

Abstract
We conducted a retrospective observational study to describe a cohort and identify the prognostic factors in adult-onset Still disease (AOSD). Patients enrolled in this retrospective chart review fulfilled either Yamaguchi or Fautrel criteria. Candidate variables were analyzed with logistic unadjusted and adjusted regression models.
Fifty-seven patients were seen in the internal medicine (75%) and rheumatology (25%) departments over a mean period of 8.4 years. The median time to diagnosis was 4 months. The course of AOSD was monocyclic in 17 patients, polycyclic in 25, and chronic in 15. The assessment of glycosylated ferritin (GF) in 37 patients was correlated with early diagnosis. Nine 18F-fluorodeoxyglucose positron emission tomography (18FDG-PET) scans identified the lymph nodes and glands as the main sites of hypermetabolism. Complications were frequent (n = 19), including reactive hemophagocytic syndrome (n = 8). None of the 3 deaths could be attributed to AOSD. Corticosteroid dependence, as predicted by a low GF level, occurred in 23 patients (45%). A quarter of the patients received tumor necrosis factor-α blockers or anakinra with good tolerance. Fever >39.5°C was predictive of monocyclic AOSD, while arthritis and thrombocytopenia were associated with chronic and complicated AOSD, respectively. The youngest patients had the highest risks of resistance to first-line treatments.
AOSD remains difficult to diagnose. Mortality is low despite frequent complications. GF and 18FDG-PET scans were of value in the diagnostic approach. The condition in highly symptomatic patients evolved to systemic AOSD, whereas more progressive patterns with arthritis predicted chronic AOSD.
Abbreviations: AE = adverse event, AOSD = adult-onset Still disease, BM = bone marrow, CI = confidence interval, CRP = C-reactive protein, CSs = corticosteroids, CT = computed tomography, DMARDs = disease-modifying antirheumatic drugs, ESR = erythrocyte sedimentation rate, 18FDG-PET = 18F-fluorodeoxyglucose positron emission tomography, GF = glycosylated ferritin, IL = interleukin, IVIg = polyvalent intravenous immunoglobulins, MTX = methotrexate, NSAIDs = nonsteroidal antiinflammatory drugs, OR = odds ratio, PMN = polymorphonuclear neutrophils, RA = receptor antagonist, RHS = reactive hemophagocytic syndrome, SD = standard deviation, SF = serum ferritin, TNF-α = tumor necrosis factor α
INTRODUCTION
First described in 1971 by Bywaters,2 adult-onset Still disease (AOSD) is an uncommon systemic inflammatory disorder of unknown etiology. Its prevalence is estimated to be less than 1 case per 100,000 people, and it affects predominantly young adults.30 The precise pathogenesis of this disease remains unknown, but it seems that genetically predisposed hosts develop autoinflammatory disorders triggered by macrophage cell activation and TH1 cytokines such as interleukin (IL)-1, IL-2, IL-6, IL-18, tumor necrosis factor (TNF)-α, and interferon γ.31
The main features of AOSD are a high spiking fever, evanescent rash, sore throat, polyarthralgia, lymphadenopathy, hepatosplenomegaly, serositis, and leukocytosis, as well as elevated liver enzymes, polymorphonuclear neutrophils (PMN), erythrocyte sedimentation rate (ESR), and serum ferritin (SF). Despite the diagnostic value attributed to high SF associated with low glycosylated fraction of ferritin (<20%), the diagnosis of AOSD remains one of exclusion.16 The clinical course of the disease may have 1 of 3 patterns: a self-limiting or monocyclic systemic course, an intermittent or polycyclic systemic course, and a chronic articular course.41 The treatment of AOSD remains empirical. Nonsteroidal antiinflammatory drugs (NSAIDs); corticosteroids (CSs); disease-modifying antirheumatic drugs (DMARDs), such as methotrexate (MTX); and polyvalent intravenous immunoglobulins (IVIg) are usually used.11
The recent use of biologic agents in AOSD has been shown capable of improving considerably the condition of different subgroups of patients.10,36 Data have shown a most impressive response with anakinra in patients with systemic disease, whereas TNF-α blockers and tocilizumab had better results in chronic AOSD.35 Within this context, determining whether the clinical and laboratory features found at diagnosis can predict the outcome of AOSD would be of great value in patient management. Thus, we conducted the present study to describe a cohort of AOSD patients, identify the baseline prognostic factors that influence the clinical course of the disease, and monitor the response to treatment and the appearance of complications.
PATIENTS AND METHODS
Patients
This retrospective study received institutional review board approval. All patients registered as having AOSD from 1998 to 2010 were identified through a review of the database of the Medical Information Department of Hospices Civils de Lyon. Patients were included if they fulfilled either Yamaguchi42 or Fautrel16 criteria. The exclusion criteria were an onset of the disease before 16 years old and insufficient medical record data.
Data
Clinical and laboratory data were collected and analyzed by the same investigator (MGV) using a standardized form. The collected data included the following: 1) clinical features; 2) laboratory features including blood cell count, serum electrolytes, coagulation parameters, SF and glycosylated ferritin (GF), ESR, C-reactive protein (CRP), liver enzymes, triglycerides, rheumatoid factor, antinuclear autoantibody, serum protein electrophoresis; 3) pathology laboratory findings; and, 4) imaging features: computed tomography (CT) and 18F-fluorodeoxyglucose positron emission tomography (18FDG-PET).
All prescribed treatments were chronologically listed with their doses, durations, results, adverse events (AEs), and, when available, the reasons for treatment failure. The doses of CSs were expressed in milligrams of equivalent prednisone/day. The disease was considered CS dependent when a patient suffered from AOSD recurrence despite a maintenance dose >15 mg/d without tapering.41
The study considered 3 clinical AOSD courses: 1) monocyclic, defined as a single episode that faded subsequently and was followed by persistent good health after 1 year or more of follow-up;41 2) polycyclic, defined as a complete remission followed by 1 or more exacerbations; and 3) chronic, defined as persistently active disease, usually associated with polyarthritis.8,37
Controlled disease was defined as clinically asymptomatic AOSD with no biologic inflammatory syndrome at 1-year follow-up.36 Complicated AOSD included 1 or more of the following conditions: acute fulminant hepatitis, disseminated intravascular coagulation, thrombotic microangiopathy, reactive hemophagocytic syndrome (RHS) as defined by HLH-2004 criteria,19 shock, multiple organ failure, myocarditis, complicated pericarditis (tamponade, followed by restrictive pericarditis), severe sepsis, acute respiratory distress syndrome in adults, or AA amyloidosis.36
Statistical Analysis
The sociodemographic, clinical, laboratory, and other variables were expressed as numbers and percentages when considered as categorical data and as means and standard deviations when considered as continuous data. The variables were first displayed per type of clinical course (Table 1). The differences between the 3 subgroups were tested overall. In order to limit the inflation of alpha level with such a sample size, a few relevant variables were tested using the Fisher exact test for categorical variables and the Kruskal-Wallis test for continuous variables. Then, continuous variables were standardized before regression analyses.
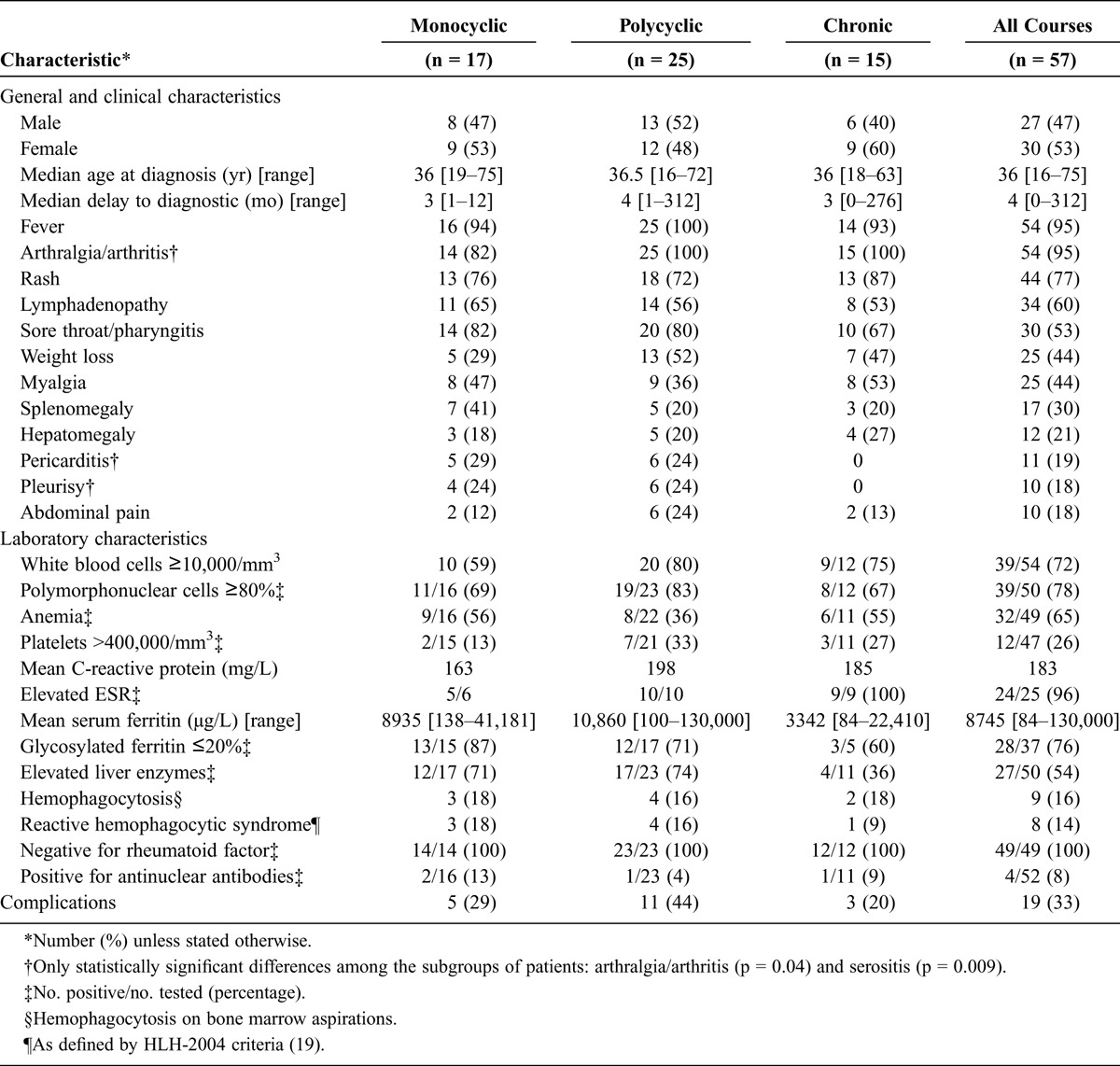
TABLE 1.
Patient Characteristics at Time of Diagnosis According to the Clinical Course of the Disease
To identify the variables that predict the progression from 1 clinical course to another, we selected a set of candidate variables stemming from the initial visit: arthritis; fever >39.5°C; 1 of the following, lymphadenopathy, splenomegaly, or hepatomegaly; serositis; elevated liver enzymes; PMN; and GF. The risk of having a monocyclic, polycyclic, or chronic form at the end of follow-up was quantified, and the odds ratios (ORs) of each compared to the chronic form for monocyclic and polycyclic forms and compared to the monocyclic form for chronic forms were estimated by logistic regression. Unadjusted and adjusted models first were fitted. Then, a final adjusted regression model was built with the variables found significant in the unadjusted and adjusted analyses together with a few relevant confounding variables. The results were expressed in ORs and 95% confidence intervals (95% CIs).
The analyses were performed with R software (R Foundation for Statistical Computing, Vienna, Austria; http://cran.r-project.org/).
RESULTS
Clinical Features
Fifty-seven patients (27 men and 30 women) with AOSD were identified: 53 fulfilled the Fautrel16 classification criteria and 55 fulfilled the Yamaguchi42 criteria. These patients were followed in an internal medicine department (43/57, 75%) or in a rheumatology department (14/57, 25%) over a mean period of 8.4 years (range, 1.2–48.6 yr). Most patients were of European (44/57) or North African (7/57) origin. The median age at AOSD diagnosis was 36 years (range, 16–75 yr) (see Table 1). The median delay to diagnosis was 4 months (range, 1–312 mo). At AOSD diagnosis, 10 patients (18%) were aged over 55 years. The disease-onset manifestations were fever (95%), weight loss (44%), articular symptoms (arthralgia and/or arthritis; 95%), radiologic joint erosions (7%), rash (77%), sore throat or pharyngitis (53%), lymphadenopathy (60%), splenomegaly (30%), hepatomegaly (21%), pleurisy (18%), pericarditis (19%), or myalgia (44%). Two patients developed symptoms consistent with Still disease when aged between 16 and 18 years old.
The clinical courses in the 57 AOSD patients were monocyclic in 17 patients (30%), polycyclic in 25 (44%), and chronic in the remaining 15 (26%). Among the cases of chronic AOSD, 12 were chronic arthritis, 2 were isolated inflammatory syndromes, and 1 was chronic hepatitis not induced by the treatment (see Table 1).
Laboratory Features
Leukocytosis (white blood cell count ≥10,000/mm3) was present at diagnosis in 41 (72%) patients (mean ± SD, 13.93 ± 7.05/mm3) and was composed of ≥80% PMN in 44 (77%) (11.98 ± 6.14/mm3). Anemia occurred in 65% of patients, and thrombocytosis (>400,000/mm3) in 26%. Fibrinogen was increased (>4 g/L) in 79%, and CRP in 98% (mean ± SD, 183.4 ± 108.5 mg/L). SF was increased in 82% of patients (mean ± SD, 8745 ± 19,867 μg/L; range, 80–130,000). GF was measured at diagnosis in 37 patients (65%) and found ≤20% of SF in 28 of them (76%). During the remission of 5 patients, the GF normalized in 4 (up to 31%; 40%; 45%, and 50%) but remained low at 19% in the fifth. Thirty-three patients among the 50 (66%) who had liver function tests assessed at diagnosis exhibited abnormalities. Twenty-seven had cytolysis with or without cholestasis (mean aspartate aminotransferase, 362 IU/L; and mean alanine aminotransferase, 528 IU/L). Rheumatoid factor was absent in all patients. Fifty-two patients were tested for antinuclear autoantibodies: 4 patients were positive but none for soluble nuclear antigen and anti-double-stranded DNA antibodies. Triglycerides were high in 14 of the 22 patients in whom they were assayed. Bone marrow aspiration was carried out in 33 patients, including 9 with hemophagocytosis; in 8 of the latter, the diagnostic criteria for RHS were met (see Table 1).
Forty-five pathology examinations were performed: 19 on bone marrow, 9 on liver biopsies; 7 on lymph node biopsies; 5 on skin biopsies; and 1 each on pericardial, pleural, lung, synovial, and salivary gland biopsies. The repeatedly found results were nonspecific inflammatory reactions on 9 bone marrow biopsies, hemophagocytosis on 6 bone marrow biopsies, and 5 normal bone marrow biopsies.
Of the 22 thoraco-abdomino-pelvic CT scans available, 7 were normal; 5 showed pulmonary involvement (pneumonia, ventilatory disorders, micronodules); 7 deep lymph node enlargement (subdiaphragmatic in 4 cases, mediastinal in 5 cases); and 5 showed splenomegaly and/or hepatomegaly. Six serous effusions were identified on CT.
Among the 9 18FDG-PET scans performed, 2 were normal; 4 showed hypermetabolic lymph nodes; 3 showed hypermetabolism of the salivary glands, 1 of the testis, 1 of the pericardium, 1 of the gastric fundus, and 1 of the liver. The median standardized uptake value was 4 (range, 1.8–8.0).
Complications
Nineteen patients had complications: 8 patients had RHS (of whom 6 had RHS as presenting syndrome at AOSD diagnosis), 4 had myocarditis, 4 had acute respiratory distress syndrome, 3 had cardiac tamponade, 2 had cardiopulmonary shock, 2 had multiple organ failure, 2 had fulminant hepatitis, and 1 patient had disseminated intravascular coagulation. Although complications seemed to be more prevalent in polycyclic AOSD, the difference between groups was not statistically significant (see Table 1). Three deaths occurred, but none could be directly attributed to AOSD or its complications: 1 myocardial infarction, 1 accidental death, and 1 death due to a non-Hodgkin lymphocytic lymphoma that occurred 20 years after AOSD onset during disease remission.
Treatment
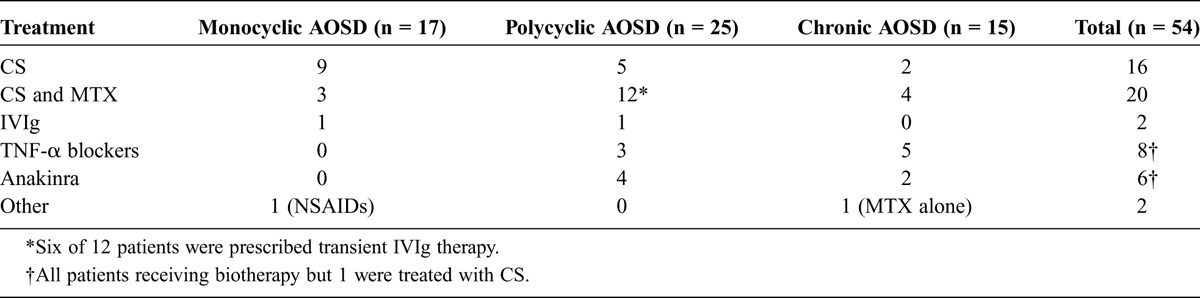
Three patients had monocyclic AOSD that regressed spontaneously without treatment, and 54 patients received a first-line treatment. The different lines of treatment are detailed in Table 2, and treatments that led to remission of the disease are described in Table 3.
TABLE 2.
Distribution and Adverse Events of Treatment Lines in 57 Patients With AOSD
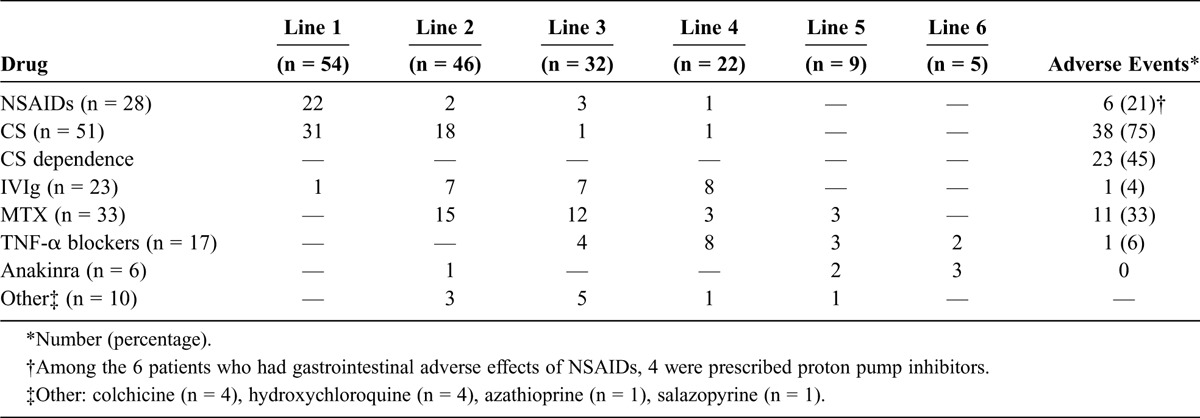
TABLE 3.
Treatments in Use at AOSD Remission
Twenty-eight patients received NSAIDs: indomethacin in 6 patients, ketoprofen in 9, aspirin in 7, diclofenac in 3, and other drugs in 3. NSAIDs were used as first-line treatment in 22 patients and controlled the disease in only 5 (18%). Among the 6 patients who experienced gastrointestinal AEs, 4 were prescribed proton pump inhibitors.
Of 51 patients treated with CSs, 49 received them as a first- or second-line treatment. CSs were first intravenous in 11 patients. The initial dose of oral CSs was 1 mg/kg per day in 36 patients (70%), <1 mg/kg per day in 12 patients (24%), and >1 mg/kg per day in 3 patients (6%). Thirty-eight of 51 patients (75%) developed various AEs such as Cushing syndrome (n = 19), osteoporosis (n = 8), aseptic osteonecrosis (n = 5), CS-induced diabetes (n = 4), high blood pressure (n = 4), cataract (n = 3), psychiatric disorders (n = 3), and infectious diseases (n = 2). Twenty-three of 51 patients (45%) developed a CS-dependent disease. In the present study, the use of <1 mg/kg per day initial dose was not found to be associated with dependence on high-dose CSs, complicated AOSD, resistance to treatment, or progression to a more severe disease course (data not shown).
Thirty-three patients (58%) required MTX as DMARD. Eleven developed AEs (33%), mainly elevated liver enzymes (n = 5), low blood cell counts (n = 3), or cough (n = 2). MTX was the last-line treatment in 14 of 33 patients (42%).
IVIg was sufficient to control the disease in 4 of 23 cases. It was more frequently prescribed in polycyclic or chronic AOSD (n = 20) and in complicated AOSD (n = 12). One patient developed an acute renal failure after IVIg infusion. One woman received IVIg as first-line treatment during pregnancy without any complication.
Treatment with biologic agents was prescribed 23 times in 15 of 57 patients (26%). These 15 patients had polycyclic or chronic AOSD, complicated in 7 of 15 patients and/or CS-dependent in 7 of 15 patients. The characteristics of these patients were not significantly different from those of patients who did not receive these targeted therapies (data not shown).
Of 17 prescriptions for TNF-α blockers, 8 (47%) succeeded in controlling the disease: infliximab (n = 4/8), etanercept (n = 3/8), and adalimumab (n = 1/1). TNF-α blockers were effective in 5 of 9 (56%) chronic AOSD cases and 2 of 6 (33%) cases of polycyclic AOSD, but this difference in effectiveness was not significant (p = 0.6). Among 3 switches between TNF-α blockers, only 1 was efficient. Anakinra, an IL-1-receptor antagonist (IL-1-RA), was prescribed 6 times and was always the last line of treatment; it led to remission in 5 of 6 patients after a mean follow-up of 27.8 months (range, 14–36 mo). Among the 4 patients with polycyclic AOSD who received anakinra, 2 had experienced RHS. The treatment was effective in all but 1 who suffered from recurrence of the disease after 11 months on anakinra and required drug discontinuation. Two patients with chronic AOSD were effectively treated with anakinra. In all 6 patients, there was no AE other than a local inflammatory reaction. In this small sample, there was no significant difference in the effectiveness of anakinra between the polycyclic and the chronic course of AOSD.
Prognostic Factors
Patients with GF levels assessed during the diagnostic process had significantly shorter times to diagnosis compared to those without (OR, 3.33; p = 0.039). Furthermore, univariate analysis showed that GF testing was correlated only with the monocyclic course of AOSD (OR, 5.97; 95% CI, 1.14–60.65; p = 0.018). In the unadjusted analysis, a short delay to diagnosis (≤2 mo) was rather predictive of a monocyclic course of AOSD compared with other severe courses (OR, 5.31; 95% CI, 1.386–22.676; p = 0.007). However, this was not confirmed by multivariate analysis.
The unadjusted analysis of the variables collected at diagnosis revealed 2 determinants of a monocyclic course of AOSD: fever >39.5°C and abnormal liver function tests, whereas the presence of arthritis was negatively correlated with a monocyclic course. The adjusted multivariate analysis confirmed the predictive role of high fever in the occurrence of monocyclic AOSD (Table 4).
TABLE 4.
Prognostic Factors of Outcome in 57 Patients With AOSD
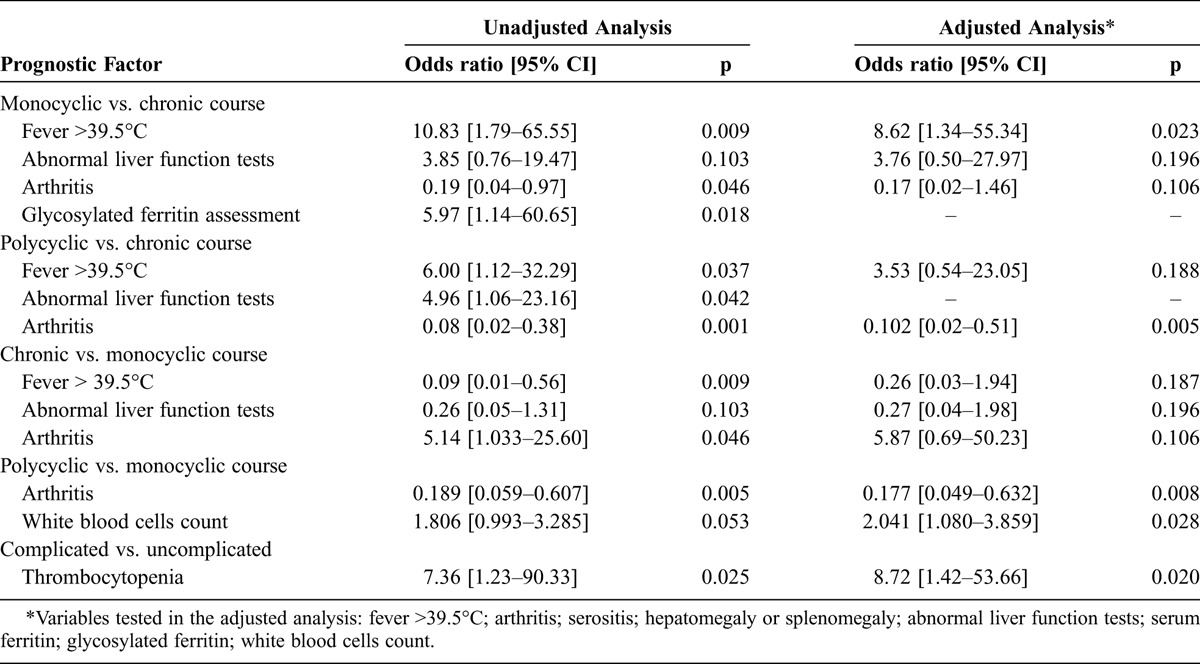
In the unadjusted analysis, the prognostic determinants of a polycyclic course were also fever >39.5°C and abnormal liver function tests, whereas arthritis was a protective factor. In the adjusted multivariate analysis, only arthritis as a protective factor remained (see Table 4).
When these 2 systemic courses of the disease were compared, arthritis was rather negatively correlated with a polycyclic course, whereas white blood cell count appeared higher in patients with progression to a polycyclic course than in patients with progression to a monocyclic course (see Table 4).
In the unadjusted analysis, the absence of serositis and normal liver function tests seemed to predict a chronic course of AOSD. The mean initial SF was 3342 μg/L in patients who experienced chronic AOSD and 9893 μg/L in those who experienced a systemic—monocyclic or polycyclic—course; the difference was hardly significant (p = 0.053). However, these differences were not significant in the adjusted analysis. Unadjusted and adjusted analyses indicated that the presence of arthritis at diagnosis was a strong predictor of a chronic disease course (see Table 4). The same was true for radiologic joint erosion (OR, 14.21; 95% CI, 1.236–763.542; p = 0.004 in the unadjusted analysis only).
Only thrombocytopenia remained a significant predictor of occurrence of complications in the adjusted regression (OR, 8.72; 95% CI, 1.42–53.66; p = 0.020).
Response to Treatment
Not surprisingly, steroid dependence was higher in polycyclic and chronic AOSD (OR, 8.01; 95% CI, 0.79–137.84; p = 0.007). In the multivariate adjusted analysis, low GF seemed to be predictive of high-dose CS dependence (OR, 0.3, 95% CI, 0.07–1.26; p = 0.10 per each SD increment in GF level, SD = 14.16; mean GF, 16.7%). In the unadjusted analysis only, splenomegaly seemed to protect from steroid dependence (OR, 0.28; 95% CI, 0.79–8.87; p = 0.074), and an age at diagnosis more than the average age (38 yr) appeared predictive of resistance to the first 2 lines of treatment (OR, 0.6; 95% CI, 0.34–1.06; p = 0.078).
DISCUSSION
In the present retrospective study, we analyzed the empirical treatment of AOSD, the prognostic factors of the disease, and new data such as 18FDG-PET scan characteristics in 57 patients over an important mean follow-up of 8.4 years. Compared to previous studies, the present study is original because it included mostly white patients, was carried out recently, and used biologic agents.
The current findings from white AOSD patients are consistent with those previously reported in the main literature.3,4,6,16,27,33,37,44 The delay to final diagnosis was highly variable, which indicates possible diagnostic difficulties. Weight loss was more common in the current study (44%) than in previous studies (18% for Pay et al;33 19% for Cagatay et al3). This may be the result of underreporting, but also of higher proportions of systemic AOSD in the current study compared to others (74% here vs. 60% on average in the literature). This also stands for polycyclic AOSD (44% here vs. 23% on average). These differences may also be due to a referral bias and longer follow-up times in the present cohort. In fact, most patients in the current study were seen in the internal medicine department (not in rheumatology as usual) for fever of unknown origin. This might have increased the number of systemic polycyclic patterns. In addition, the long follow-up times enabled us to see more relapses; thus, more polycyclic courses than in other studies.
The interest in SF and GF is mainly supported by a retrospective study by Fautrel et al.14 However, in a series of 14 patients, Vignes et al39 assumed that high SF could be a marker of activity in the acute phase of AOSD (because it rapidly normalized with treatment and remission), whereas GF remained low at 3 years (at 16% on average), which could be used to diagnose AOSD under treatment or during remission. This could not be confirmed in the current study because only 1 of the 5 GF assayed during remission within the first 3 years of follow-up remained <20% (precisely, 19%).
We describe here the largest series, to our knowledge, of 18FDG-PET scans on 9 AOSD patients and report that the lymph nodes and glands were the main sites of hypermetabolism. This involvement of lymphoid tissues may force to rule out lymphoma. In case of fever of unknown origin, 18FDG-PET may support the diagnosis of AOSD by allowing the rejection of hypotheses of localized infection, solid tumor, or other noninfectious inflammatory diseases.7,25 Moreover, 18FDG-PET may be useful in monitoring the disease progression and responses to treatment,5 although this has to be confirmed by prospective cohorts.
The treatment of AOSD remains largely empirical, relying so far on a few prospective or retrospective studies and not on double-blinded randomized trials with suitable sample sizes. Some considerations about treatment are listed below.
First, although NSAIDs are traditionally considered as the first-line therapy, they were effective in controlling the disease in 18% of the present cohort patients. Indomethacin was deemed more effective than salicylates but was prescribed in less than a quarter (21%) of patients. Moreover, AEs, mainly gastrointestinal, were prevalent (21%). Thus, as suggested by Franchini et al,18 NSAIDs constitute a supportive treatment that could be administered during the diagnostic process before confirmation of AOSD and then reserved for use if CSs are insufficient.
Second, in the current study neither the dose nor the route of administration had an impact on the therapeutic response to CS and the outcome of AOSD. This contrasts with the report of Kong et al,27 where patients treated with prednisone (or its equivalent) ≥40 mg (0.8 mg/kg) achieved quicker remission and had fewer relapses compared with those who received a lower dosage. As in the literature, the tolerance in the current study was poor, with 75% of AEs during treatment. In addition, we confirm the high prevalence of steroid dependence in AOSD (45% here and 42% reported by Kim et al24). As one would expect and as already described,17,24,43 CS dependence was significantly higher in the polycyclic and chronic forms than in monocyclic AOSD, and is an additional factor of poor prognosis in the former patterns. However, in contrast to the results reported by Kim et al,24 in the current study splenomegaly appeared to be a protective factor against steroid dependence. We have shown that young patients were at risk of resistance to the first 2 lines of treatment, which often include CS, and that a low GF level would predict steroid dependence. Accordingly, it makes sense to pay particular attention to age, splenomegaly, elevated ESR, and low GF before an early introduction of sparing treatments such as MTX.
Third, regarding MTX, in 1999 Fautrel et al13 published a retrospective study on 26 AOSD patients highlighting the beneficial role of MTX as a second-line treatment in case of CS dependence and as a sparing treatment. As in most of studies on AOSD, MTX was the most prescribed DMARD and had good tolerance.
Fourth, 23 of 57 patients (40%) received transient IVIg therapy. Tolerance was excellent but remission of the disease was obtained in only 4 cases (17%). Data on IVIg in AOSD are scarce. In 2 open-label studies, IVIg was shown to be effective in 8 of 14 patients early in the disease course.34,40 For Kim et al,24 IVIg treatment had no consequence on the course or the prognosis of AOSD. As with patients with RHS,12 IVIg may be useful in life-threatening situations.
Fifth, a growing body of literature has documented the efficacy of several biologic agents in the treatment of CS- and DMARD-refractory AOSD. In the current series, 15 patients (26%) did not achieve satisfactory control of the disease with CSs and DMARDs and were thus given various biologic agents, which is comparable to the study of Franchini et al.18 TNF-α blockers were efficient in controlling the disease in only 47% of our prescriptions. TNF-α blockers seemed to be more effective in chronic than in polycyclic courses of the disease.35 Previous results on TNF-α blockers were retrospective and rather heterogeneous. In the largest previous study15 that we know of, 25 prescriptions of anti-TNF-α (infliximab and etanercept) were administered to 20 AOSD patients resistant to conventional DMARDs. After a mean follow-up of 15 months, only 5 prescriptions led to remission, whereas 4 were ineffective, and 4 led to AEs. Altogether, more than 120 patients were treated with a TNF-α blocker and, as in the present study, complete remissions were irregular and often transient.15,21,24,33 Switching from 1 TNF-α blocker to another was beneficial in only a few cases; in the current study, this was observed only once. Finally, these treatments may cause worsening of the disease because of RHS occurrence.23
In the present study, the use of anakinra resulted in a rapid and complete remission in 5 of 6 patients. Several case reports and 2 small case series have shown spectacular effects of anakinra in AOSD,22,28 which has been confirmed by 2 prospective clinical trials. In the first, Laskari et al28 reported on 25 AOSD patients, 9 of whom were given anakinra alone, and 16, anakinra and a DMARD. As in our patients, the clinical efficacy was rapid (0.2 mo) and frequent (84% at 3 mo; 80% at end of follow-up), with a favorable agent safety profile.36 In the second, more recent trial, Nordström et al32 compared the outcomes of 12 AOSD patients treated with anakinra and 10 patients treated with a DMARD (all taking prednisolone ≥10 mg/d) early in the course of AOSD to avoid the adverse effects of corticotherapy. With no statistically significant differences in characteristics between the 2 groups, greater improvements were seen in the anakinra group.
None of our patients received anti-IL-6 receptor, which has been considered for treating AOSD. A recent review of 35 patients reported that 86% of tocilizumab-treated patients experienced prompt articular improvement, and 96% experienced a disappearance of systemic symptoms.10 Regarding safety, tocilizumab was well tolerated, but severe effects such as macrophage activation syndrome39,40 are possible and require ongoing vigilance.
In the present series, one-third of the patients experienced life-threatening complications of AOSD, none of which led to death. The most common complication was RHS. We report 8 of 57 (14%) cases of AOSD-associated RHS, which is similar to the incidence reported in 2 other series;1,20 however, in those series, the time relationship between RHS and AOSD was not reported. A complicated onset of AOSD is overrepresented in the current study compared with previous data (33% vs. 5%–11%, respectively).3,6,38 This may be the consequence of 1) the high proportion of polycyclic AOSD; 2) the recruitment in internal medicine department compared with rheumatology departments in other series; 3) the recruitment in a university hospital, which often increases the proportion of severe cases; and 4) a longer follow-up period than in previous studies.
None of the 3 deaths during the study period could be attributed to Still disease. This is in agreement with the benign and nonfatal nature of the disease. Nevertheless, authors of 2 recent studies in Asian patients24,43 reported high mortality rates (9.26% and 10%, respectively) due to infection and disease progression, but only a small proportion of the patients had received biologic agents. Multicentric studies are needed to check whether the prognosis of AOSD is significantly influenced by patient ethnicity.
In the present study, we sought the clinical and laboratory features of good prognosis (monocyclic disease) and poor prognosis (polycyclic, chronic articular disease, treatment failure, and complications). Arthritis at diagnosis was a significant predictor of disease chronicity. This reinforces the few reports that confer a poor prognosis to the presence of polyarthritis.6,8 In addition, as already observed,6 joint erosion (as shown by standard radiography) seemed to predict chronicity. However, there could have been an overlap with seronegative rheumatoid arthritis. Moreover, we showed that high fever was a good predictor of a systemic—particularly monocyclic—disease course. To our knowledge, this has not been reported before.
According to our data, thrombocytopenia at diagnosis predicted the occurrence of complications, but in fact this is confounding, because the most prevalent complication in our patients was RHS, in which thrombocytopenia is almost constant.
It is noteworthy that our results suggest an early diagnosis improves the prognosis: indeed, in the unadjusted analyses, a ≤2-month delay was predictive of a monocyclic course. Moreover, GF level testing during the diagnostic process was significantly associated with earlier diagnoses and seemed to be predictive of a monocyclic course. However, that testing could be a confounding factor because GF may have been more frequently assessed in highly symptomatic patients who evolved spontaneously toward a monocyclic course. Thus, we may recommend early and frequent GF assays to help early diagnosis and early treatment.
Several studies have suggested the use of SF as prognostic factor in AOSD.6,27,29,43 In our unadjusted analysis results, a mild increase in SF was associated with a chronic course of AOSD. In 2009, in a retrospective study of 61 Chinese AOSD patients, Zeng et al43 found that an elevated SF level was an unfavorable prognostic factor. Two more recent studies showed that high SF levels were significantly associated with a high rate of relapse.6,27 In a study by Lee et al29 in 2009, only the middle range of the adjusted SF level (area under the curve divided by the number of hospitalization days; that is, 784.1–4120.0 ng/mL) had a significant predictive value for disease chronicity. The present study results are consistent with this, which seems close to the clinical reality. Indeed, higher SF levels (>8000 or 10,000 ng/mL) do not seem predictive of a chronic course, but were associated with the “highly symptomatic” type of AOSD we describe below, which is monocyclic or associated with RHS.20
Among other laboratory features, the white blood cell count seems to be an interesting predictor of patient outcome. Kong et al27 found that increased white blood cell counts (≥30,000/mm3) were associated with AOSD relapses. Other studies revealed that inflammation markers, such as elevated serum ESR or CRP, were significantly associated with poor prognoses24 or higher relapse rates.27
The present study provides important information on the clinical, laboratory, therapeutic, and prognostic features of AOSD, but has a number of limitations. First, the study was retrospective, noncomparative, and had to contend with some degree of missing clinical and laboratory investigation data. A prospective study would provide much clearer and exhaustive information. The second limitation is that the study was carried out in a single university hospital and might have included a higher percentage of severe cases than would be expected in primary settings. Thus, the results are applicable only to secondary or tertiary AOSD practices and to AOSD cases seen in internal medicine departments. Moreover, AOSD management differs widely between countries and even physicians. Therefore, we await additional studies from other institutions in different countries to enrich and confirm the present results.
Conclusion
The results of this second retrospective study on French AOSD patients are consistent with previous results. AOSD diagnosis remains difficult. GF level testing seems to be underused, whereas it would shorten the delay to diagnosis and help in predicting high-dose CS dependence (which is prevalent in AOSD). The hypermetabolism on 18FDG-PET scan focuses on the lymphoid tissues and glands. The usefulness of the treatment sequence CS, MTX, then IL-1-RA before TNF-α blockers should be confirmed by prospective studies. The long mean follow-up period (8.4 yr) allowed us to study various prognostic factors. On the basis of the initial clinical and laboratory data, we suggest distinguishing 2 phenotypes of the disease, which progress quite differently: 1) a highly symptomatic onset with high fever, serositis, elevated liver enzymes, and very high SF with arthralgia but less commonly arthritis, which would evolve to systemic—monocyclic or polycyclic—AOSD; and 2) a more progressive onset with arthritis and radiologic joint erosions and poor systemic symptoms, which would progress to a chronic course. Overall, our results indicate that AOSD is a relatively benign disease and that most deaths may be related to the AEs of long-term treatment. Prospective studies in independent cohorts are needed to confirm these results.
Acknowledgments
The authors thank Pr. R. Chapurlat, Dr. S. Dargent, Pr. P. Lantelme, Pr. G. Llorca, Pr. D. Peyramond, Pr. J. Tebib, Pr. E. Vignon, Pr. D. Vital-Durand, and Pr. F. Zoulim for their valuable comments.
Footnotes
Financial support and conflicts of interest: The author listed here has received financial support (personal or institutional) from the listed companies, unrelated to the present work: PS: Pfizer, LFB, and GlaxoSmithKline. No funding source or sponsor has been involved in the current study. The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Arlet J-B, Le Thi Huong D, Marinho A, Amoura Z, Wechsler B, Papo T, Piette J-C. Reactive haemophagocytic syndrome in adult-onset Still’s disease: a report of six patients and a review of the literature. Ann Rheum Dis. 2006;65:1596–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bywaters EG. Still’s disease in the adult. Ann Rheum Dis. 1971;30:121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cagatay Y, Gul A, Cagatay A, Kamali S, Karadeniz A, Inanc M, Ocal L, Aral O, Konice M. Adult-onset Still’s disease. Int J Clin Pract. 2007;63:1050–1055. [DOI] [PubMed] [Google Scholar]
- 4.Chen P-D, Yu S-L, Chen S, Weng X-H. Retrospective study of 61 patients with adult-onset Still’s disease admitted with fever of unknown origin in China. Clin Rheumatol. 2012;31:175–181. [DOI] [PubMed] [Google Scholar]
- 5.Choe J-Y, Chung DS, Park S-H, Kwon H-H, Kim S-K. Clinical significance of 18F-fluoro-dexoxyglucose positron emission tomography in patients with adult-onset Still’s disease: report of two cases and review of literatures. Rheumatol Int. 2010;30:1673–1676. [DOI] [PubMed] [Google Scholar]
- 6.Colina M, Zucchini W, Ciancio G, Orzincolo C, Trotta F, Govoni M. The evolution of adult-onset Still disease: an observational and comparative study in a cohort of 76 Italian patients. Semin Arthritis Rheum. 2011;41:279–285. [DOI] [PubMed] [Google Scholar]
- 7.Crouzet J, Boudousq V, Lechiche C, Pouget JP, Kotzki PO, Collombier L, Lavigne JP, Sotto A. Place of (18)F-FDG-PET with computed tomography in the diagnostic algorithm of patients with fever of unknown origin. Eur J Clin Microbiol Infect Dis. 2012;31:1727–1733. [DOI] [PubMed] [Google Scholar]
- 8.Cush JJ, Medsger TA Jr, Christy WC, Herbert DC, Cooperstein LA. Adult-onset Still’s disease. Clinical course and outcome. Arthritis Rheum. 1987;30:186–194. [DOI] [PubMed] [Google Scholar]
- 9.De Bandt M, Saint-Marcoux B. Tocilizumab for multirefractory adult-onset Still’s disease. Ann Rheum Dis. 2009;68:153–154. [DOI] [PubMed] [Google Scholar]
- 10.De Boysson H, Fevrier J, Nicolle A, Auzary C, Geffray L. Tocilizumab in the treatment of the adult-onset Still’s disease: current clinical evidence. Clin Rheumatol. 2013;32:141–147. [DOI] [PubMed] [Google Scholar]
- 11.Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still’s disease. Ann Rheum Dis. 2006;65:564–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Emmenegger U, Frey U, Reimers A, Fux C, Semela D, Cottagnoud P, Spaeth PJ, Neftel KA. Hyperferritinemia as indicator for intravenous immunoglobulin treatment in reactive macrophage activation syndromes. Am J Hematol. 2001;68:4–10. [DOI] [PubMed] [Google Scholar]
- 13.Fautrel B, Borget C, Rozenberg S, Meyer O, Le Loet X, Masson C, Koeger AC, Kahn MF, Bourgeois P. Corticosteroid sparing effect of low dose methotrexate treatment in adult Still’s disease. J Rheumatol. 1999;26:373–378. [PubMed] [Google Scholar]
- 14.Fautrel B, Le Moel G, Saint-Marcoux B, Taupin P, Vignes S, Rozenberg S, Koeger AC, Meyer O, Guillevin L, Piette JC, Bourgeois P. Diagnostic value of ferritin and glycosylated ferritin in adult onset Still’s disease. J Rheumatol. 2001;28:322–329. [PubMed] [Google Scholar]
- 15.Fautrel B, Sibilia J, Mariette X, Combe B. Tumour necrosis factor alpha blocking agents in refractory adult Still’s disease: an observational study of 20 cases. Ann Rheum Dis. 2005;64:262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fautrel B, Zing E, Golmard J-L, Le Moelle G, Bissery A, Riou C, Rozenberg S, Piette J-C, Bourgeois P. Proposal for a new set of classification criteria for adult-onset Still disease. Medicine (Baltimore). 2002;81:194–200. [DOI] [PubMed] [Google Scholar]
- 17.Fraisse T-C, Degraeve F, Riviere S, Le Quellec A. [Evolution and prognosis of adult onset Still’s disease. A monocentric study of 17 patients]. Rev Med Interne. 2006;27:658–664. [DOI] [PubMed] [Google Scholar]
- 18.Franchini S, Dagna L, Salvo F, Aiello P, Baldissera E, Sabbadini MG. Efficacy of traditional and biologic agents in different clinical phenotypes of adult-onset Still’s disease. Arthritis Rheum. 2010;62:2530–2535. [DOI] [PubMed] [Google Scholar]
- 19.Henter J-I, Horne A, Arico M, Egeler RM, Filipovic AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J, Janka G. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–131. [DOI] [PubMed] [Google Scholar]
- 20.Hot A, Toh M-L, Coppere B, Perard L, Girard-Madoux MH, Mausservey C, Desmurs-Clavel H, Ffrench M, Ninet J. Reactive hemophagocytic syndrome in adult-onset Still disease: clinical features and long-term outcome: a case–control study of 8 patients. Medicine (Baltimore). 2010;89:37–46. [DOI] [PubMed] [Google Scholar]
- 21.Husni ME, Maier AL, Mease PJ, Overman SS, Fraser P, Gravallese EM, Weinblatt ME. Etanercept in the treatment of adult patients with Still’s disease. Arthritis Rheum. 2002;46:1171–1176. [DOI] [PubMed] [Google Scholar]
- 22.Kalliolias GD, Georgiou PE, Antonopoulos IA, Andonopoulos AP, Liossis S-NC. Anakinra treatment in patients with adult-onset Still’s disease is fast, effective, safe and steroid sparing: experience from an uncontrolled trial. Ann Rheum Dis. 2007;66:842–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaneko K, Kaburaki M, Muraoka S, Tanaka N, Yamamoto T, Kusunoki Y, Abe H, Endo H, Kawai S. Exacerbation of adult-onset Still’s disease, possibly related to elevation of serum tumor necrosis factor-alpha after etanercept administration. Int J Rheum Dis. 2010;13:e67–69. [DOI] [PubMed] [Google Scholar]
- 24.Kim H-A, Sung J-M, Suh C-H. Therapeutic responses and prognosis in adult-onset Still’s disease. Rheumatol Int. 2012;32:1291–1298. [DOI] [PubMed] [Google Scholar]
- 25.Kim YJ, Kim SI, Hong K-W, Kang MW. Diagnostic value of (18)F-FDG PET/CT in patients with fever of unknown origin. Intern Med J. 2012;42:834–837. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi M, Takahashi Y, Yamashita H, Kaneko H, Mimori A. Benefit and a possible risk of tocilizumab therapy for adult-onset Still’s disease accompanied by macrophage-activation syndrome. Mod Rheumatol. 2011;21:92–96. [DOI] [PubMed] [Google Scholar]
- 27.Kong X-D, Xu D, Zhang W, Zhao Y, Zeng X, Zhang F. Clinical features and prognosis in adult-onset Still’s disease: a study of 104 cases. Clin Rheumatol. 2010;29:1015–1019. [DOI] [PubMed] [Google Scholar]
- 28.Laskari K, Tzioufas AG, Moutsopoulos HM. Efficacy and long-term follow-up of IL-1R inhibitor anakinra in adults with Still’s disease: a case-series study. Arthritis Res Ther. 2011;13:R91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee S-W, Park Y-B, Song J-S, Lee S-K. The mid-range of the adjusted level of ferritin can predict the chronic course in patients with adult onset Still’s disease. J Rheumatol. 2009;36:156–162. [DOI] [PubMed] [Google Scholar]
- 30.Magadur-Joly G, Billaud E, Barrier JH, Pennec YL, Masson C, Renou P, Prost A. Epidemiology of adult Still’s disease: estimate of the incidence by a retrospective study in west France. Ann Rheum Dis. 1995;54:587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mavragani CP, Spyridakis EG, Koutsilieris M. Adult-onset Still’s disease: from pathophysiology to targeted therapies. Int J Inflam. 2012;2012:879020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nordstrom D, Knight A, Luukkainen R, van Vollenhoven R, Rantalaiho V, Kajalainen A, Brun JG, Proven A, Ljung L, Kautiainen H, Pettersson T. Beneficial effect of interleukin 1 inhibition with anakinra in adult-onset Still’s disease. An open, randomized, multicenter study. J Rheumatol. 2012;39:2008–2011. [DOI] [PubMed] [Google Scholar]
- 33.Pay S, Turkcapar N, Kalyoncu M, Simsek I, Beyan E, Ertenli I, Osturk MA, Duzgun N, Erdem H, Ozbalkan Z. A multicenter study of patients with adult-onset Still’s disease compared with systemic juvenile idiopathic arthritis. Clin Rheumatol. 2006;25:639–644. [DOI] [PubMed] [Google Scholar]
- 34.Permal S, Wechsler B, Cabane J, Perrot S, Blum L, Imbert JC. [Treatment of Still disease in adults with intravenous immunoglobulins]. Rev Med Interne. 1995;16:250–254. [DOI] [PubMed] [Google Scholar]
- 35.Pouchot J, Arlet J-B. Biological treatment in adult-onset Still’s disease. Best Pract Res Clin Rheumatol. 2012;26:477–487. [DOI] [PubMed] [Google Scholar]
- 36.Pouchot J, Fautrel B. Maladie de Still de l’adulte. In: Guillevin L, Meyer O, Sibilia J, eds. Traite des Maladies et Syndromes Systemiques. 5th ed Paris: Flammarion; 2008: 1249–1263. [Google Scholar]
- 37.Pouchot J, Sampalis JS, Beaudet F, Carette S, Decary F, Salusinsky-Sternbach M, Hill RO, Gutkowski A, Harth M, Myhal D. Adult Still’s disease: manifestations, disease course, and outcome in 62 patients. Medicine (Baltimore). 1991;70:118–136. [PubMed] [Google Scholar]
- 38.Uppal SS, Al-Mutairi M, Hayat S, Abraham M, Malaviya A. Ten years of clinical experience with adult onset Still’s disease: is the outcome improving- Clin Rheumatol. 2007;26:1055–1060. [DOI] [PubMed] [Google Scholar]
- 39.Vignes S. Percentage of glycosylated serum ferritin remains low throughout the course of adult onset Still’s disease. Ann Rheum Dis. 2000;59:347–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vignes S, Wechsler B, Amoura Z, Papo T, Frances C, Huong DL, Veyssier P, Godeau P, Piette JC. Intravenous immunoglobulin in adult Still’s disease refractory to non-steroidal anti-inflammatory drugs. Clin Exp Rheumatol. 1998;16:295–298. [PubMed] [Google Scholar]
- 41.Wouters JM, Van de Putte LB. Adult-onset Still’s disease; clinical and laboratory features, treatment and progress of 45 cases. Q J Med. 1986;61:1055–1065. [PubMed] [Google Scholar]
- 42.Yamaguchi M, Ohta A, Tsunematsu T, Kasukawa R, Mizushima Y, Kashiwagi H, Kashiwazaki S, Tanimoto K, Matsumoto Y, Ota T. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992;19:424–430. [PubMed] [Google Scholar]
- 43.Zeng T, Zou Y-Q, Wu M-F, Yang C-D. Clinical features and prognosis of adult-onset Still’s disease: 61 cases from China. J Rheumatol. 2009;36:1026–1031. [DOI] [PubMed] [Google Scholar]
- 44.Zhu G, Liu G, Liu Y, Xie Q, Shi G. Liver abnormalities in adult onset Still’s disease: a retrospective study of 77 Chinese patients. J Clin Rheumatol. 2009;15:284–288. [DOI] [PubMed] [Google Scholar]