

Abstract
Pancreatic ductal adenocarcinoma (PDAC) has the poorest prognosis of all malignancies and is largely resistant to standard therapy. Novel treatments against PDAC are desperately needed. Anti-Gal is the most abundant natural antibody in humans, comprising about 1% of immunoglobulins and is also naturally produced in apes and Old World monkeys. The anti-Gal ligand is a carbohydrate antigen called “α-gal epitopes” with the structure Galα1-3Galβ1-4GlcNAc-R. These epitopes are expressed as major carbohydrate antigens in non-primate mammals, prosimians, and New World monkeys. Anti-Gal is exploited in cancer vaccines to increase the immunogenicity of antigen-presenting cells (APCs). Cancer cells or PDAC tumor lysates are processed to express α-gal epitopes. Vaccination with these components results in in vivo opsonization by anti-Gal IgG in PDAC patients. The Fc portion of the vaccine-bound anti-Gal interacts with Fcγ receptors of APCs, inducing uptake of the vaccine components, transport of the vaccine tumor membranes to draining lymph nodes, and processing and presentation of tumor-associated antigens (TAAs). Cancer vaccines expressing α-gal epitopes elicit strong antibody production against multiple TAAs contained in PDAC cells and induce activation of multiple tumor-specific T cells. Here, we review new areas of clinical importance related to the α-gal epitope/anti-Gal antibody reaction and the advantages in immunotherapy against PDAC.
Keywords: Pancreatic cancer, Immunotherapy, Cancer antigen, MUC1, α-gal epitopes, Cancer vaccine, Cancer stem cell, Carbohydrate research
Core tip: The goal of cancer immunotherapy is to elicit an immune response against autologous tumors and to induce multiple T cell clones against multiple tumor-associated antigens. To establish effective, next-generation immunotherapy toward pancreatic ductal adenocarcinoma (PDAC), we focus on the strong interaction between the natural human antibody, anti-Gal, and carbohydrate antigens called “α-gal epitopes”. Here, we review the literature on the distribution of natural anti-Gal antibody and its ligand in mammals and characterization of the immunosuppressive microenvironment of PDAC tumors, which is a major obstacle against effective clinical immunotherapies. We also discuss immunotherapeutic strategies using the α-gal epitope/anti-Gal antibody reaction.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is common worldwide, and its incidence is gradually increasing in the United States, with an estimated 43920 new cases and 37390 deaths in 2012[1]. Surgical resection is the only known curative treatment for PDAC[2], and patients who develop a recurrence usually present with the recurrence between 9 and 12 mo after resection[3]. The median survival of PDAC patients following surgery is 15-20 mo, with a 5-year survival rate of approximately 20%[3,4]. Accordingly, the median survival of patients with locally advanced, unresectable PDAC is very poor[2,5]. Currently, only a few chemotherapeutic agents have been shown to be effective against PDAC, including gemcitabine and a combination of fluorouracil, leucovorin, oxaliplatin, and irinotecan, which is called the FOLFIRINOX regimen[6,7]. Unfortunately, the survival of patients treated with these regimens is marginal.
From the point of view of the PDAC microenvironment, reciprocal interactions between cancer cells and host cells including fibroblasts and inflammatory and vascular endothelial cells orchestrate a microenvironment that is immunosuppressive, fibrotic, and poorly vascular[8-10]. This desmoplastic reaction that surrounds PDAC lesions constitutes a major obstacle to the efficacy of therapy[11]. Indeed, cytotoxic drugs poorly penetrate this dense stromal matrix. Hence, novel therapeutic approaches against PDAC are urgently needed. As immunotherapies act differently than conventional therapies, including chemotherapy or radiation therapy, they are a promising alternative treatment modality for this deadly disease.
Here, we review relevant immunotherapies and address the basic problems with cancer immunotherapy. We detail our recent strategy for vaccination with tumor antigens exploiting the interaction between α-gal epitopes and anti-Gal antibody. The ligand for anti-Gal is a carbohydrate antigen called α-gal epitope with the structure Galα1-3Galβ1-4GlcNAc-R, which is on carbohydrate chains of glycolipids and glycoproteins[12]. Furthermore, we also discuss our novel immunotherapy approach that targets pancreatic cancer stem cells (CSCs) using stem cell markers that are engineered to express α-gal epitopes.
Epidemiology and clinical management of pancreatic cancer
PDAC is the fifth leading cause of cancer-related death in the developed world, with more than 260000 deaths annually worldwide[13]. Surgical resection (resectable disease) is the only curative treatment. However, the 5-year survival rate after surgical resection is only 5.5%-21%[2]. Radiation therapy (as combined modality therapy for locally advanced/unresectable disease) and chemotherapy (as adjuvant treatment for both locally advanced/unresectable and metastatic disease) have become a part of the armamentarium of therapy for PDAC[14,15]. However, most patients present with advanced, unresectable disease, and even those that undergo successful surgical resection have high recurrence rates, with an average overall survival of 16-18 mo[14,15]. Chemotherapeutic options include gemcitabine-based therapy[16] and more recently, FOLFIRINOX in select patients with a favorable performance status[6,7]. We and others have reported encouraging survival rates following preoperative gemcitabine-based chemoradiotherapy in patients with potentially resectable PDAC[17-19]. Despite modest improvements in mortality and quality of life, the benefits of treatment remain limited, and cures are rare.
The poor prognosis of PDAC is related to a combination of late detection and relatively ineffective standards of care. Several promising drugs that target important characteristics of malignancy, such as angiogenesis, proliferation, and metastasis, have failed to provide clinically relevant benefits and have provided only trivial improvements in disease-free survival and overall survival rates.
As immunotherapies act differently than standard treatments (chemotherapy and radiation therapy), they represent a promising alterative treatment modality for this deadly disease. Immunotherapies use techniques such as vaccination that is designed to activate the patient’s immune system with tumor-associated antigens (TAAs) expressed in PDAC cells. The immune system that has been activated by vaccination can recognize TAAs and eradicate cancer cells. Although several clinical studies have documented evidence of treatment-induced, antigen-specific immune responses, few, if any, protective immune responses have been observed in patients with metastatic disease[20]. In addition, vaccination against TAAs is an attractive approach as an adjuvant-setting treatment after surgery when tumor-induced immune suppression is minimal[21,22]. Effective anticancer functions of the immune system require cytotoxic CD8+ T cells, Tn helper-1 (Th-1) cells, mature dendritic cells (DCs), activated pro-inflammatory macrophages (M1), and natural killer cells. However, PDAC cells induce both local and systemic immune dysfunction, thus avoiding detection by the immune system[8-10,23].
Immune cells in PDAC promote an immunosuppressive, anti-inflammatory environment, which is a major obstacle in clinical immunotherapy
At the level of cancer cells, PDAC cells induce both local and systemic immune dysfunction via at least three mechanisms involving modulation of the immune system and avoidance of detection by effector cells: (1) contact-dependent factors [i.e., expression of immune system checkpoint ligands such as ligand for programmed death-1 (PD-L1)]; (2) secretion of soluble immunosuppressive factors such as interleukin (IL)-10, transforming growth factor (TGF)-β, and vascular endothelial growth factor; and (3) interference with major histocompatibility complex (MHC) class I peptide presentation by downregulation of MHC class I expression, disabling antigen degradation, or preventing antigen insertion into the MHC class I groove (Figure 1).
Figure 1.
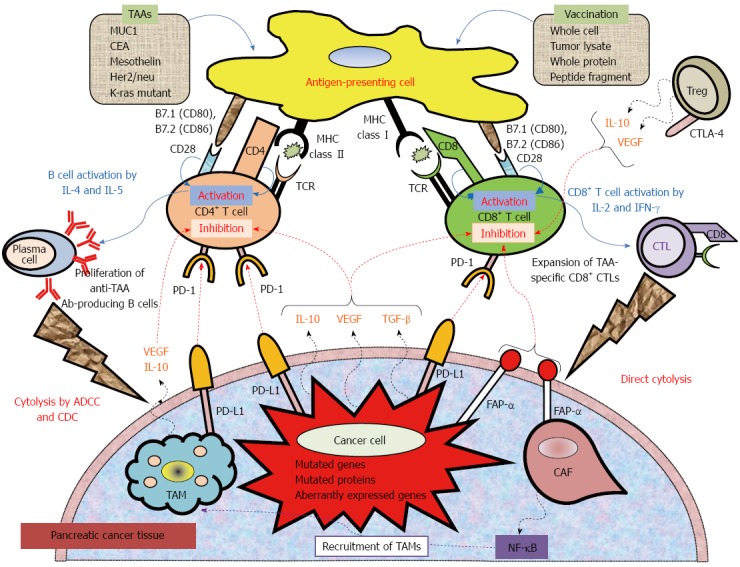
Mechanistic basis for vaccine-based immunotherapy, and immune co-stimulatory, co-inhibitory ligands and receptors, and soluble immune modulating factors involved in T cell activation and inhibition. Anticancer immunotherapy aims to harness the natural ability of the immune system to recognize and react against potential TAAs. Peptide-based, protein-based, or whole cell-based vaccines rely on identified immune-dominant TAA epitopes to stimulate anticancer T-cell responses. Antigen-presenting cells (APCs), including dendritic cells, capture antigens obtained from vaccinations. After intracellular processing, antigen peptides are loaded onto major histocompatibility complex (MHC class I or II) molecules on the surface of the APC. Specific T cells encounter these MHC-peptide complexes in conjugation with a co-stimulatory signal. The activated T cells proliferate and secrete cytokines (IL-2, IL-4, IL-5, INF-γ), resulting in the production of a cascade of immune effector cells. Immunotherapeutic strategies that inhibit immune checkpoints such as those mediated by CTLA-4 and PD-1 reduce the barriers that vaccines must overcome to trigger therapeutically relevant anticancer immune responses. Recently, preclinical and clinical studies have demonstrated that combining immune-modulating agents such as cyclophosphamide (CY) and checkpoint inhibitors (anti-CTLA-4, anti-PD-1, anti-PD-L1) with vaccine strategies can enhance anticancer immune responses as well as block the tolerizing mechanisms that would otherwise inhibit these responses. ADCC: Antibody-dependent cell-mediated cytotoxicity; CAF: Cancer-associated fibroblast; CDC: Complement-dependent cytolysis; CTLA-4: Cytotoxic T lymphocyte antigen-4; FAP-α: Fibroblast activation protein-α; TAM: Tumor-associated macrophage; TCR: T cell receptor; PD-1: Programmed cell death protein 1; PD-L1: Programmed death-ligand.
The tumor microenvironment of PDAC consists of not only cancer cells but also immune suppressive cells such as cancer-associated fibroblasts (CAFs), tolerogenic DCs, myeloid-derived suppressor cells (MDSCs), immunosuppressive tumor-associated macrophages (TAMs), and regulatory T cells (Tregs) (Figure 1). These immunosuppressive cells in PDAC can inhibit the anti-tumor immunity that is induced by vaccines. Accumulation of these immunosuppressive cells may be closely related to the extent of disease and may contribute to the failure to provide clinically relevant benefits. CAFs secrete fibroblast activation protein (FAP-α), which further suppresses effector T cells by interfering with tumor necrosis factor- and interferon-γ-related activation[24,25]. FAP-α is overexpressed in both the PDAC stroma and on PDAC cells[26], and anti-FAP-α monoclonal antibodies are currently in clinical development. MDSCs are immature myeloid cells that suppress both innate and adoptive immunity[27]. Factors contributing to their action in immunity include sequestration of cysteine, expression of high levels of arginase, impairment of T cell homing to lymph nodes, and secretion of TGF-β. These factors inhibit the function of effector T cells and natural killer cells and promote the development of Tregs. Patients with PDAC have increased numbers of MDSCs in their circulation compared to healthy controls, and MDSC numbers are correlated with levels of the Th-2 cytokine IL-13 and Treg cell numbers[28,29]. An increased number of circulating MDSCs is an independent poor prognostic factor in PDAC patients[28,29]. Furthermore, TAMs interact with the immune system via multiple mechanisms such as secretion of IL-10 and TGF-β and expression of immune inhibitory ligands such as PD-L1. In PDAC, TAMs are significantly increased in tumor tissue[30,31]. Patients with PDAC have increased numbers of Tregs, both in the circulation and in tumor tissues. By expression of cytotoxic T lymphocyte antigen-4 and secretion of IL-10 and TGF-β, Tregs suppress the exaggerated immune responses induced by vaccination[32,33]. Conversely, a low Treg percentage in the circulation 1 year after surgical resection is correlated with improved survival[34]. Taken together, these cellular subtypes, including CAFs, MDSCs, TAMs, and Tregs, are potent obstacles against effective clinical immunotherapies.
Reciprocal distribution of the natural anti-Gal antibody and its ligand, α-gal epitopes, in mammals
Anti-Gal is the most abundant antibody in humans, comprising about 1% of immunoglobulins, and is present as IgG, IgM, and IgA isotypes[35,36]. Anti-Gal is continuously produced throughout life as an immunological response to antigenic stimulation by bacteria of the normal flora, including Klebsiella pneumonia, Escherichia coli, and Serratiamarcecens[35,36]. As many as 1% of human B cells are capable of producing anti-Gal[12], most of which are quiescent; only those in the gastrointestinal tract produce this antibody in response to continuous antigenic stimulation by gastrointestinal bacteria. Anti-Gal in humans is encoded by several heavy-chain genes, primarily of the VH3 immunoglobulin gene family[12,37]. The distribution of anti-Gal in mammals is unique (Figure 2). Anti-Gal is produced only in humans and Old World primates (monkeys of Asia and Africa). In contrast, all other mammals including non-primate mammals (e.g., kangaroos, mice, rats, pigs, dogs, horses, lions, and dolphins), prosimians (e.g., lemurs), and New World monkeys (monkeys of central and south America) produce only the specific ligand for anti-Gal and not the antibody (Figure 2). The ligand for anti-Gal is a carbohydrate antigen called the α-gal epitope with the structure Galα1-3Galβ1-4GlcNAc-R, which is present on carbohydrate chains of glycolipids and glycoproteins[12]. In 1968, Eto et al[38] were the first to isolate the glycolipid ceramidepentahexoside, which contains the non-reducing terminal sequence Galα1-3Galβ1-4GlcNAc-R, from rabbit red blood cells (RBCs). Subsequently, the structure of rabbit RBC ceramidepentahexoside was further characterized by Stellner et al[39] in 1973. The synthesis of α-gal epitopes in mammals is catalyzed by the glycosylation enzyme α1, 3 galactosyltransferase (α1, 3 GT). As α-gal epitopes are abundant in both marsupials and placental mammals and absent in non-mammalian vertebrates (fish, reptiles, and birds), the α1, 3 GT gene and α-gal epitope appeared in mammalian evolution at least 140 million years ago (Figure 2). The continued prevention of anti-Gal production in mammals by natural selection throughout this evolutionary period may have resulted in the elimination of anti-Gal-encoding immunoglobulin genes from the mammalian genome[12].
Figure 2.

Reciprocal evolution of α1, 3 galactosyltransferase (α1, 3 GT) enzyme activity, α-gal epitopes, and anti-Gal antibody in mammals. α-gal epitopes have been synthesized in mammals by α1, 3 galactosyltransferase (α1, 3 GT) for more than 125 million years, since before the divergence of placental mammals and marsupials. All non-mammalian vertebrates lack α1, 3 GT and do not express α-gal epitopes. Expression of this epitope was suppressed in ancestral Old World primates after they diverged from New World monkeys, and probably after apes and monkeys diverged from each other. Suppression of α-gal epitopes was followed by production of natural anti-Gal antibody, which is absent in non-primate mammals, prosimians, and New World monkeys.
The only known exceptions to anti-Gal production in mammals are in Old World monkeys, apes, and humans[12], which all have an inactive α1, 3 GT gene as the result of a few single-base deletions that generate premature stop codons that truncate the enzyme molecule, resulting in an inactive protein[12]. Based on the sequence of the α1, 3 GT pseudogene in Old World primates and humans, inactivation of the α1, 3 GT gene in ancestral Old World primates may have occurred 20-28 million years ago[12] (Figure 2), and the inactivation may have been associated with a major catastrophic epidemiological event that affected only ancestral Old World primates[12]. New World monkeys and lemurs were not subjected to this selective pressure because they evolved in geographical areas that were separated from the Old World land mass by oceanic barriers. Primates with an inactivated α1, 3 GT gene lack the α-gal epitope and thus are not immunotolerant to it. The anti-Gal antibody, if produced following inactivation of the α1, 3 GT gene, may provide immune protection to ancestral Old World primates against pathogens endemic to the Old World land mass that were detrimental to primates that expressed α-gal epitopes[12]. Several pathogens, including enveloped viruses[12], bacteria[12], and protozoa[12], express α-gal epitopes and can be destroyed by anti-Gal binding.
Anti-Gal antibody interacts specifically with α-gal epitopes on glycolipids and glycoproteins. Anti-Gal was initially discovered on RBCs of patients with β-thalassemia, on normal human senescent RBCs[12,40], and on sickle cell anemia RBCs. A cryptic antigen capable of binding anti-Gal may be present on human RBCs that are about 120 d old or on thalassemia and sickle cell anemia RBCs on which this antigen is present on younger RBCs[12,40]. The amount of this cryptic antigen on RBCs is very low, resulting in markedly high binding of anti-Gal, which is detrimental[41].
Although anti-Gal contributes to a number of pathological phenomena, this antibody is ubiquitous in humans. Furthermore, anti-Gal activity is found in cancer patients with solid tumors, including colon cancer, ovarian cancer, and PDAC and in patients with B cell lymphoma; anti-Gal activity is similar in patients with various types of cancer and healthy individuals[40]. Anti-Gal may be amenable to exploitation in a number of clinical settings such as cancer immunotherapy, as described in this review.
Interaction of anti-Gal/α-gal epitopes as a barrier in clinical xenotransplantation
Xenotransplantation, or transplantation of organs and tissues from animals such as pigs into humans, is of considerable clinical importance because the number of human organ donors is insufficient[42,43]. Pigs are considered to be the most suitable organ donors because their organs are similar in size and function as many human organs[42,43]. However, pig cells express very high levels of α-gal epitopes[35]. Anti-Gal in xenograft recipients binds to α-gal epitopes on the endothelial cells of xenografts and induces complement-dependent cytolysis followed by platelet aggregation, occlusion of small blood vessels, collapse of the vascular bed, and hyperacute rejection of the xenograft within 0.5-24 h (Figure 3)[35]. An additional complicating factor in xenotransplantation is associated with the important finding that approximately 1% of B cells in humans produce anti-Gal[35,36]. When a xenograft is transplanted into humans, the released α-gal glycoproteins activate these quiescent B cells to produce anti-Gal. The anti-Gal IgG titer increases by approximately 100-fold due to increases in both the concentration and affinity of the antibody[41]. In noteworthy studies performed by Groth and Galili, they clearly demonstrated such an increase in anti-Gal activity in patients with diabetes who received both an allogeneic kidney and fetal pig islets. This increase in anti-Gal occurred despite immunosuppressive treatment that was potent enough to prevent rejection of the kidney allograft[40,44]. This elicited anti-Gal IgG activity is likely to mediate destruction of xenograft cells by antibody-dependent cell-mediated cytotoxicity.
Figure 3.
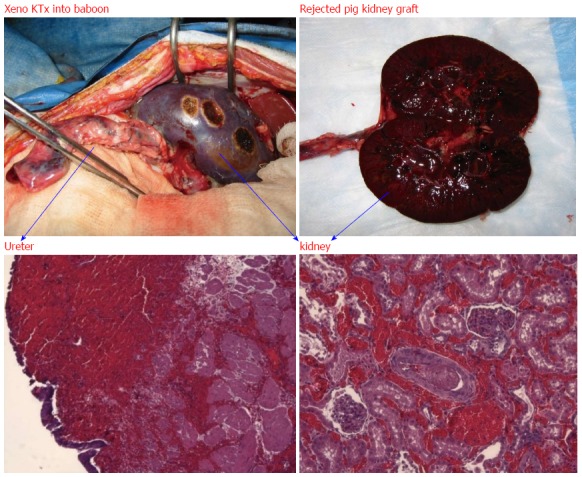
Hyperacute rejection of α-gal+/+ pig kidney xenografted into a baboon (1 d after kidney transplantation). The interaction of natural baboon anti-Gal antibody with millions of α-gal epitopes expressed on the pig cell surface causes strong xenograft rejection. The in vivo binding of anti-Gal antibody to α-gal epitopes on transplanted pig heart or kidney is the main cause of hyperacute rejection of such grafts in humans and Old World monkeys. The recent generation of α1, 3 GT knockout pigs that lack α-gal epitopes has resulted in the elimination of this immunological barrier.
Fortunately, this immunological barrier was overcome in 2003 by the generation of α1, 3 GT knockout pigs, which lack α-gal epitopes[45,46]. Accordingly, heart and kidney xenografts from these knockout pigs transplanted into monkeys survived for several months[42,46-48]. The detrimental anti-Gal/α-gal epitope interaction that occurs in xenotransplantation may be harnessed for beneficial purposes in other clinical areas such as immunotherapy.
Principals of PDAC treatment with immunotherapy
Because currently available therapies have significant limitations, PDAC is an ideal setting for the development of novel treatment modalities such as immunotherapy. However, certain obstacles must be overcome for immunotherapeutic regimens against PDAC to be successful.
Tumor cell vaccines have been considered for use in immunotherapy. The simplest vaccine approach that has been applied in PDAC is inoculation of individuals with irradiated tumor cells (i.e., whole cancer cell-based vaccines). This approach has the following advantages[49-51]. Whole cancer cell-based vaccines circumvent the need for targeting a selected TAA as they rely on irradiated tumor cells that by definition express a panel of TAAs. In this setting, allogeneic preparations overcome the technical difficulties that may be posed by the production of autologous vaccines, which require the isolation of a sufficient amount of malignant tissue from patients. Whole cell-based vaccines also provide non-biased immunization of lymphocytes and sera against TAAs, resulting in the generation of a reagent that may be used to identify immunologically relevant TAAs for use in the design of antigen-specific vaccination strategies.
In general, cytotoxic T cell lymphocytes play a critical role in the immunological cascade that ultimately results in the lysis of cancer cells in a TAA-specific manner[23]. Receptors on the surface of T cells bind to TAAs or peptide fragments that are bound to MHC class I molecules, which are present on the surface of professional antigen-presenting cells (APCs) such as DCs and macrophages. T cell activation, however, also requires the presence of costimulatory molecules (e.g., B7.1, B7.2), which can be provided only by professional APCs[52]. The interaction of the T cell receptor on naïve T cells with the TAA on tumors without the delivery of a costimulatory nonspecific signal (Signal 2) is thought to result in the T cell entering a state of long-term unresponsiveness to the TAA, called anergy[53-55]. Once T cells are activated, helper T cells are recruited that secrete cytokines such as IL-2 and granulocyte macrophage colony-stimulating factor, which further enhances T cell activation and proliferation (Figure 1). Accordingly, T cells require these two signals to become fully activated[56]. Despite these immunological responses to the presence of PDAC cells, effective immunity does not develop against PDAC cells because of impaired tumor recognition by immune cells, poor immunogenicity of TAAs, and the presence of an immunosuppressive milieu in the PDAC tumor microenvironment, which includes CAFs, MDSCs, TAMs, and Tregs (Figure 1).
Another reason for the absolute requirement for effective uptake of whole cell-based vaccines by APCs is that activation of TAA-specific T cells does not occur at the vaccination site, but rather takes place within the draining lymph nodes of the vaccination sites or in the spleen. Only after they are activated can tumor-specific T cells leave the lymph nodes or spleen to seek and destroy cancer cells that express the TAAs. For such activation to occur, the whole cell-based vaccine must be transported from the vaccination site by APCs to lymph nodes or the spleen[57,58]. Transportation of vaccines occurs only after effective uptake of the vaccine by APCs[57,58].
Improving APC targeting through formation of immune complexes containing α-gal epitopes and anti-Gal
As described above, TAA molecules expressed on whole cell-based vaccines are not modified to express markers that allow effective recognition by APCs. This section describes how whole cell-based vaccines can be directed to APCs through formation of immune complexes that interact with Fcγ receptors (FcγRs) on APCs. The carbohydrate make-up of whole cell-based vaccines can be modified to include expression of α-gal epitopes (Figure 4). These epitopes are recognized by naturally abundant anti-Gal antibodies that opsonize the whole cell-based vaccines, and the resulting immune complex enhances the immunogenicity of the whole cell-based vaccine. APCs, including macrophages, skin Langerhans cells, and blood-derived DCs, all express FcγRs (e.g., Fcγ RI/CD64, Fcγ RII/CD32, Fcγ RIII/CD66). These FcγRs bind and mediate the internalization of opsonized cells (i.e., cells with bound IgG molecules), cell membranes, or molecules (all defined as cancer antigens) via the Fc portion of the opsonizing IgG antibody[59-61]. This results in enhancement of the immunogenicity of the antigen that is complexed with an IgG antibody. Thus, vaccination of cancer patients with a tumor cell vaccine that is modified to express α-gal epitopes should result in in situ binding of the patient’s anti-Gal IgG molecules to α-gal epitopes on the vaccinating cell membrane. This targets the vaccines to APCs by interaction of the Fc portion of the anti-Gal antibody on the vaccinating cell membrane with FcγRs on the APCs[62,63]. This interaction induces the uptake of the whole cell-based vaccine by APCs, which subsequently transport the vaccinating tumor membranes to the draining lymph nodes or spleen.
Figure 4.
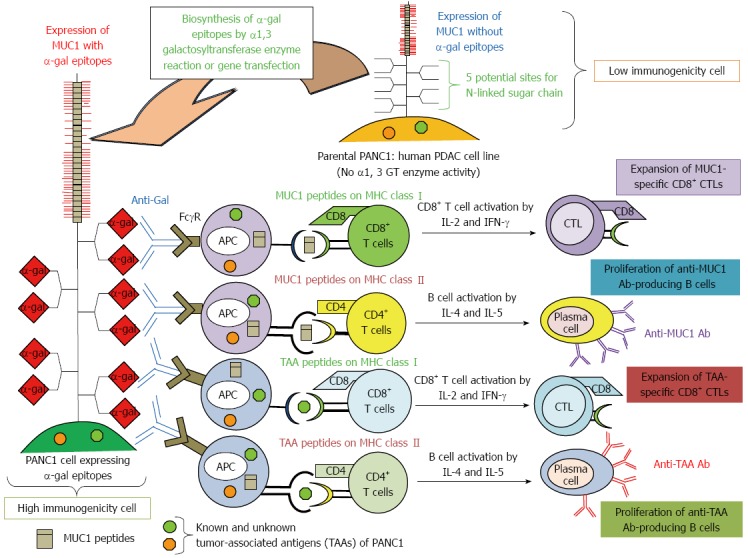
Increased immunogenicity of known and unknown tumor-associated antigens and MUC1 engineered to express α-gal epitopes. Immunity towards known and unknown tumor-associated antigens (TAAs), including MUC1, in PDAC patients is relatively weak, and presentation of these TAAs to the immune system is poor due low immunogenicity. We tested the effects of vaccination using immunogenetically enhanced known and unknown TAAs and MUC1 with expression of α-gal epitopes on production of antibodies for MUC1 and other TAAs derived from PDAC cells, as well as induction of tumor-specific T cell activation.
In our previous study[64], we investigated the beneficial effects of whole cell-based vaccines with α-gal epitope-expressing pancreatic cancer cells in the induction of tumor-specific B- and T-cell responses, in vivo prevention of tumor growth, and improvement in survival[64]. We employed a human pancreatic cell line, PANC1, which endogenously expresses Mucin1 (MUC1) in the whole cell-based vaccine. MUC1 can be used as a tumor marker and is a potential target for PDAC immunotherapy. However, vaccination with MUC1 peptides fails to stimulate an immune response against PDAC because immunity toward TAAs, including MUC1, in PDAC patients is relatively weak, and the presentation of these TAAs to the immune system is poor due to their low immunogenicity (Figure 4). To increase the immunogenicity of the PANC1 whole cell-based vaccine, which includes unknown TAAs and the MUC1 antigen against APCs, we modified these cells to express α-gal epitopes by transfection of the mouse α1, 3 GT gene (designated here as α-gal PANC1) (Figure 4). This modified whole cell-based vaccine takes advantage of anti-Gal antibodies, resulting in increased uptake of TAAs contained in the tumor cell vaccine in an antibody-dependent manner. Simultaneously, MUC1 can also be engineered to express α-gal epitopes, because the MUC1 molecule has five potential sites for N-glycans and can bind anti-Gal in situ at the vaccination sites (Figure 4).
In Figure 5A, we show a schematic illustration of an experimental protocol. The anti-Gal antibody as a natural antibody is not present in naïve α1, 3 GT knockout mice. Repeated immunizations with pig kidney fragments result in the appearance of anti-Gal antibodies, with an anti-Gal IgG titer that is similar to that observed in a large proportion of samples of human serum. In vitro analysis of the immune response showed that three vaccinations with α-gal PANC1 elicited a strong anti-MUC1 IgG response, whereas vaccination with whole parental PANC1 cells did not elicit such an antibody response (Figure 5B). Furthermore, α-gal PANC1 whole cell-based vaccines induced a protective immune response against a tumor challenge with the MUC1-expressing B16F10 melanoma cell line (Figure 5C). The beneficial effects of α-gal PANC1 whole cell-based vaccines are illustrated by prolonged survival after tumor challenge.
Figure 5.
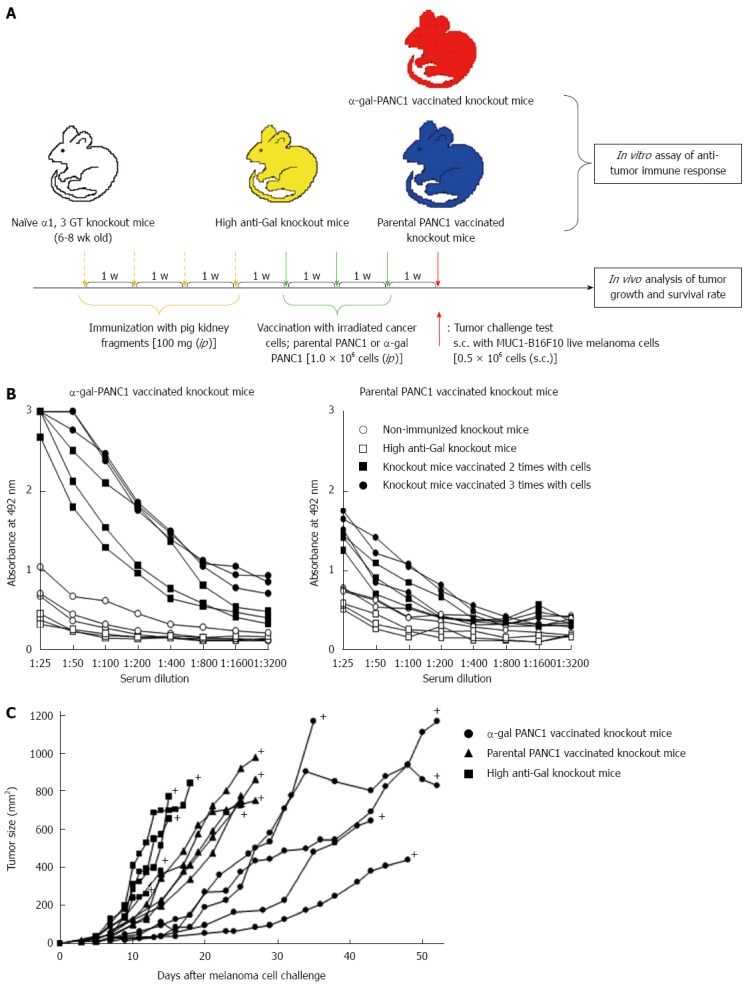
Experimental design for in vitro and in vivo studies and anti-MUC1 IgG antibody production assessed with an enzyme-linked immunosorbent assay. A: Schematic illustration of the experimental protocol; B: Anti-MUC1 IgG production in knockout mice vaccinated with α-gal PANC1, and anti-MUC1 IgG production in knockout mice vaccinated with parental PANC1; C: Size of subcutaneous tumors after challenge with MUC1-B16F10 cells. +: Death.
PDAC tumor lysates that are engineered to express α-gal epitopes can target pancreatic CSCs
In previous sections, we described the in vitro and in vivo effects of whole cell-based vaccination with α-gal epitope-expressing pancreatic cancer cells[64]. However, the effect was somewhat weak as shown in Figure 5C. To further develop an effective immunotherapy for PDAC, we hypothesized that the tumor lysate is a more suitable source of TAAs for vaccination because it contains several known and unknown antigens expressed in cancer cells and stromal cells that can elicit a broad-spectrum anti-tumor immune response (Figure 6). Moreover, the primary PDAC tumor tissue contains a subset of putative pancreatic CSCs[65-69] (Figure 6). These pancreatic CSCs are resistant to the standard cytotoxic agent gemcitabine and show enhanced metastatic potential[65-69]. Additionally, inducing an immune response against pancreatic CSCs, which constitute only 1% of all cancer cells, is often difficult[65-69].
Figure 6.
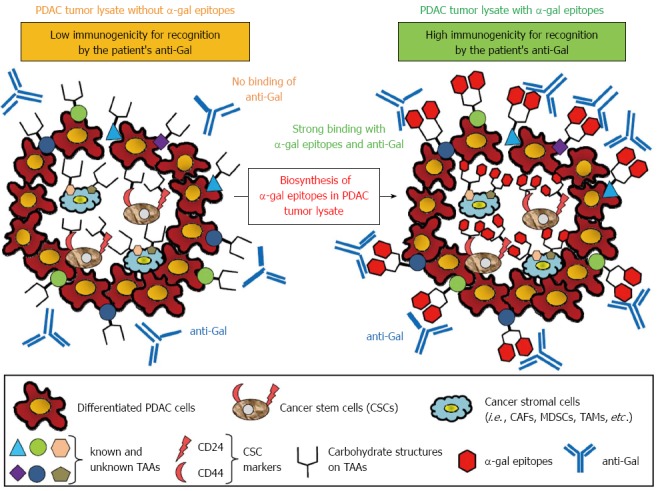
Concept of effective vaccination with α-gal tumor lysate against pancreatic ductal adenocarcinoma. A tumor lysate is a more suitable source of tumor-associated antigens (TAAs) because it contains several known and unknown antigens in cancer cells and stromal cells that can elicit a broad-spectrum anti-tumor immune response. Moreover, the primary tumor of pancreatic adenocarcinoma contains a subset of pancreatic cancer cells with stem cell properties (i.e., pancreatic cancer stem cells: pancreatic CSCs). To increase the immunogenicity of known and unknown TAAs, CSC markers, or TAAs contained in cancer stromal cells to antigen-presenting cells, anti-Gal bound to α-gal-expressing TAAs could be a suitable strategy.
In the newest study in our institute, we prepared a polyvalent tumor lysate vaccine that was engineered to express α-gal epitopes on primary PDAC tumors (designated α-gal tumor lysate vaccine)[70]. Accordingly, α-gal tumor lysate vaccines should be able to increase the immunogenicity of the broad spectrum of TAAs, which are present in differentiated pancreatic cancer cells, pancreatic CSCs, and stromal cells (Figure 6). As shown in Figures 7 and 8, we investigated the beneficial effects of the α-gal tumor lysate vaccine using adoptive transfer models. The tumor growth of live PDAC cells, which include differentiated pancreatic cancer cells and pancreatic CSCs, in non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice was examined. The experimental design of the adoptive transfer model using NOD/SCID mice is shown in Figure 7. High anti-Gal knockout mice were generated as described in a previous study[41]. Subsequently, these mice were vaccinated with either the parental tumor lysate or an α-gal tumor lysate vaccine. To compare the effectiveness of the α-gal whole cell-based vaccine with that of the α-gal tumor lysate vaccine, the NOD/SCID mice were given ip injections of an α-gal whole cell-based vaccine consisting of 1 × 106 cells irradiated with 50 Gy in a manner similar to the tumor lysate vaccine. One week after the last vaccination, splenocytes were prepared from successfully vaccinated donor knockout mice. For adoptive transfer, these isolated splenocytes were transferred by ip injection into NOD/SCID mice. One day after adoptive transfer, all NOD/SCID mice were challenged with either live PDAC cells or live pancreatic CSCs (i.e., CD44+CD24+ PANC1 cells). These mice were examined for both tumor growth and survival (Figure 7). Regarding the size of subcutaneous tumors after a challenge with live PDAC cells (Figure 8A), untreated control mice, parental tumor lysate-vaccinated, and α-gal whole cell-vaccinated mice developed large tumors, whereas no tumor growth was noted in the α-gal tumor lysate-vaccinated mice[70]. Regarding the size of subcutaneous tumors after a challenge with live pancreatic CSCs, control mice, parental tumor lysate-vaccinated, and α-gal whole cell-vaccinated mice developed large tumors, but tumorigenesis by pancreatic CSCs was completely prevented in all α-gal tumor lysate-vaccinated mice (Figure 8B). With the exception of the α-gal tumor lysate group, no significant differences were found in the time to appearance of palpable tumors after tumor challenge among these three groups, including the α-gal whole-cell group. Moreover, vaccination with the parental tumor lysate and with α-gal whole-cell did not prolong the survival time after tumor challenge with pancreatic CSCs, whereas vaccination using the α-gal tumor lysate significantly improved survival after tumor challenge[70]. Taken together, in vivo anti-tumor effects induced by α-gal tumor lysate vaccination were markedly stronger than those with either the parental tumor lysate or α-gal whole-cell. The reason for the powerful induction of anti-tumor effects by α-gal tumor lysate vaccination was clearly shown with flow cytometry (Figure 8C). Sera from both α-gal whole-cell and α-gal tumor lysate groups more strongly bound to both CD44+CD24+ (pancreatic CSCs) and CD44-CD24- PANC1 cells (differentiated PDAC cells) than to those from the parental tumor lysate group. Importantly, vaccination with the α-gal tumor lysate induced better antibody production against both PANC1 cell populations than α-gal whole cell-based vaccination (Figure 8C).
Figure 7.
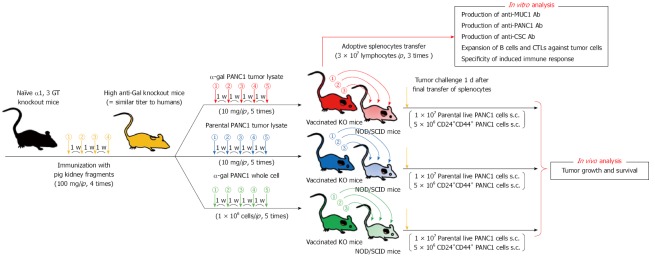
Experimental design of the adoptive transfer model using non-obese diabetic/severe combined immunodeficiency mice. Experimental design of in vitro and in vivo studies was shown. For in vivo study, the adaptive transfer model, using non-obese diabetic/severe combined immunodeficiency mice as donors was employed.
Figure 8.
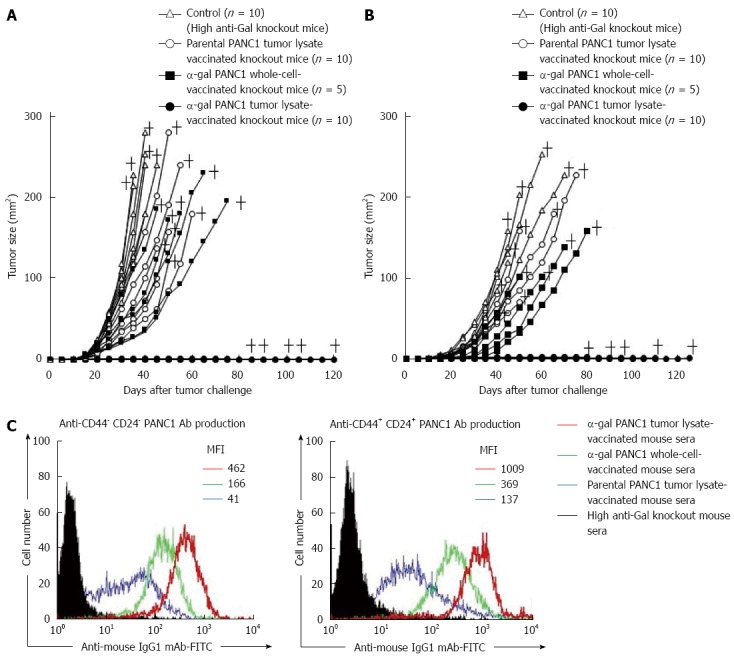
In vivo tumor growth in adoptively transferred non-obese diabetic/severe combined immunodeficiency mice challenged with either live PANC1 cells or live CD44+CD24+ PANC1 cells, and production of antibodies against differentiated cancer cells and cancer stem cells. A: We monitored tumor growth in splenocyte-transferred mice. No tumors were noted in the α-gal tumor lysate-vaccinated mice. No significant differences in the time to appearance of a palpable tumor after tumor challenge were observed in the untreated control group and parental tumor lysate group (untreated: 10.6 ± 2.5 d; parental tumor lysate: 11.9 ± 2.1 d). In contrast, the development of tumors in the α-gal whole cell vaccination group was significantly delayed compared with the untreated and parental tumor lysate groups (α-gal whole-cell: 16.0 ± 2.8 d, P = 0.018 vs control; P = 0.004 vs parental tumor lysate). In the untreated control group, the maximum tumor size was 100 mm2 within 29 to 34 d (mean: 31.4 ± 2.1 d). In comparison, tumor growth to a similar size was markedly delayed in both the parental tumor lysate group (40.3 ± 6.9 d, P = 0.007 vs control) and α-gal whole-cell group (45.6 ± 8.3 d, P = 0.0013 vs control). +; death; B: The tumorigenesis of pancreatic CSCs was completely prevented in all α-gal tumor lysate-vaccinated mice. With the exception of the α-gal tumor lysate group, no significant differences were seen in the time to appearance of palpable tumors after tumor challenge among the groups (untreated: 13.1 ± 3.3 d; parental tumor lysate: 14.4 ± 3.4 d; α-gal whole-cell: 17.0 ± 3.8 d). The tumor size reached 100 mm2 in 40.6 ± 1.8 and 48.0 ± 4.4 d in the untreated and parental tumor lysate groups, respectively. However, tumor growth to a similar size was significantly delayed in the α-gal whole-cell group (60.5 ± 7.9 d; P < 0.001 vs control; P = 0.033 vs parental tumor lysate). +; death; C: Production of either anti-CD44-CD24- PANC1 (i.e., differentiated pancreatic cancer cells) antibodies or anti-CD44+CD24+ PANC1 (i.e., pancreatic cancer stem cells) antibodies in sera of vaccinated knockout mice assessed with flow cytometry. Closed histogram; stained cells with sera from non-vaccinated knockout mice, open histogram; stained cells with sera from vaccinated knockout mice. MFI: Mean fluorescence intensity.
We conclude that the use of a tumor lysate vaccine that is engineered to express α-gal epitopes can elicit a durable and broadly protective immune response to subtypic PDAC cells, and that such vaccination may be a strategy for a universal cancer vaccine that will cure patients with PDAC.
Conclusion and future perspectives
The inability of the immune system to mount an antitumor response in PDAC despite an influx of lymphocytes has led to an abundance of therapeutic approaches aiming to modify and potentiate the immune reaction. Immunotherapeutic strategies have shown defined activity in the tumor and its microenvironment. Despite an antitumor response, application of these mechanisms has not yet become a component of standard therapy, largely due to the transient, unpredictable nature of the result of treatment and the lack of advanced phase studies.
Most PDAC patients have advanced unresectable disease at the time of diagnosis. Therefore, the high tumor burden of unresectable disease may overcome the effects of immune system stimulation that is induced by immunotherapy. Moreover, genomic instability confers resistance, and a fibrotic, hypoxic, hypovascular, and immunosuppressive tumor microenvironment may limit the potential benefits of immunotherapeutic applications of mono-TAA-based immunization. Accordingly, any vaccine used in patients with PDAC should contain multiple TAAs, that is, a polyvalent tumor lysate vaccine prepared from autologous tumors. However, immunotherapy alone is limited by the number of cytotoxic T lymphocytes that can penetrate a large and established pancreatic tumor. The most encouraging results of immunotherapy in PDAC have been in adjuvant settings, such as post-surgery[21,22]. In clinical application for resectable disease, we plan to employ as vaccinating material, autologous tumor lysates prepared from surgically resected PDAC that is enzymatically processed in vitro to express α-gal epitopes (Figure 9). However, the tumor mass resected from the pancreas is often small and limited, and the vast majority of PDAC patients are diagnosed as inoperable because they present with incurable metastatic disease. To overcome the problem of limited tissue, we propose using tumors generated in mice for vaccination material as described in this article (Figure 9). Furthermore, the role of combination therapy with either systemic chemotherapy or radiation therapy with immunotherapy using α-gal tumor lysate vaccination should also be assessed due to evidence that synergistic effects may occur when both therapies are administered simultaneously (Figure 9). We sincerely hope that the use of a tumor lysate vaccine that is engineered to express α-gal epitopes will elicit a strong immune response toward all PDAC cells, including differentiated PDAC cells and PDAC CSCs, and will improve the prognosis for patients with PDAC. For clinical application of this effective immunotherapy, we need to assess the toxicity and safety of injection of α-gal tumor lysates in humans. Although further studies are required, we should earnestly and simultaneously engage in both clinical studies involving α-gal tumor lysate vaccination and safety studies for this novel immunotherapy against the deadly disease, PDAC.
Figure 9.
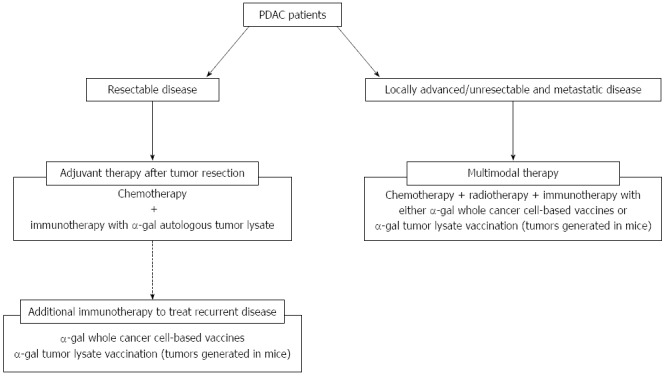
Treatment strategy using cancer immunotherapy utilizing α-gal epitope/anti-Gal antibody reaction for pancreatic ductal adenocarcinoma patients. The clinical implications of this cancer immunotherapy model are shown. For patients with resectable disease, we plan to employ autologous tumor lysates prepared from surgically resected PDAC that is enzymatically engineered to express α-gal epitopes. For patients with recurrent disease after surgery, additional immunotherapy with either α-gal whole cancer cell-based vaccines or α-gal tumor lysate vaccination (tumors generated in mice) should be assessed. For patients with unresectable and metastatic disease, multimodal therapy, including cancer immunotherapy using either α-gal whole cancer cell-based vaccines or α-gal tumor lysate vaccination (tumors generated in mice) should be conducted.
ACKNOWLEDGMENTS
We thank Professor Kazuhiko Yamada from the Graduate School of Medical and Dental Sciences, Kagoshima University (Kagoshima, Japan) for generously providing the photographs of hyperacute rejection of the pig kidney xenograft. We thank Dr. Kristine De La Torre PhD, a professional medical editor of forte (Tokyo, Japan) for the careful reading and editing of the manuscript.
Footnotes
Supported by Ministry of Education, Sports and Culture of Japan to M. T., No. 25462129.
Conflict-of-interest statement: The authors have no conflict of interest related to the manuscript.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 9, 2015
First decision: June 2, 2015
Article in press: August 31, 2015
P- Reviewer: Sperti C, Thomas SM S- Editor: Ma YJ L- Editor: A E- Editor: Ma S
References
- 1.American Cancer Society. Cancer Facts & Figures 2012. Atlanta: American Cancer Society; 2012. [Google Scholar]
- 2.Cress RD, Yin D, Clarke L, Bold R, Holly EA. Survival among patients with adenocarcinoma of the pancreas: a population-based study (United States) Cancer Causes Control. 2006;17:403–409. doi: 10.1007/s10552-005-0539-4. [DOI] [PubMed] [Google Scholar]
- 3.Winter JM, Cameron JL, Campbell KA, Arnold MA, Chang DC, Coleman J, Hodgin MB, Sauter PK, Hruban RH, Riall TS, et al. 1423 pancreaticoduodenectomies for pancreatic cancer: A single-institution experience. J Gastrointest Surg. 2006;10:1199–210; discussion 1210-1. doi: 10.1016/j.gassur.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 4.Garcea G, Dennison AR, Pattenden CJ, Neal CP, Sutton CD, Berry DP. Survival following curative resection for pancreatic ductal adenocarcinoma. A systematic review of the literature. JOP. 2008;9:99–132. [PubMed] [Google Scholar]
- 5.Roy R, Maraveyas A. Chemoradiation in pancreatic adenocarcinoma: a literature review. Oncologist. 2010;15:259–269. doi: 10.1634/theoncologist.2009-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 7.Oettle H, Post S, Neuhaus P, Gellert K, Langrehr J, Ridwelski K, Schramm H, Fahlke J, Zuelke C, Burkart C, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA. 2007;297:267–277. doi: 10.1001/jama.297.3.267. [DOI] [PubMed] [Google Scholar]
- 8.Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18:4266–4276. doi: 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 10.Sideras K, Braat H, Kwekkeboom J, van Eijck CH, Peppelenbosch MP, Sleijfer S, Bruno M. Role of the immune system in pancreatic cancer progression and immune modulating treatment strategies. Cancer Treat Rev. 2014;40:513–522. doi: 10.1016/j.ctrv.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 11.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galili U. Anti-Gal: an abundant human natural antibody of multiple pathogeneses and clinical benefits. Immunology. 2013;140:1–11. doi: 10.1111/imm.12110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 14.Bakkevold KE, Kambestad B. Long-term survival following radical and palliative treatment of patients with carcinoma of the pancreas and papilla of Vater--the prognostic factors influencing the long-term results. A prospective multicentre study. Eur J Surg Oncol. 1993;19:147–161. [PubMed] [Google Scholar]
- 15.Riall TS, Cameron JL, Lillemoe KD, Winter JM, Campbell KA, Hruban RH, Chang D, Yeo CJ. Resected periampullary adenocarcinoma: 5-year survivors and their 6- to 10-year follow-up. Surgery. 2006;140:764–772. doi: 10.1016/j.surg.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 16.Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 17.Eguchi H, Nagano H, Tanemura M, Takeda Y, Marubashi S, Kobayashi S, Kawamoto K, Wada H, Hama N, Akita H, et al. Preoperative chemoradiotherapy, surgery and adjuvant therapy for resectable pancreatic cancer. Hepatogastroenterology. 2013;60:904–911. doi: 10.5754/hge12974. [DOI] [PubMed] [Google Scholar]
- 18.Evans DB, Varadhachary GR, Crane CH, Sun CC, Lee JE, Pisters PW, Vauthey JN, Wang H, Cleary KR, Staerkel GA, et al. Preoperative gemcitabine-based chemoradiation for patients with resectable adenocarcinoma of the pancreatic head. J Clin Oncol. 2008;26:3496–3502. doi: 10.1200/JCO.2007.15.8634. [DOI] [PubMed] [Google Scholar]
- 19.Ohigashi H, Ishikawa O, Eguchi H, Takahashi H, Gotoh K, Yamada T, Yano M, Nakaizumi A, Uehara H, Tomita Y, et al. Feasibility and efficacy of combination therapy with preoperative full-dose gemcitabine, concurrent three-dimensional conformal radiation, surgery, and postoperative liver perfusion chemotherapy for T3-pancreatic cancer. Ann Surg. 2009;250:88–95. doi: 10.1097/SLA.0b013e3181ad65cc. [DOI] [PubMed] [Google Scholar]
- 20.Wedén S, Klemp M, Gladhaug IP, Møller M, Eriksen JA, Gaudernack G, Buanes T. Long-term follow-up of patients with resected pancreatic cancer following vaccination against mutant K-ras. Int J Cancer. 2011;128:1120–1128. doi: 10.1002/ijc.25449. [DOI] [PubMed] [Google Scholar]
- 21.Shakhar G, Ben-Eliyahu S. Potential prophylactic measures against postoperative immunosuppression: could they reduce recurrence rates in oncological patients? Ann Surg Oncol. 2003;10:972–992. doi: 10.1245/aso.2003.02.007. [DOI] [PubMed] [Google Scholar]
- 22.Weighardt H, Heidecke CD, Emmanuilidis K, Maier S, Bartels H, Siewert JR, Holzmann B. Sepsis after major visceral surgery is associated with sustained and interferon-gamma-resistant defects of monocyte cytokine production. Surgery. 2000;127:309–315. doi: 10.1067/msy.2000.104118. [DOI] [PubMed] [Google Scholar]
- 23.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 24.Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, Gopinathan A, Tuveson DA, Fearon DT. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330:827–830. doi: 10.1126/science.1195300. [DOI] [PubMed] [Google Scholar]
- 25.Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell. 2010;17:135–147. doi: 10.1016/j.ccr.2009.12.041. [DOI] [PubMed] [Google Scholar]
- 26.Shi M, Yu DH, Chen Y, Zhao CY, Zhang J, Liu QH, Ni CR, Zhu MH. Expression of fibroblast activation protein in human pancreatic adenocarcinoma and its clinicopathological significance. World J Gastroenterol. 2012;18:840–846. doi: 10.3748/wjg.v18.i8.840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ostrand-Rosenberg S. Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol Immunother. 2010;59:1593–1600. doi: 10.1007/s00262-010-0855-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. 2011;60:1419–1430. doi: 10.1007/s00262-011-1028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao F, Obermann S, von Wasielewski R, Haile L, Manns MP, Korangy F, Greten TF. Increase in frequency of myeloid-derived suppressor cells in mice with spontaneous pancreatic carcinoma. Immunology. 2009;128:141–149. doi: 10.1111/j.1365-2567.2009.03105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Esposito I, Menicagli M, Funel N, Bergmann F, Boggi U, Mosca F, Bevilacqua G, Campani D. Inflammatory cells contribute to the generation of an angiogenic phenotype in pancreatic ductal adenocarcinoma. J Clin Pathol. 2004;57:630–636. doi: 10.1136/jcp.2003.014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurahara H, Shinchi H, Mataki Y, Maemura K, Noma H, Kubo F, Sakoda M, Ueno S, Natsugoe S, Takao S. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J Surg Res. 2011;167:e211–e219. doi: 10.1016/j.jss.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 32.Ikemoto T, Yamaguchi T, Morine Y, Imura S, Soejima Y, Fujii M, Maekawa Y, Yasutomo K, Shimada M. Clinical roles of increased populations of Foxp3+CD4+ T cells in peripheral blood from advanced pancreatic cancer patients. Pancreas. 2006;33:386–390. doi: 10.1097/01.mpa.0000240275.68279.13. [DOI] [PubMed] [Google Scholar]
- 33.Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res. 2006;12:5423–5434. doi: 10.1158/1078-0432.CCR-06-0369. [DOI] [PubMed] [Google Scholar]
- 34.Yamamoto T, Yanagimoto H, Satoi S, Toyokawa H, Hirooka S, Yamaki S, Yui R, Yamao J, Kim S, Kwon AH. Circulating CD4+CD25+ regulatory T cells in patients with pancreatic cancer. Pancreas. 2012;41:409–415. doi: 10.1097/MPA.0b013e3182373a66. [DOI] [PubMed] [Google Scholar]
- 35.Galili U. The alpha-gal epitope and the anti-Gal antibody in xenotransplantation and in cancer immunotherapy. Immunol Cell Biol. 2005;83:674–686. doi: 10.1111/j.1440-1711.2005.01366.x. [DOI] [PubMed] [Google Scholar]
- 36.Macher BA, Galili U. The Galalpha1,3Galbeta1,4GlcNAc-R (alpha-Gal) epitope: a carbohydrate of unique evolution and clinical relevance. Biochim Biophys Acta. 2008;1780:75–88. doi: 10.1016/j.bbagen.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kearns-Jonker M, Swensson J, Ghiuzeli C, Chu W, Osame Y, Starnes V, Cramer DV. The human antibody response to porcine xenoantigens is encoded by IGHV3-11 and IGHV3-74 IgVH germline progenitors. J Immunol. 1999;163:4399–4412. [PubMed] [Google Scholar]
- 38.Eto T, Ichikawa Y, Nishimura K, Ando S, Yamakawa T. Chemistry of lipid of the posthemyolytic residue or stroma of erythrocytes. XVI. Occurrence of ceramide pentasaccharide in the membrane of erythrocytes and reticulocytes of rabbit. J Biochem. 1968;64:205–213. doi: 10.1093/oxfordjournals.jbchem.a128881. [DOI] [PubMed] [Google Scholar]
- 39.Stellner K, Saito H, Hakomori SI. Determination of aminosugar linkages in glycolipids by methylation. Aminosugar linkages of ceramide pentasaccharides of rabbit erythrocytes and of Forssman antigen. Arch Biochem Biophys. 1973;155:464–472. doi: 10.1016/0003-9861(73)90138-0. [DOI] [PubMed] [Google Scholar]
- 40.Galili U. Discovery of the natural anti-Gal antibody and its past and future relevance to medicine. Xenotransplantation. 2013;20:138–147. doi: 10.1111/xen.12034. [DOI] [PubMed] [Google Scholar]
- 41.Tanemura M, Yin D, Chong AS, Galili U. Differential immune responses to alpha-gal epitopes on xenografts and allografts: implications for accommodation in xenotransplantation. J Clin Invest. 2000;105:301–310. doi: 10.1172/JCI7358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ekser B, Ezzelarab M, Hara H, van der Windt DJ, Wijkstrom M, Bottino R, Trucco M, Cooper DK. Clinical xenotransplantation: the next medical revolution? Lancet. 2012;379:672–683. doi: 10.1016/S0140-6736(11)61091-X. [DOI] [PubMed] [Google Scholar]
- 43.Tisato V, Cozzi E. Xenotransplantation: an overview of the field. Methods Mol Biol. 2012;885:1–16. doi: 10.1007/978-1-61779-845-0_1. [DOI] [PubMed] [Google Scholar]
- 44.Groth CG, Korsgren O, Tibell A, Tollemar J, Möller E, Bolinder J, Ostman J, Reinholt FP, Hellerström C, Andersson A. Transplantation of porcine fetal pancreas to diabetic patients. Lancet. 1994;344:1402–1404. doi: 10.1016/s0140-6736(94)90570-3. [DOI] [PubMed] [Google Scholar]
- 45.Lai L, Kolber-Simonds D, Park KW, Cheong HT, Greenstein JL, Im GS, Samuel M, Bonk A, Rieke A, Day BN, et al. Production of alpha-1,3-galactosyltransferase knockout pigs by nuclear transfer cloning. Science. 2002;295:1089–1092. doi: 10.1126/science.1068228. [DOI] [PubMed] [Google Scholar]
- 46.Phelps CJ, Koike C, Vaught TD, Boone J, Wells KD, Chen SH, Ball S, Specht SM, Polejaeva IA, Monahan JA, et al. Production of alpha 1,3-galactosyltransferase-deficient pigs. Science. 2003;299:411–414. doi: 10.1126/science.1078942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamada K, Yazawa K, Shimizu A, Iwanaga T, Hisashi Y, Nuhn M, O’Malley P, Nobori S, Vagefi PA, Patience C, et al. Marked prolongation of porcine renal xenograft survival in baboons through the use of alpha1,3-galactosyltransferase gene-knockout donors and the cotransplantation of vascularized thymic tissue. Nat Med. 2005;11:32–34. doi: 10.1038/nm1172. [DOI] [PubMed] [Google Scholar]
- 48.Kuwaki K, Tseng YL, Dor FJ, Shimizu A, Houser SL, Sanderson TM, Lancos CJ, Prabharasuth DD, Cheng J, Moran K, et al. Heart transplantation in baboons using alpha1,3-galactosyltransferase gene-knockout pigs as donors: initial experience. Nat Med. 2005;11:29–31. doi: 10.1038/nm1171. [DOI] [PubMed] [Google Scholar]
- 49.Saito H, Dubsky P, Dantin C, Finn OJ, Banchereau J, Palucka AK. Cross-priming of cyclin B1, MUC-1 and survivin-specific CD8+ T cells by dendritic cells loaded with killed allogeneic breast cancer cells. Breast Cancer Res. 2006;8:R65. doi: 10.1186/bcr1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thomas AM, Santarsiero LM, Lutz ER, Armstrong TD, Chen YC, Huang LQ, Laheru DA, Goggins M, Hruban RH, Jaffee EM. Mesothelin-specific CD8(+) T cell responses provide evidence of in vivo cross-priming by antigen-presenting cells in vaccinated pancreatic cancer patients. J Exp Med. 2004;200:297–306. doi: 10.1084/jem.20031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jaffee EM, Hruban RH, Biedrzycki B, Laheru D, Schepers K, Sauter PR, Goemann M, Coleman J, Grochow L, Donehower RC, et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J Clin Oncol. 2001;19:145–156. doi: 10.1200/JCO.2001.19.1.145. [DOI] [PubMed] [Google Scholar]
- 52.Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol. 2003;3:939–951. doi: 10.1038/nri1248. [DOI] [PubMed] [Google Scholar]
- 53.Schwartz RH. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell. 1992;71:1065–1068. doi: 10.1016/s0092-8674(05)80055-8. [DOI] [PubMed] [Google Scholar]
- 54.Lanzavecchia A. Mechanisms of antigen uptake for presentation. Curr Opin Immunol. 1996;8:348–354. doi: 10.1016/s0952-7915(96)80124-5. [DOI] [PubMed] [Google Scholar]
- 55.Grabbe S, Beissert S, Schwarz T, Granstein RD. Dendritic cells as initiators of tumor immune responses: a possible strategy for tumor immunotherapy? Immunol Today. 1995;16:117–121. doi: 10.1016/0167-5699(95)80125-1. [DOI] [PubMed] [Google Scholar]
- 56.Ermann J, Fathman CG. Costimulatory signals controlling regulatory T cells. Proc Natl Acad Sci USA. 2003;100:15292–15293. doi: 10.1073/pnas.0307001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bear AS, Cruz CR, Foster AE. T cells as vehicles for cancer vaccination. J Biomed Biotechnol. 2011;2011:417403. doi: 10.1155/2011/417403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zinkernagel RM, Ehl S, Aichele P, Oehen S, Kündig T, Hengartner H. Antigen localisation regulates immune responses in a dose- and time-dependent fashion: a geographical view of immune reactivity. Immunol Rev. 1997;156:199–209. doi: 10.1111/j.1600-065x.1997.tb00969.x. [DOI] [PubMed] [Google Scholar]
- 59.Hogarth PM, Pietersz GA. Fc receptor-targeted therapies for the treatment of inflammation, cancer and beyond. Nat Rev Drug Discov. 2012;11:311–331. doi: 10.1038/nrd2909. [DOI] [PubMed] [Google Scholar]
- 60.Kohrt HE, Houot R, Marabelle A, Cho HJ, Osman K, Goldstein M, Levy R, Brody J. Combination strategies to enhance antitumor ADCC. Immunotherapy. 2012;4:511–527. doi: 10.2217/imt.12.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fanger NA, Wardwell K, Shen L, Tedder TF, Guyre PM. Type I (CD64) and type II (CD32) Fc gamma receptor-mediated phagocytosis by human blood dendritic cells. J Immunol. 1996;157:541–548. [PubMed] [Google Scholar]
- 62.Abdel-Motal UM, Wigglesworth K, Galili U. Intratumoral injection of alpha-gal glycolipids induces a protective anti-tumor T cell response which overcomes Treg activity. Cancer Immunol Immunother. 2009;58:1545–1556. doi: 10.1007/s00262-009-0662-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abdel-Motal UM, Wigglesworth K, Galili U. Mechanism for increased immunogenicity of vaccines that form in vivo immune complexes with the natural anti-Gal antibody. Vaccine. 2009;27:3072–3082. doi: 10.1016/j.vaccine.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 64.Deguchi T, Tanemura M, Miyoshi E, Nagano H, Machida T, Ohmura Y, Kobayashi S, Marubashi S, Eguchi H, Takeda Y, et al. Increased immunogenicity of tumor-associated antigen, mucin 1, engineered to express alpha-gal epitopes: a novel approach to immunotherapy in pancreatic cancer. Cancer Res. 2010;70:5259–5269. doi: 10.1158/0008-5472.CAN-09-4313. [DOI] [PubMed] [Google Scholar]
- 65.Wang Z, Li Y, Ahmad A, Banerjee S, Azmi AS, Kong D, Sarkar FH. Pancreatic cancer: understanding and overcoming chemoresistance. Nat Rev Gastroenterol Hepatol. 2011;8:27–33. doi: 10.1038/nrgastro.2010.188. [DOI] [PubMed] [Google Scholar]
- 66.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 67.Terao N, Takamatsu S, Minehira T, Sobajima T, Nakayama K, Kamada Y, Miyoshi E. Fucosylation is a common glycosylation type in pancreatic cancer stem cell-like phenotypes. World J Gastroenterol. 2015;21:3876–3887. doi: 10.3748/wjg.v21.i13.3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yin T, Shi P, Gou S, Shen Q, Wang C. Dendritic cells loaded with pancreatic Cancer Stem Cells (CSCs) lysates induce antitumor immune killing effect in vitro. PLoS One. 2014;9:e114581. doi: 10.1371/journal.pone.0114581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hou YC, Chao YJ, Tung HL, Wang HC, Shan YS. Coexpression of CD44-positive/CD133-positive cancer stem cells and CD204-positive tumor-associated macrophages is a predictor of survival in pancreatic ductal adenocarcinoma. Cancer. 2014;120:2766–2777. doi: 10.1002/cncr.28774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tanida T, Tanemura M, Miyoshi E, Nagano H, Furukawa K, Nonaka Y, Akita H, Hama N, Wada H, Kawamoto K, et al. Pancreatic cancer immunotherapy using a tumor lysate vaccine, engineered to express α-gal epitopes, targets pancreatic cancer stem cells. Int J Oncol. 2015;46:78–90. doi: 10.3892/ijo.2014.2717. [DOI] [PubMed] [Google Scholar]