1097/MEG.0000000000000104. [DOI] [PubMed] [Google Scholar]
- 28.Aytaç E, Büyüktaş D, Baysal B, Atar M, Yıldız M, Baca B, Karahasanoğlu T, Çelik A, Seymen HO, Hamzaoğlu İ. Visual evoked potentials and pulse wave velocity in inflammatory bowel disease. Turk J Gastroenterol. 2015;26:15–19. doi: 10.5152/tjg.2015.4349. [DOI] [PubMed] [Google Scholar]
- 29.Zanoli L, Rastelli S, Inserra G, Lentini P, Valvo E, Calcagno E, Boutouyrie P, Laurent S, Castellino P. Increased arterial stiffness in inflammatory bowel diseases is dependent upon inflammation and reduced by immunomodulatory drugs. Atherosclerosis. 2014;234:346–351. doi: 10.1016/j.atherosclerosis.2014.03.023. [DOI] [PubMed] [Google Scholar]
- 30.Theocharidou E, Mavroudi M, Soufleris K, Griva T, Giouleme O, Athyros VG, Karagiannis A. Theocharidou E, Mavroudi M, Soufleris K, Griva T, Giouleme O, Athyros VG, Karagiannis A. Aortic stiffness in patients with inflammatory bowel diseases. Hellenic J Atherosclerosis. 2013;4:200–207. [Google Scholar]
- 31.Theocharidou E, Tellis CC, Mavroudi M, Soufleris K, Gossios TD, Giouleme O, Athyros VG, Tselepis AD, Karagiannis A. Lipoprotein-associated phospholipase A2 and arterial stiffness evaluation in patients with inflammatory bowel diseases. J Crohns Colitis. 2014;8:936–944. doi: 10.1016/j.crohns.2014.01.016. [DOI] [PubMed] [Google Scholar]
- 32.Wilkinson IB, Qasem A, McEniery CM, Webb DJ, Avolio AP, Cockcroft JR. Nitric oxide regulates local arterial distensibility in vivo. Circulation. 2002;105:213–217. doi: 10.1161/hc0202.101970. [DOI] [PubMed] [Google Scholar]
- 33.McEniery CM, Qasem A, Schmitt M, Avolio AP, Cockcroft JR, Wilkinson IB. Endothelin-1 regulates arterial pulse wave velocity in vivo. J Am Coll Cardiol. 2003;42:1975–1981. doi: 10.1016/j.jacc.2003.06.016. [DOI] [PubMed] [Google Scholar]
- 34.Chichester: John Wiley & Sons, 1995 [Google Scholar]
- 35.Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol. 2005;25:932–943. doi: 10.1161/01.ATV.0000160548.78317.29. [DOI] [PubMed] [Google Scholar]
- 36.Jacob MP. Extracellular matrix remodeling and matrix metalloproteinases in the vascular wall during aging and in pathological conditions. Biomed Pharmacother. 2003;57:195–202. doi: 10.1016/s0753-3322(03)00065-9. [DOI] [PubMed] [Google Scholar]
- 37.Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 2002;90:251–262. [PubMed] [Google Scholar]
- 38.Wang M, Zhang J, Jiang LQ, Spinetti G, Pintus G, Monticone R, Kolodgie FD, Virmani R, Lakatta EG. Proinflammatory profile within the grossly normal aged human aortic wall. Hypertension. 2007;50:219–227. doi: 10.1161/HYPERTENSIONAHA.107.089409. [DOI] [PubMed] [Google Scholar]
- 39.Floege J, Ketteler M. Vascular calcification in patients with end-stage renal disease. Nephrol Dial Transplant. 2004;19 Suppl 5:V59–V66. doi: 10.1093/ndt/gfh1058. [DOI] [PubMed] [Google Scholar]
- 40.Geerling BJ, Badart-Smook A, Stockbrügger RW, Brummer RJ. Comprehensive nutritional status in recently diagnosed patients with inflammatory bowel disease compared with population controls. Eur J Clin Nutr. 2000;54:514–521. doi: 10.1038/sj.ejcn.1601049. [DOI] [PubMed] [Google Scholar]
- 41.Levy E, Rizwan Y, Thibault L, Lepage G, Brunet S, Bouthillier L, Seidman E. Altered lipid profile, lipoprotein composition, and oxidant and antioxidant status in pediatric Crohn disease. Am J Clin Nutr. 2000;71:807–815. doi: 10.1093/ajcn/71.3.807. [DOI] [PubMed] [Google Scholar]
- 42.Jahnsen J, Falch JA, Mowinckel P, Aadland E. Body composition in patients with inflammatory bowel disease: a population-based study. Am J Gastroenterol. 2003;98:1556–1562. doi: 10.1111/j.1572-0241.2003.07520.x. [DOI] [PubMed] [Google Scholar]
- 43.Yarur AJ, Deshpande AR, Pechman DM, Tamariz L, Abreu MT, Sussman DA. Inflammatory bowel disease is associated with an increased incidence of cardiovascular events. Am J Gastroenterol. 2011;106:741–747. doi: 10.1038/ajg.2011.63. [DOI] [PubMed] [Google Scholar]
- 44.Dorn SD, Sandler RS. Inflammatory bowel disease is not a risk factor for cardiovascular disease mortality: results from a systematic review and meta-analysis. Am J Gastroenterol. 2007;102:662–667. doi: 10.1111/j.1572-0241.2006.01018.x. [DOI] [PubMed] [Google Scholar]
- 45.Haapamäki J, Roine RP, Turunen U, Färkkilä MA, Arkkila PE. Increased risk for coronary heart disease, asthma, and connective tissue diseases in inflammatory bowel disease. J Crohns Colitis. 2011;5:41–47. doi: 10.1016/j.crohns.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 46.Zanoli L, Inserra G, Castellino P. Increased cardiovascular risk in subjects with a low prevalence of classic cardiovascular risk factors: The inflammatory bowel disease paradox. Trends Cardiovasc Med. 2015:Epub ahead of print. doi: 10.1016/j.tcm.2015.04.001. [DOI] [PubMed] [Google Scholar]