

Abstract
Microbiota in human alimentary tract plays important roles for homeostatic maintenance of the body. Compositional difference of gut microbiota is tightly associated with susceptibility of many diseases, including inflammatory diseases, obesity, diabetes mellitus, cancer, and atherosclerosis. “Dysbiosis” refers to a state of imbalance among the colonies of microorganisms within the body, which brings abnormal increase of specific minor components and decrease in the normally dominant species. Since stomach secrets strong acid for its digestive role, this organ has long been thought a sterile organ. However, the discovery of Helicobacter pylori (H. pylori) has changed the concept. This bacterium has proven to cause gastritis, peptic ulcer, and gastric cancer. However, recent cross-sectional studies revealed that H. pylori carriers had a decreased risk of developing immunological diseases, such as asthma. H. pylori coinfection also suppresses inflammatory bowel diseases. This review describes human gastric microbiota by discussing its mutual interaction and pathogenic enrollment. Gastric “dysbiosis” may affect host inflammatory response and play important role for gastric pathogenesis. We will topically discuss enrollment of dysbiosis for genesis of gastric cancer as well as for disruption of immunological homeostasis affecting oncogenic resistance.
Keywords: Stomach, Microbiota, Dysbiosis, Helicobacter pylori, Epstein-Barr virus
Core tip: The imbalance of microflora in the gut induces dysbiosis. Altered gut microflora is known to be associated with inflammatory diseases, obesity, diabetes, cancer, and atherosclerosis. Little is known about gastric microflora, which will also interacts with bacteria, viruses and funguses. In this review, we discuss that dysbiosis in the stomach may disrupt immunological homeostasis, reduce of carcinogenic resistance, and induce gastric cancer.
INTRODUCTION
Various microbes, from commensal to pathogenic, reside in the human body. Not only they are interacting with their host, but also these different microorganisms (bacteria, yeast, viruses, parasites, etc.) are interacting with each other. This interaction sometimes causes dysbiosis, which refers to microbial imbalance inside the body. Dysbiosis in the digestive tract sometimes exacerbate bowel disease[1].
Microbial colonies found in human body are normally beneficial, but are parasitic, commensal, or symbiotic. These appropriately sized microbial colonies assist necessary functions in digestion. The beneficial bacterial colonies also protect the body from the penetration of pathogenic microbes by competing with pathogens for space and nutrition. Dysbiosis in bacteria refers to increased levels of harmful bacteria and reduced levels of the beneficial bacteria.
The microbial interaction also occurs between bacterium and other microbes, such as virus and fungus. Bacteria and viruses residing together can work sometimes synergistically to enhance pathogenesis. Gene expression of viruses showing latent infection, such as Kaposi’s sarcoma-associated herpesvirus, Epstein-Barr virus (EBV), and HIV, is influenced by epigenetic modifications induced by bacteria[2]. Latent infection of these viruses can be disrupted by bacterial products and viral production will be reactivated (Figure 1). In HIV-positive persons, immunosuppression promotes growth of other opportunistic organisms that contributes progression of acquired immune deficiency syndrome (AIDS). In addition, bacterium-virus interactions should be involved in oncogenic process. Both Helicobacter pylori (H. pylori) and EBV are associated with gastric cancer, respectively[3,4]. Since H. pylori spreads in many human populations and its roles for stomach cancer development is well accepted. Infection of EBV into gastric epitherial cells also develops gastric cancer in fewer than 10% of total cases, which often associates with lymphoplasmacytic infiltrate.
Figure 1.
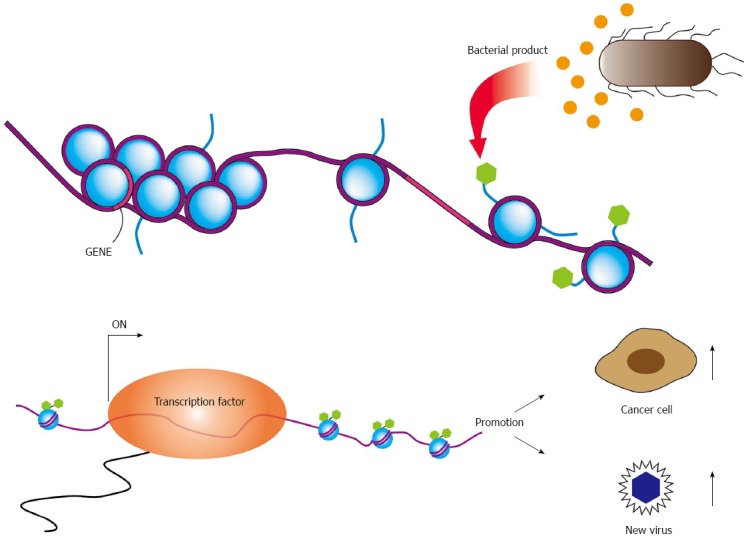
Epigenetic modifications to promoters. Epigenetic modifications to viral or cellular promoters regulate expression of human and viral genes. Bacterial products and or proinflammatory cytokines activate epigenetic marks on viral or cellular promoters, which can promote viral production as well as stimulate the transcription of viral oncogenes. These epigenetic modifiers also stimulate cellular proliferation. The reactivation of a latent virus results not only production of virion, but also may drive cellular transformation.
This paper will be focused on dysbiotic infection in the stomach. Although not as many as lower alimentary tract, some microbes reside in the stomach. Some are derived from dietary intake, others are from oral, nasopharyngeal, and tracheal swallows. Duodenal reflux will also bring microbes to the stomach. Dysbiosis in the stomach will bring imbalance to immunological homeostasis, which may take some part in inflammatory response and will be involved with pathogenesis. The effects of coexisting bacterial-bacterial and bacterial-viral coinfections should be considered for pathogenesis of gastric diseases.
GASTRIC ACIDITY, H. PYLORI, AND OTHER BACTERIA
The gastric juice represents a barrier to microbes in saliva and ingested food, mostly due to the degenerative activity of hydrochloric acid[5]. If this bactericidal activity is weakened by an elevation of gastric pH, microbes will be allowed to survive in the stomach. It is reported that 80% of healthy subjects between 80 to 91 years old showed hypochlorhydria with pH 6.6. These people posessed 105 to 108 colony forming units per ml of bacteria in fasting gastric aspirate[6]. The strong association between diminished gastric acid secretion and the presence of opportunistic enteric pathogens was clearly observed in AIDS patients[7]. The gastric barrier to infection has more significant meaning to hosts of whom immunological defense is weakened.
We showed when H. pylori was mixed with an acid resistant isolate of Kingella denitrificans (K. denitrificans), a commensal of the human respiratory tract, survival of H. pylori in acidic condition was increased compared with the single culture of H. pylori[8]. Binding of the acid resistant K. denitrificans with H. pylori seemingly coated the bacterial body to allow survival of H. pylori in the acidic condition. Another studies have revealed that commensal and foreign microbes may interact intimately with gut epithelium to influence host signaling pathways that regulate metabolic and stress responses[9,10]. The colonization of commensal microbes in gastric epithelium may affect the carcinogenic potentials of H. pylori by modulating CagA-mediated regulation of oncogenic signals.
GASTRIC MICROBIOTA
Thick mucus layer, acidic gastric juice and peristaltic movement in the stomach have raised the dogma that “the stomach is a sterile organ”. However, the dogma quickly changes after the discovery of Campylobacter pyloridis in 1982, which is renamed into H. pylori in 1984[11]. H. pylori can colonise the stomach by producing urease to survive under acidic condition. Soon after the discovery of H. pylori, another type of bacteria such as Vellomella, Lactobacillus and Clostridium are found as transient bacteria that reside in the stomach[12]. However, the ability of the transient bacteria to crosslink with the host and penetrate the mucosa layer is drawing people’s attention.
Recently, the development of culture-independent molecular technologies based on 16s rRNA has revealed that five abundant genera microbiota other than H. pylori reside in the stomach. They are Neisseria, Haemophilus, Prevotella, Streptococcus, and Porphyromonas[13-16].
Dysbiosis of the gastric microbiota has been implicated in immune system regulation and enhancing disease symptoms. Several researchers showed the gastric microbiota arose from patients infected with H. pylori are different from uninfected people[17,18]. Osaki et al[19] also described the prolonged exposure to H. pylori infection has altered the composition of the microbiota in rodent stomachs. These findings suggested an interaction between H. pylori and the gastric microbial community[8]. Though the mechanism of H. pylori in altering the gastric microbiota remains unclear, possible explanation is that the induction of host antimicrobial peptides, such as β-defensin 2[20] or cecropin-like peptide, may directly kill another microbiota[21].
All of these findings had shed a light that dysbiosis of gastric microbiota might related to the susceptibility to gastric inflammation and tumorigenicity in patients with H. pylori infection. H. pylori infection also initiate the inflammatory cascades that induce physiological changes that reduces the gastric secretion from parietal cells and elevation of pH in the stomach. The elevation of pH eventually resulted in the colonisation of another microrganisms in the stomach[22-25]. Engstrand et al[26] reported that gastric cancer development may related to the alteration of gastric microbiota. The commensal microbes can communicate with dysbiotic pathogens such as Salmonella typhimurium that have the ability to alter gastrointestinal homeostasis to pathogenic inflammation. However, it should be further investigated whether infections with commensals are associated with the susceptibility to gastric inflammation and tumorigenicity in patients with H. pylori infection.
INFECTION AND GASTRIC CANCER
H. pylori is a primary causative agent not only for peptic ulcer diseases and chronic gastritis, but also for gastric cancer. Other than H. pylori, EBV is also known to cause gastric cancer. EBV-associated gastric carcinoma (EBVaGC) comprises about 10% of all gastric carcinomas worldwide[27,28]. H. pylori infection has been linked to CpG hypermethylation of tumor suppressor genes, including Runx3, E-cadherin, and p16[29-32]. EBV infection was correlated with overexpression of DNA methyltransferase 1 in gastric cancers[33]. And EBVaGCs have a unique pattern of methylation linked to the downregulation of p16 but not MLH1[34,35]. High methylation frequencies of several tumor suppressor genes, APC, PTEN, and RASSF1A, and cell adhesion molecules, THBS1 and E-cadherin, were also reported in EBVaGC. The posttranscriptional modification might change the epithelial phenotype, important for generating gastric microbial niche, however, it is too early to discuss effect of such alteration for gastric microbiota. On the other hand, several reports describe synergy between H. pylori and EBV for the genesis of gastric cancer. Firstly, individuals co-infected with H. pylori and EBV significantly possessed severe inflammatory lesions than persons with a single H. pylori infection[36]. It has also been shown that H. pylori infection was associated with EBV reactivation in patients with gastric symptoms[37]. Lastly, reactivation of EBV in latently infected gastric epithelial cells was induced by monochloramine, a product of H.pylori infection[38]. These observations suggest that coinfection of the two pathogens possibly heighten risk of gastric cancer[39,40].
H. pylori-related gastritis frequently initiates in the antrum. On the other hand, EBVaGC tumors are frequently located near the mucosal atrophic border, where mild to moderate atrophy is common[41]. Both EBV and H. pylori could be abundantly detected in the same mucosa of patients suffering with moderate chronic atrophic gastritis, where inflammatory cell infiltration is abundant. However, neither microbe could be frequently detected in the mucosa with marked atrophic gastritis, where inflammatory cell infiltration is scarce[34]. The observation strongly suggested inflammation caused by bacterial infection might promote generation of cancer associated with EBV infection[4,35].
Gastric remnant cancer arises after distal gastrectomy for benign disease, which includes refractory gastric or duodenal ulcer disease and recurrent ulcer with gastric outlet obstruction. The incidence of gastric remnant cancer ranges from 1% to 7% of all gastric carcinomas[42]. Gastric remnant carcinoma is characteristically associated with EBV infection in high frequency (25% to 41.2%). It is considered that the reflux of bile and pancreatic juice causes regenerative atypia and cell proliferation in epithelial cells[43]. In Billroth-II anastomoses, atrophic change of remnant gastritis is frequently accompanied by EBV-positive gastric remnant carcinoma[34,44].
EBV efficiently drives proliferation of human primary B cells in vitro, which subsequently transforms B cells. B-cell proliferation is also driven by ligands of Toll-like receptors (TLRs). Proliferation of EBV-infected B cells and their capability to interact with immune effector cells may be directly influenced by components of bacteria or other microbes present at the site of infection[45,46]. Oral commensal Porphyromonas gingivalis produces butyric acid, which may reactivate epigenetic silencing by increasing H3 and H4 acetylation[47]. It is well known that EBV transactivator, BZLF1 same as ZEBRA or Zta, which reactivates latent infection of EBV to lytic replicative infection, can be induced by treatment of latently EBV-infected cells with butyric acid[48]. These reports suggest dysbiotic bacterial infection activates latently infected viruses, which exacerbate microbial infection (Figure 2). These observations strongly remind us of the idea that cancer associated with inflammation.
Figure 2.
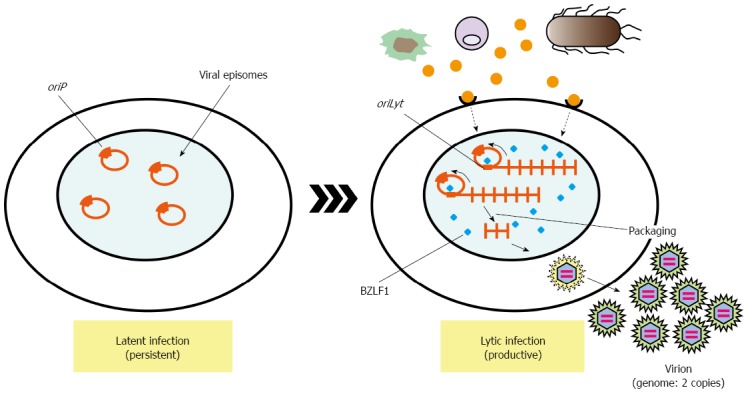
Lytic activation of Epstein-Barr virus by inflammatory product. After primary infection of EBV, the infected cells undergo prelatent cycles in which only immediate-early and early genes are expressed with no viral production. This transient lytic state is silenced, and latent infection is persistently established by expressing only limited numbers of latent genes. The latent infection may undergo the lytic cycle, in which viral late gene expression, viral genome replication, and production of the progeny virus (virion) can be observed. BZLF1 is a molecular switch for EBV reactivation from latent infection. And various signaling pathways activate cis-acting elements in the BZLF1 promoter[47, 48]. Although viral latent gene such as LMP1 can also strongly expressed on lytic infection, which sometimes promote cell proliferation by enhancing cell signaling, modulating immune system, and inducing genomic instability. oriP is a latent origin for viral genome replication. BZLF1 is a transactivator of virus replication, which forms homodimers and binds to oriLyt, origin for EBV DNA replication in lytic infection. EBV: Epstein-Barr virus.
POST-INFECTIOUS IMMUNE-DISORDER IN UPPER GASTROINTESTINAL TRACT
Infection and immune dysregulation in intestinal tract
Exposure to acute gastrointestinal infection induces persistent low grade mucosal inflammation, which sometimes leads to onset of post-infectious irritable bowel syndrome (PI-IBS)[49-53]. Organisms such as Campylobacter, Salmonella, Escherichia coli (E. coli), and Shigella are common pathogens involved in the development of PI-IBS. Immune disorders found in PI-IBS patients are characterized by mucosal infiltration of immune cells, including macrophages, T cells, mast cells, and eosinophils, as well as increased production of various cytokines[49,50,52,54-58]. TLR-dependent innate immunity is also activated along with persistent low grade gut inflammation following acute gastroenteritis (AGE), which may be associated with dysbiosis of gut microbiota[59-62].
Functional disorders following AGE in upper gastrointestinal tract
Functional dyspepsia (FD), a main functional disorder in the upper gastrointestinal tract, can also develop in previously asymptomatic individuals following an episode of microbial infection-related AGE. This type of FD is currently recognized as post-infectious FD (PI-FD)[63,64]. Tack et al[65] reported that 55 (17%) cases from 400 FD patients had episodes of AGE, while PI-FD onset was not correlated with the rate of H. pylori infection. A prospective observational study evaluated the incidence of FD development in patients with Salmonella infection-induced AGE after 1 year. The FD incidence was significantly higher in the infection cohort (13.4%) as compared to the non-infection cohort (2%)[66]. The systematic review including meta-analysis findings was performed at more than 6 months after AGE. The mean prevalence of FD following AGE was 9.55% in adult populations. The summary odds ratio for development of PI-FD was 2.54 (95%CI: 1.76-3.65)[67]. The pathogens Salmonella spp., E. coli O157, Campylobacter jejuni, Giardia lamblia, and Norovirus have all been associated with the development of PI-FD.
Altered populations of epithelial and mucosal immune cells in upper gastrointestinal tract following AGE
Although AGE may be one of the crucial causes in the development of PI-FD, its pathogenesis has not been fully investigated. AGE was shown to induce persistent low-grade mucosal inflammation via altered immune functions in the upper gastrointestinal tract (Table 1). Kindt et al[68] reported aggregation of CD3+ T cells, decrease of CD4+ T cells, and increase of CD68+ macrophages along the muscularis mucosae of duodenum in PI-FD patients. Increased infiltration of CC chemokine receptor-2+/CD68+ macrophages and eosinophils in duodenal mucosa was also found in certain populations of FD patients[69]. The number of mast cells and enterochromaffin cells in gastric mucosa was significantly increased in PI-FD patients than FD patients with no episodes of AGE[70]. In addition, increased number of mast cells and enterochromaffin cells is often found in the colonic mucosa of PI-IBS patients. Apart from bacterial and viral infections, the incidence of PI-FD was increased in patients with a history of parasitic Giardia infection. Moreover, the number of cholecystokinin-producing enterochromaffin cells was increased, but the number of serotonin-producing enterochromaffin cells was decreased in the duodenal mucosa of giardiasis patients[71]. The pathogenesis of PI-FD may be influenced by altered populations of immune cells as well as serotonin metabolism in the upper gastrointestinal tract. However, the detailed mechanisms of PI-FD remain to be fully clarified.
Table 1.
Altered populations of epithelial and mucosal immune cells in post-infectious functional dyspepsia patients
| Ref. | Location | Cell population | Changes |
| Kindt et al[68] | Duodenum | CD3+ T cells | Aggregated |
| CD4+ T cells | Decreased | ||
| CD68+ cells (macrophages) | Increased | ||
| Futagami et al[69] | Duodenum | CCR2 +/CD68+ cells (macrophages) | Increased |
| Eosinophils | Increased | ||
| Li et al[70] | Stomach | Mast cells | Increased |
| EC cells | Increased | ||
| Dizdar et al[71] | Duodenum | EC cells (5-HT-producing) | Decreased |
| EC cells (CCK-producing) | Increased |
EC: Enterochromaffin; CCR: CC chemokine receptor; 5-HT: Serotonin; CCK: Cholecystokinin.
Is post-infectious immune-disorder in the upper gastrointestinal tract associated with dysbiosis?
Dysbiosis of the gut microbiota has shown to be associated with the pathogenesis of intestinal inflammatory and functional disorders. AGE certainly plays an important role in the pathogenesis of PI-FD through an immune disorder in the upper gastrointestinal tract. However, it remains largely unknown whether AGE directly induces dysbiosis or only influences the process of development of PI-FD. Inflammasomes regulate gut microbiota by co-functioning with various inflammatory signals from cytokines, such as interleukin-1β and 18, as well as with signals from TLR-4 and TLR-9 innate immune receptors[72]. AGE-associated induction of dysbiosis may be regulated by such processes, however, further investigations are required to elucidate the role of infection-induced dysbiosis and its association with functional disorders in the upper gastrointestinal tract.
CONCLUSION
It has been proven recently that not only long-term dietary intake, but also short-term dietary intake alters human gut microbiome[73]. The animal-based diet significantly increased the levels of fecal deoxycholic concentrations, which is the product of microbial metabolism and promotes liver cancer[74]. Moreover, the animal-based diet significantly increased sulphite-reducing bacteria which might increase inflammation to intestinal tissue through H2S production[75].
Human disease can also be developed from an imbalance between commensal bacteria and fungi[76]. Candida albicans (C. albicans) extensively distributes on human skin and mucosal surfaces, such as the oral cavity, the gastrointestinal tract, and the lower female reproductive tract. Because of this, the fungus is most frequently implicated in mixed bacterial-fungal infections. Enhancement of bacterial virulence by C. albicans has been described in studies assessing the virulence of mixed C. albicans and Staphylococcus aureus infection in mice[77].
Bacteria, virus, fungus, and some parasites are affecting each other to reside and propagate in human alimentary tract. Their opportunistic imbalance often provides illness to human beings. Our body had better keep benign microbiota and refrain from having dysbiotic microbiota. Though little in known, further investigation will surely tell us the way how to keep symbiotic relation with gastric microbiota.
ACKNOWLEDGMENTS
We thank to Ms. Sayuri Hamada for preparation of figures. The authors have declared no conflicts of interest.
Footnotes
Supported by (in part) Grant-in-Aid for Scientific Research from the Ministry of Education; Culture, Science and Technology of Japan, No. 26460546 (to Yoshiyama H).
Conflict-of-interest statement: The authors have no conflict of interest to report.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 20, 2015
First decision: June 23, 2015
Article in press: September 15, 2015
P- Reviewer: Kim JM S- Editor: Ma YJ L- Editor: A E- Editor: Zhang DN
References
- 1.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doolittle JM, Webster-Cyriaque J. Polymicrobial infection and bacterium-mediated epigenetic modification of DNA tumor viruses contribute to pathogenesis. MBio. 2014;5:e01015–e01014. doi: 10.1128/mBio.01015-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pagano JS, Blaser M, Buendia MA, Damania B, Khalili K, Raab-Traub N, Roizman B. Infectious agents and cancer: criteria for a causal relation. Semin Cancer Biol. 2004;14:453–471. doi: 10.1016/j.semcancer.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 4.Rickinson AB. Co-infections, inflammation and oncogenesis: future directions for EBV research. Semin Cancer Biol. 2014;26:99–115. doi: 10.1016/j.semcancer.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 5.Yoshiyama H, Nakazawa T. Unique mechanism of Helicobacter pylori for colonizing the gastric mucus. Microbes Infect. 2000;2:55–60. doi: 10.1016/s1286-4579(00)00285-9. [DOI] [PubMed] [Google Scholar]
- 6.Husebye E, Skar V, Høverstad T, Melby K. Fasting hypochlorhydria with gram positive gastric flora is highly prevalent in healthy old people. Gut. 1992;33:1331–1337. doi: 10.1136/gut.33.10.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belitsos PC, Greenson JK, Yardley JH, Sisler JR, Bartlett JG. Association of gastric hypoacidity with opportunistic enteric infections in patients with AIDS. J Infect Dis. 1992;166:277–284. doi: 10.1093/infdis/166.2.277. [DOI] [PubMed] [Google Scholar]
- 8.Okamoto T, Hayashi Y, Mizuno H, Yanai H, Nishikawa J, Nakazawa T, Iizasa H, Jinushi M, Sakaida I, Yoshiyama H. Colonization of an acid resistant Kingella denitrificans in the stomach may contribute to gastric dysbiosis by Helicobacter pylori. J Infect Chemother. 2014;20:169–174. doi: 10.1016/j.jiac.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 9.Shin SC, Kim SH, You H, Kim B, Kim AC, Lee KA, Yoon JH, Ryu JH, Lee WJ. Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science. 2011;334:670–674. doi: 10.1126/science.1212782. [DOI] [PubMed] [Google Scholar]
- 10.Patwa LG, Fan TJ, Tchaptchet S, Liu Y, Lussier YA, Sartor RB, Hansen JJ. Chronic intestinal inflammation induces stress-response genes in commensal Escherichia coli. Gastroenterology. 2011;141:1842–51.e1-10. doi: 10.1053/j.gastro.2011.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nardone G, Compare D. The human gastric microbiota: Is it time to rethink the pathogenesis of stomach diseases? United European Gastroenterol J. 2015;3:255–260. doi: 10.1177/2050640614566846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zilberstein B, Quintanilha AG, Santos MA, Pajecki D, Moura EG, Alves PR, Maluf Filho F, de Souza JA, Gama-Rodrigues J. Digestive tract microbiota in healthy volunteers. Clinics (Sao Paulo) 2007;62:47–54. doi: 10.1590/s1807-59322007000100008. [DOI] [PubMed] [Google Scholar]
- 13.Fraher MH, O’Toole PW, Quigley EM. Techniques used to characterize the gut microbiota: a guide for the clinician. Nat Rev Gastroenterol Hepatol. 2012;9:312–322. doi: 10.1038/nrgastro.2012.44. [DOI] [PubMed] [Google Scholar]
- 14.Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, Perez-Perez G, Blaser MJ, Relman DA. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci USA. 2006;103:732–737. doi: 10.1073/pnas.0506655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li XX, Wong GL, To KF, Wong VW, Lai LH, Chow DK, Lau JY, Sung JJ, Ding C. Bacterial microbiota profiling in gastritis without Helicobacter pylori infection or non-steroidal anti-inflammatory drug use. PLoS One. 2009;4:e7985. doi: 10.1371/journal.pone.0007985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delgado S, Cabrera-Rubio R, Mira A, Suárez A, Mayo B. Microbiological survey of the human gastric ecosystem using culturing and pyrosequencing methods. Microb Ecol. 2013;65:763–772. doi: 10.1007/s00248-013-0192-5. [DOI] [PubMed] [Google Scholar]
- 17.Andersson AF, Lindberg M, Jakobsson H, Bäckhed F, Nyrén P, Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One. 2008;3:e2836. doi: 10.1371/journal.pone.0002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maldonado-Contreras A, Goldfarb KC, Godoy-Vitorino F, Karaoz U, Contreras M, Blaser MJ, Brodie EL, Dominguez-Bello MG. Structure of the human gastric bacterial community in relation to Helicobacter pylori status. ISME J. 2011;5:574–579. doi: 10.1038/ismej.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osaki T, Matsuki T, Asahara T, Zaman C, Hanawa T, Yonezawa H, Kurata S, Woo TD, Nomoto K, Kamiya S. Comparative analysis of gastric bacterial microbiota in Mongolian gerbils after long-term infection with Helicobacter pylori. Microb Pathog. 2012;53:12–18. doi: 10.1016/j.micpath.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 20.Hornsby MJ, Huff JL, Kays RJ, Canfield DR, Bevins CL, Solnick JV. Helicobacter pylori induces an antimicrobial response in rhesus macaques in a cag pathogenicity island-dependent manner. Gastroenterology. 2008;134:1049–1057. doi: 10.1053/j.gastro.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pütsep K, Normark S, Boman HG. The origin of cecropins; implications from synthetic peptides derived from ribosomal protein L1. FEBS Lett. 1999;451:249–252. doi: 10.1016/s0014-5793(99)00582-7. [DOI] [PubMed] [Google Scholar]
- 22.Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest. 2004;113:321–333. doi: 10.1172/JCI20925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Müller A, Solnick JV. Inflammation, immunity, and vaccine development for Helicobacter pylori. Helicobacter. 2011;16 Suppl 1:26–32. doi: 10.1111/j.1523-5378.2011.00877.x. [DOI] [PubMed] [Google Scholar]
- 24.Oh JD, Kling-Bäckhed H, Giannakis M, Engstrand LG, Gordon JI. Interactions between gastric epithelial stem cells and Helicobacter pylori in the setting of chronic atrophic gastritis. Curr Opin Microbiol. 2006;9:21–27. doi: 10.1016/j.mib.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 25.Wroblewski LE, Peek RM. Targeted disruption of the epithelial-barrier by Helicobacter pylori. Cell Commun Signal. 2011;9:29. doi: 10.1186/1478-811X-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Engstrand L, Lindberg M. Helicobacter pylori and the gastric microbiota. Best Pract Res Clin Gastroenterol. 2013;27:39–45. doi: 10.1016/j.bpg.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 27.Takada K. Epstein-Barr virus and gastric carcinoma. Mol Pathol. 2000;53:255–261. doi: 10.1136/mp.53.5.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iizasa H, Nanbo A, Nishikawa J, Jinushi M, Yoshiyama H. Epstein-Barr Virus (EBV)-associated gastric carcinoma. Viruses. 2012;4:3420–3439. doi: 10.3390/v4123420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kitajima Y, Ohtaka K, Mitsuno M, Tanaka M, Sato S, Nakafusa Y, Miyazaki K. Helicobacter pylori infection is an independent risk factor for Runx3 methylation in gastric cancer. Oncol Rep. 2008;19:197–202. [PubMed] [Google Scholar]
- 30.Grady WM, Willis J, Guilford PJ, Dunbier AK, Toro TT, Lynch H, Wiesner G, Ferguson K, Eng C, Park JG, et al. Methylation of the CDH1 promoter as the second genetic hit in hereditary diffuse gastric cancer. Nat Genet. 2000;26:16–17. doi: 10.1038/79120. [DOI] [PubMed] [Google Scholar]
- 31.Ferrasi AC, Pinheiro NA, Rabenhorst SH, Caballero OL, Rodrigues MA, de Carvalho F, Leite CV, Ferreira MV, Barros MA, Pardini MI. Helicobacter pylori and EBV in gastric carcinomas: methylation status and microsatellite instability. World J Gastroenterol. 2010;16:312–319. doi: 10.3748/wjg.v16.i3.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan AO, Lam SK, Wong BC, Wong WM, Yuen MF, Yeung YH, Hui WM, Rashid A, Kwong YL. Promoter methylation of E-cadherin gene in gastric mucosa associated with Helicobacter pylori infection and in gastric cancer. Gut. 2003;52:502–506. doi: 10.1136/gut.52.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Etoh T, Kanai Y, Ushijima S, Nakagawa T, Nakanishi Y, Sasako M, Kitano S, Hirohashi S. Increased DNA methyltransferase 1 (DNMT1) protein expression correlates significantly with poorer tumor differentiation and frequent DNA hypermethylation of multiple CpG islands in gastric cancers. Am J Pathol. 2004;164:689–699. doi: 10.1016/S0002-9440(10)63156-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaneda A, Matsusaka K, Aburatani H, Fukayama M. Epstein-Barr virus infection as an epigenetic driver of tumorigenesis. Cancer Res. 2012;72:3445–3450. doi: 10.1158/0008-5472.CAN-11-3919. [DOI] [PubMed] [Google Scholar]
- 35.Nishikawa J, Yoshiyama H, Iizasa H, Kanehiro Y, Nakamura M, Nishimura J, Saito M, Okamoto T, Sakai K, Suehiro Y, et al. Epstein-barr virus in gastric carcinoma. Cancers (Basel) 2014;6:2259–2274. doi: 10.3390/cancers6042259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cárdenas-Mondragón MG, Carreón-Talavera R, Camorlinga-Ponce M, Gomez-Delgado A, Torres J, Fuentes-Pananá EM. Epstein Barr virus and Helicobacter pylori co-infection are positively associated with severe gastritis in pediatric patients. PLoS One. 2013;8:e62850. doi: 10.1371/journal.pone.0062850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shukla SK, Prasad KN, Tripathi A, Ghoshal UC, Krishnani N, Husain N. Expression profile of latent and lytic transcripts of epstein-barr virus in patients with gastroduodenal diseases: a study from northern India. J Med Virol. 2012;84:1289–1297. doi: 10.1002/jmv.23322. [DOI] [PubMed] [Google Scholar]
- 38.Minoura-Etoh J, Gotoh K, Sato R, Ogata M, Kaku N, Fujioka T, Nishizono A. Helicobacter pylori-associated oxidant monochloramine induces reactivation of Epstein-Barr virus (EBV) in gastric epithelial cells latently infected with EBV. J Med Microbiol. 2006;55:905–911. doi: 10.1099/jmm.0.46580-0. [DOI] [PubMed] [Google Scholar]
- 39.Hirano A, Yanai H, Shimizu N, Okamoto T, Matsubara Y, Yamamoto K, Okita K. Evaluation of epstein-barr virus DNA load in gastric mucosa with chronic atrophic gastritis using a real-time quantitative PCR assay. Int J Gastrointest Cancer. 2003;34:87–94. doi: 10.1385/IJGC:34:2-3:087. [DOI] [PubMed] [Google Scholar]
- 40.Matsusaka K, Funata S, Fukayama M, Kaneda A. DNA methylation in gastric cancer, related to Helicobacter pylori and Epstein-Barr virus. World J Gastroenterol. 2014;20:3916–3926. doi: 10.3748/wjg.v20.i14.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yanai H, Murakami T, Yoshiyama H, Takeuchi H, Nishikawa J, Nakamura H, Okita K, Miura O, Shimizu N, Takada K. Epstein-Barr virus-associated gastric carcinoma and atrophic gastritis. J Clin Gastroenterol. 1999;29:39–43. doi: 10.1097/00004836-199907000-00010. [DOI] [PubMed] [Google Scholar]
- 42.Lagergren J, Lindam A, Mason RM. Gastric stump cancer after distal gastrectomy for benign gastric ulcer in a population-based study. Int J Cancer. 2012;131:E1048–E1052. doi: 10.1002/ijc.27614. [DOI] [PubMed] [Google Scholar]
- 43.Yamamoto N, Tokunaga M, Uemura Y, Tanaka S, Shirahama H, Nakamura T, Land CE, Sato E. Epstein-Barr virus and gastric remnant cancer. Cancer. 1994;74:805–809. doi: 10.1002/1097-0142(19940801)74:3<805::aid-cncr2820740304>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 44.Nishikawa J, Yanai H, Hirano A, Okamoto T, Nakamura H, Matsusaki K, Kawano T, Miura O, Okita K. High prevalence of Epstein-Barr virus in gastric remnant carcinoma after Billroth-II reconstruction. Scand J Gastroenterol. 2002;37:825–829. [PubMed] [Google Scholar]
- 45.Iskra S, Kalla M, Delecluse HJ, Hammerschmidt W, Moosmann A. Toll-like receptor agonists synergistically increase proliferation and activation of B cells by Epstein-Barr virus. J Virol. 2010;84:3612–3623. doi: 10.1128/JVI.01400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ueda S, Uchiyama S, Azzi T, Gysin C, Berger C, Bernasconi M, Harabuchi Y, Zinkernagel AS, Nadal D. Oropharyngeal group A streptococcal colonization disrupts latent Epstein-Barr virus infection. J Infect Dis. 2014;209:255–264. doi: 10.1093/infdis/jit428. [DOI] [PubMed] [Google Scholar]
- 47.Imai K, Inoue H, Tamura M, Cueno ME, Inoue H, Takeichi O, Kusama K, Saito I, Ochiai K. The periodontal pathogen Porphyromonas gingivalis induces the Epstein-Barr virus lytic switch transactivator ZEBRA by histone modification. Biochimie. 2012;94:839–846. doi: 10.1016/j.biochi.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 48.Takada K, Shimizu N, Sakuma S, Ono Y. trans activation of the latent Epstein-Barr virus (EBV) genome after transfection of the EBV DNA fragment. J Virol. 1986;57:1016–1022. doi: 10.1128/jvi.57.3.1016-1022.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ishihara S, Tada Y, Fukuba N, Oka A, Kusunoki R, Mishima Y, Oshima N, Moriyama I, Yuki T, Kawashima K, et al. Pathogenesis of irritable bowel syndrome--review regarding associated infection and immune activation. Digestion. 2013;87:204–211. doi: 10.1159/000350054. [DOI] [PubMed] [Google Scholar]
- 50.Ishihara S, Aziz M, Oshima N, Mishima Y, Imaoka H, Moriyama I, Kinoshita Y. Irritable bowel syndrome and inflammatory bowel disease: infectious gastroenteritis-related disorders? Clin J Gastroenterol. 2009;2:9–16. doi: 10.1007/s12328-008-0051-y. [DOI] [PubMed] [Google Scholar]
- 51.Spiller R, Aziz Q, Creed F, Emmanuel A, Houghton L, Hungin P, Jones R, Kumar D, Rubin G, Trudgill N, et al. Guidelines on the irritable bowel syndrome: mechanisms and practical management. Gut. 2007;56:1770–1798. doi: 10.1136/gut.2007.119446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ohman L, Simrén M. Pathogenesis of IBS: role of inflammation, immunity and neuroimmune interactions. Nat Rev Gastroenterol Hepatol. 2010;7:163–173. doi: 10.1038/nrgastro.2010.4. [DOI] [PubMed] [Google Scholar]
- 53.El-Salhy M. Irritable bowel syndrome: diagnosis and pathogenesis. World J Gastroenterol. 2012;18:5151–5163. doi: 10.3748/wjg.v18.i37.5151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gwee KA, Collins SM, Read NW, Rajnakova A, Deng Y, Graham JC, McKendrick MW, Moochhala SM. Increased rectal mucosal expression of interleukin 1beta in recently acquired post-infectious irritable bowel syndrome. Gut. 2003;52:523–526. doi: 10.1136/gut.52.4.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barbara G, Stanghellini V, De Giorgio R, Cremon C, Cottrell GS, Santini D, Pasquinelli G, Morselli-Labate AM, Grady EF, Bunnett NW, et al. Activated mast cells in proximity to colonic nerves correlate with abdominal pain in irritable bowel syndrome. Gastroenterology. 2004;126:693–702. doi: 10.1053/j.gastro.2003.11.055. [DOI] [PubMed] [Google Scholar]
- 56.Lee KJ, Kim YB, Kim JH, Kwon HC, Kim DK, Cho SW. The alteration of enterochromaffin cell, mast cell, and lamina propria T lymphocyte numbers in irritable bowel syndrome and its relationship with psychological factors. J Gastroenterol Hepatol. 2008;23:1689–1694. doi: 10.1111/j.1440-1746.2008.05574.x. [DOI] [PubMed] [Google Scholar]
- 57.Cremon C, Gargano L, Morselli-Labate AM, Santini D, Cogliandro RF, De Giorgio R, Stanghellini V, Corinaldesi R, Barbara G. Mucosal immune activation in irritable bowel syndrome: gender-dependence and association with digestive symptoms. Am J Gastroenterol. 2009;104:392–400. doi: 10.1038/ajg.2008.94. [DOI] [PubMed] [Google Scholar]
- 58.Chen J, Zhang Y, Deng Z. Imbalanced shift of cytokine expression between T helper 1 and T helper 2 (Th1/Th2) in intestinal mucosa of patients with post-infectious irritable bowel syndrome. BMC Gastroenterol. 2012;12:91. doi: 10.1186/1471-230X-12-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brint EK, MacSharry J, Fanning A, Shanahan F, Quigley EM. Differential expression of toll-like receptors in patients with irritable bowel syndrome. Am J Gastroenterol. 2011;106:329–336. doi: 10.1038/ajg.2010.438. [DOI] [PubMed] [Google Scholar]
- 60.Belmonte L, Beutheu Youmba S, Bertiaux-Vandaële N, Antonietti M, Lecleire S, Zalar A, Gourcerol G, Leroi AM, Déchelotte P, Coëffier M, et al. Role of toll like receptors in irritable bowel syndrome: differential mucosal immune activation according to the disease subtype. PLoS One. 2012;7:e42777. doi: 10.1371/journal.pone.0042777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schoepfer AM, Schaffer T, Seibold-Schmid B, Müller S, Seibold F. Antibodies to flagellin indicate reactivity to bacterial antigens in IBS patients. Neurogastroenterol Motil. 2008;20:1110–1118. doi: 10.1111/j.1365-2982.2008.01166.x. [DOI] [PubMed] [Google Scholar]
- 62.Alonso C, Guilarte M, Vicario M, Ramos L, Ramadan Z, Antolín M, Martínez C, Rezzi S, Saperas E, Kochhar S, et al. Maladaptive intestinal epithelial responses to life stress may predispose healthy women to gut mucosal inflammation. Gastroenterology. 2008;135:163–172.e1. doi: 10.1053/j.gastro.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 63.Miwa H, Watari J, Fukui H, Oshima T, Tomita T, Sakurai J, Kondo T, Matsumoto T. Current understanding of pathogenesis of functional dyspepsia. J Gastroenterol Hepatol. 2011;26 Suppl 3:53–60. doi: 10.1111/j.1440-1746.2011.06633.x. [DOI] [PubMed] [Google Scholar]
- 64.Lee KJ, Tack J. Duodenal implications in the pathophysiology of functional dyspepsia. J Neurogastroenterol Motil. 2010;16:251–257. doi: 10.5056/jnm.2010.16.3.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tack J, Demedts I, Dehondt G, Caenepeel P, Fischler B, Zandecki M, Janssens J. Clinical and pathophysiological characteristics of acute-onset functional dyspepsia. Gastroenterology. 2002;122:1738–1747. doi: 10.1053/gast.2002.33663. [DOI] [PubMed] [Google Scholar]
- 66.Mearin F, Pérez-Oliveras M, Perelló A, Vinyet J, Ibañez A, Coderch J, Perona M. Dyspepsia and irritable bowel syndrome after a Salmonella gastroenteritis outbreak: one-year follow-up cohort study. Gastroenterology. 2005;129:98–104. doi: 10.1053/j.gastro.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 67.Futagami S, Itoh T, Sakamoto C. Systematic review with meta-analysis: post-infectious functional dyspepsia. Aliment Pharmacol Ther. 2015;41:177–188. doi: 10.1111/apt.13006. [DOI] [PubMed] [Google Scholar]
- 68.Kindt S, Van Oudenhove L, Mispelon L, Caenepeel P, Arts J, Tack J. Longitudinal and cross-sectional factors associated with long-term clinical course in functional dyspepsia: a 5-year follow-up study. Am J Gastroenterol. 2011;106:340–348. doi: 10.1038/ajg.2010.406. [DOI] [PubMed] [Google Scholar]
- 69.Futagami S, Shindo T, Kawagoe T, Horie A, Shimpuku M, Gudis K, Iwakiri K, Itoh T, Sakamoto C. Migration of eosinophils and CCR2-/CD68-double positive cells into the duodenal mucosa of patients with postinfectious functional dyspepsia. Am J Gastroenterol. 2010;105:1835–1842. doi: 10.1038/ajg.2010.151. [DOI] [PubMed] [Google Scholar]
- 70.Li X, Chen H, Lu H, Li W, Chen X, Peng Y, Ge Z. The study on the role of inflammatory cells and mediators in post-infectious functional dyspepsia. Scand J Gastroenterol. 2010;45:573–581. doi: 10.3109/00365521003632576. [DOI] [PubMed] [Google Scholar]
- 71.Dizdar V, Spiller R, Singh G, Hanevik K, Gilja OH, El-Salhy M, Hausken T. Relative importance of abnormalities of CCK and 5-HT (serotonin) in Giardia-induced post-infectious irritable bowel syndrome and functional dyspepsia. Aliment Pharmacol Ther. 2010;31:883–891. doi: 10.1111/j.1365-2036.2010.04251.x. [DOI] [PubMed] [Google Scholar]
- 72.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- 75.Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature. 2012;487:104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Peleg AY, Hogan DA, Mylonakis E. Medically important bacterial-fungal interactions. Nat Rev Microbiol. 2010;8:340–349. doi: 10.1038/nrmicro2313. [DOI] [PubMed] [Google Scholar]
- 77.Carlson E, Johnson G. Protection by Candida albicans of Staphylococcus aureus in the establishment of dual infection in mice. Infect Immun. 1985;50:655–659. doi: 10.1128/iai.50.3.655-659.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]