

Abstract
Data regarding the incidence and outcome of renal involvement in patients with inflammatory myopathies (IM) remain scarce. We assessed the incidence and causes of acute kidney injury (AKI) and chronic kidney disease (CKD) in 150 patients with dermatomyositis, polymyositis, and antisynthetase syndrome followed in 3 French referral centers. Renal involvement occurred in 35 (23.3%) patients: AKI in 16 (10.7%), and CKD in 31 (20.7%) patients. The main cause of AKI was drug or myoglobinuria-induced acute tubular necrosis. Male sex, cardiovascular risk factors, cardiac involvement, and initial proteinuria >0.3 g/d were associated with the occurrence of AKI. The outcome of patients with AKI was poor: 13 (81%) progressed to CKD and 2 (12.5%) reached end-stage renal disease. In multivariate survival analysis, age at IM onset, male sex, a history of cardiovascular events, and a previous episode of AKI were associated with the risk of CKD. We also identified 14 IM patients who underwent a kidney biopsy in 10 nephrology centers. Renal pathology disclosed a wide range of renal disorders, mainly immune-complex glomerulonephritis. We identified in 5 patients a peculiar pattern of severe acute renal vascular damage consisting mainly of edematous thickening of the intima of arterioles.
We found that AKI and CKD are frequent in patients with IM. Prevention of AKI is crucial in these patients, as AKI is a major contributor to their relatively high risk of CKD. A peculiar pattern of acute vascular damage is part of the spectrum of renal diseases associated with IM.
INTRODUCTION
Inflammatory myopathies (IM) include dermatomyositis (DM) and polymyositis (PM), 2 rare autoimmune diseases (prevalence of 7 cases/100,000) characterized by moderate to severe muscle weakness and inflammatory lesions in the muscle.19 The pathophysiology of these 2 entities is not fully understood, but they are probably related to 2 distinct mechanisms. DM, which is frequently associated with neoplasia, results from a complement-mediated microangiopathy in the muscle. Complement activation and the subsequent generation of the C5–b9 membrane attack complex (MAC) is a central player in the genesis of DM.16 MAC targets the endothelial cells and induces capillary necrosis, perivascular inflammation, microinfarcts, and destruction of muscle fibers.2,5 Complement activation also triggers cytokine and chemokine release, and the ensuing influx of B and CD4+ T lymphocytes in the perimysium. In contrast, PM is related to muscle fiber (endomysium) inflammation and necrosis due to infiltrating activated cytotoxic CD8+ cells that recognize upregulated MHC class I antigens expressed on the membrane of muscle fibers.5
Although DM and PM arise from 2 distinct mechanisms, they are both associated with the occurrence of various nonspecific myositis-associated antibodies (anti-Ssa, Ssb, RnP, DNA, Sm, PmScl, Scl70 antibodies, and antineutrophil cytoplasmic antibodies [ANCA]) and myositis-specific antibodies (the antisynthetase antibodies: anti-JO1, OJ, EJ, KS, ZO, PL12, PL7;13 Mi219, SRP, and MDA-5 antibodies2).
DM and PM lead to frequent, and, in some cases, life-threatening extramuscular complications. The most frequently affected organ is the lung (25%–45%).8 Pulmonary involvement includes nonspecific interstitial pneumonia (NSIP), usual interstitial pneumonia, bronchiolitis obliterans organizing pneumopathy, diffuse alveolar damage, and interstitial pneumonia related to neutrophil or CD8+ cell-induced alveolitis.8 Pulmonary disease may occur in the setting of “antisynthetase syndrome” (AS) that combines myositis, interstitial pneumonia, arthritis, Raynaud phenomena, cutaneous disease, mechanics’ hands, and the presence of antisynthetase antibodies. Cardiac involvement has also been reported in 5%–15% of cases.17 Data regarding the incidence and outcome of renal involvement in patients with IM remain particularly scarce, even though renal involvement is frequent in various systemic autoimmune diseases.
In the present study, we retrospectively assessed the incidence and outcome of acute kidney injury (AKI) and chronic kidney disease (CKD) in a large cohort of patients with IM. We also analyzed the spectrum of nephropathies documented by kidney biopsy performed in patients with IM.
PATIENTS AND METHODS
We identified patients with DM, PM, and AS using computerized databases in 3 French referral centers for patients with IM (internal medicine department of CHU de Nantes, Nantes, and internal medicine departments 1 and 2 at the Hôpital de la Pitié Salpêtrière, Paris). Diagnosis of DM, PM, and AS was based on the ENMC classification.12 We excluded patients in whom muscle biopsy disclosed features suggestive of a type of myositis distinct from DM/PM, and patients with myositis associated with a well-recognized autoimmune systemic disease (systemic lupus erythematosus, systemic sclerosis, sarcoidosis). Long-term clinical and laboratory (including serum creatinine [SCr] and proteinuria) follow-up of these patients was performed mainly in outpatient clinics or during day hospitalizations, as frequently as required based on their clinical status.
Clinical and Laboratory Features
We retrospectively reviewed the medical records of included patients and collected relevant data. Muscle weakness was assessed using a testing score (NFIP score: 0–5). The muscle injury was estimated using the blood creatine phosphokinase (CPK) level expressed in IU/L. Evaluation of extramuscular involvement was mainly performed in the presence of suggestive clinical signs. Pulmonary involvement was diagnosed based on the presence of clinical signs (shortness of breath, cough, etc) associated with features of interstitial pneumonia on the chest computed tomography, and/or a decrease in the diffusing capacity of the lung for carbon monoxide. Heart involvement was defined based on electrocardiogram findings (dysrhythmias, conduction defects) and cardiac echocardiography findings suggestive of infiltrative cardiomyopathy, whereas features of sensory or motor impairment in the electromyogram (demyelinating or axonal polyneuropathy) defined neurologic involvement.
Autoantibodies were detected by indirect immunofluorescence and quantitated by ELISA or Dot-Blot. Disease remission was defined by the resolution of muscle weakness and a decrease >50% in CPK level. Relapse was defined by a worsening of muscular symptoms and/or an increase in CPK level requiring a modification of treatment regimen. The resistance to initial treatment was defined by the persistence of clinical or laboratory features leading to the institution of a second-line treatment.
AKI was defined as an acute doubling of SCr level. Patients with prerenal AKI defined by a normalization of SCr following hydration with at least 500 mL of saline solution were excluded from the analysis. An estimated glomerular filtration rate (eGFR) using the MDRD formula <60 mL/min on at least 2 measurements 3 months apart defined CKD.
We identified kidney biopsies performed in patients with IM through a query sent to 10 French nephrology centers. Kidney biopsies were blindly reviewed by the same expert renal pathologist (A. Moreau).
We performed a PubMed-based search (National Library of Medicine, Bethesda, MD) for published cases of kidney biopsies in DM, PM, and AS patients using the following key words: “IM, DM, PM, AS, nephritis, renal involvement.”
Statistical Analysis and Ethical Review
Data are expressed as mean ± standard deviation for continuous covariates or as count and percentage for categorical covariates. Comparisons were performed using the t test and chi-square test (for continuous and categorical variables, respectively) or the Mann-Whitney or Fisher exact test, depending on the number of patients. Time-to-CKD analyses were performed using Cox models. Only variables significantly associated (p < 0.2) with the occurrence of CKD on univariate analysis were subsequently entered into the multiple regression model. The final model was constructed by backward stepwise selection process (p < 0.05). A prognostic score predictive of the occurrence of CKD was established. This score is equal to the sum of the risk factor values in the final survival model, multiplied by the corresponding log-hazard ratios. The time-dependent receiver operating characteristic (ROC) method was used to analyze the ability of the score to predict the occurrence of CKD, while taking into account data censoring during follow-up.11 Prediction of CKD risk was limited to 5 years, as only a few patients were followed beyond that time. We aimed to identify high-risk patients for CKD, and thus we chose a cutoff score value in order to discriminate high and low CKD risk groups with a high sensitivity (0.9). All the analyses were performed using R software.
For this retrospective study, no review board approval was required.
RESULTS
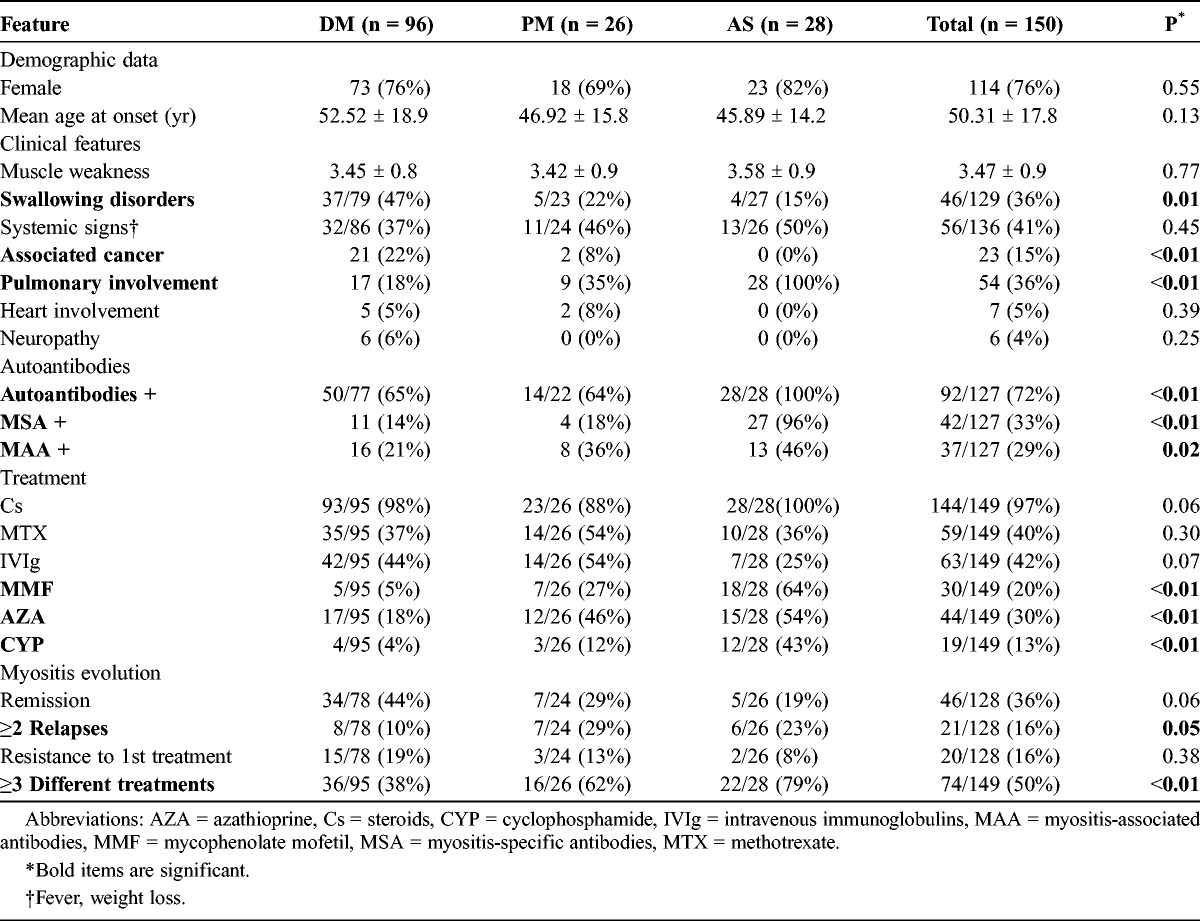
We identified 199 patients with the diagnosis of DM, PM, or AS, and excluded from the analysis 49 patients with incomplete data. Among the remaining 150 patients, 96 (64%) had DM, 26 (17%) had PM, and 28 (19%) had AS. Patient characteristics are shown in Table 1. The mean follow-up for the entire cohort was 4.1 ± 4.5 years. The cohort included mainly women (76%), and most patients had relatively severe forms of IM as evidenced by the muscle weakness score (mean, 3.47) and the number of treatments used to control the disease (74/149 patients [50%] needed 3 or more consecutive immunosuppressive treatments). As expected, the most frequently affected organ was the lung (36%).
TABLE 1.
Clinical and Laboratory Features of 150 Patients With Inflammatory Myopathies
The Spectrum of Kidney Injury
Renal involvement occurred in 35 (23.3%) patients of the entire cohort, including 21 patients (22%) with DM, 6 (23%) with PM, and 8 (29%) with AS. Renal involvement consisted of AKI in 16 (10.7%) patients and CKD in 31 (20.7%) patients. Proteinuria was >0.3 g/d in 26/86 patients (30%).
AKI in Patients With IM
AKI occurred during the first 6 months after the diagnosis of IM in 50% of patients, and during the first year in 60% of cases. AKI was related to drug toxicity in 6 patients: intravenous immunoglobulin in 4 cases, tacrolimus in 1 case, and cisplatine used to treat lung adenocarcinoma in 1 case. In 3 cases, AKI was due to myoglobinuria-related acute tubular necrosis (ATN) that was associated in 1 case with severe acute heart failure due to myositis-associated myocardiopathy. One patient developed, 2 weeks after the start of steroids (1 mg/kg), thrombotic microangiopathy (platelets, 40 G/L; hemoglobin, 5.9 g/dL, undetectable haptoglobin, presence of 3% schizocytes on blood smear) and AKI requiring dialysis. Another patient presented with AKI (SCr: 440 μmol/L), mild proteinuria (1 g/d), and microscopic hematuria 2 years after the diagnosis of DM. Kidney biopsy disclosed acute vascular lesions consisting of edematous changes of the intima of arteries and severe glomerular ischemia. In the last patient, AKI (SCr: 150 μmol/L, microscopic hematuria, proteinuria, 7.5 g/d) complicated the course of membranous nephropathy (MN) diagnosed 10 years earlier. AKI coincided with a relapse of AS, and kidney biopsy disclosed MN with superimposed crescentic glomerulonephritis in more than 50% of glomeruli. In 4 cases, the cause of AKI was unknown.
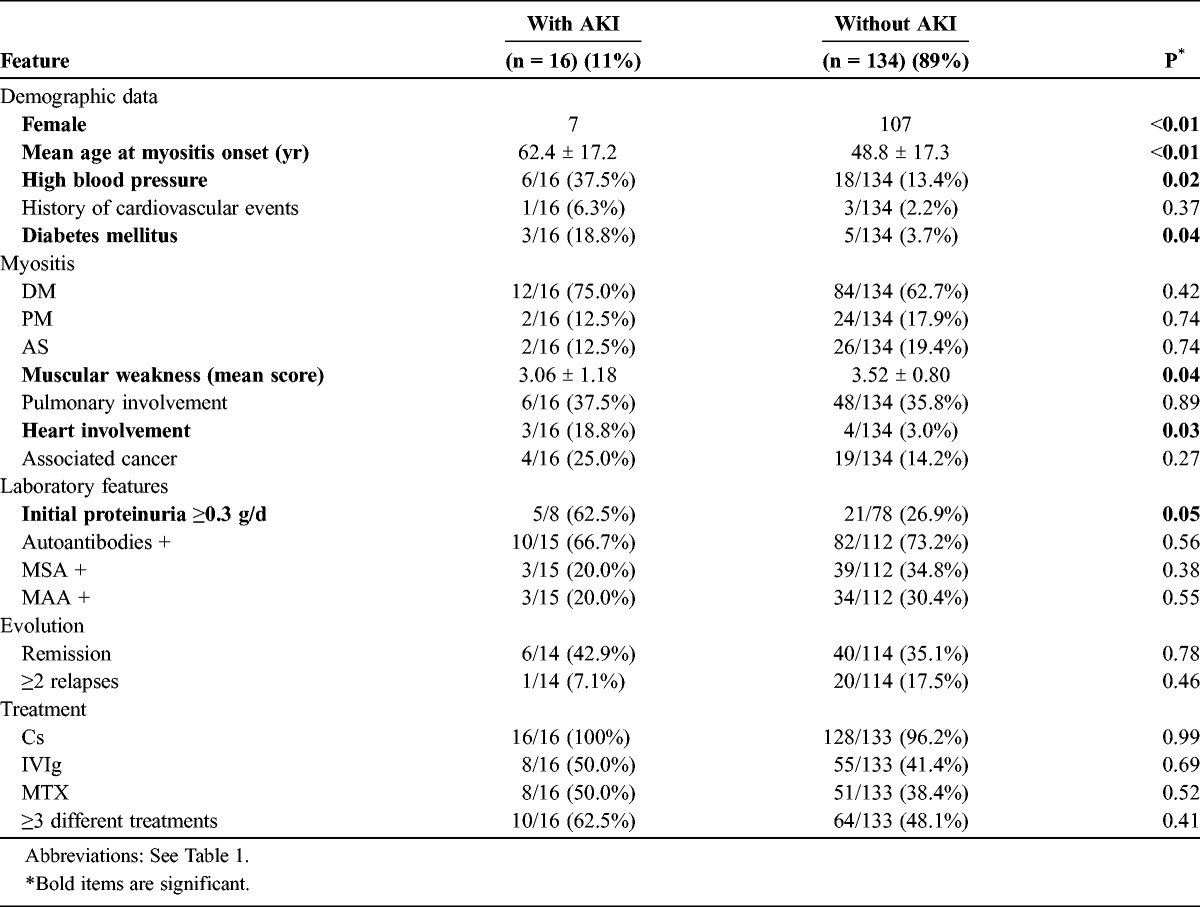
In descriptive analysis, male sex, cardiovascular risk factors, the severity of muscle injury, heart involvement, and initial proteinuria (>0.3 g/d) were associated with AKI (Table 2). In contrast, analysis of available data did not show a significant association between the type, severity, and treatment of IM; the level of CPK; and the occurrence of AKI. Survival analysis was not performed due to the relatively limited number of patients. Among patients with AKI, 13 (81%) progressed to CKD, including 2 patients (12.5%) who reached end-stage renal disease (ESRD). The 3 (19%) remaining patients recovered and retained normal renal function.
TABLE 2.
Clinical and Laboratory Features of 150 IM Patients With or Without Acute Kidney Injury (AKI)
CKD in Patients With IM
CKD was noted in 31 (20.7%) patients included in the study (mean eGFR: 44.1 mL/min ± 15.8). In 4 patients, CKD predated the onset of IM; their initial SCr values were 136 μmol/L (eGFR: 47 mL/min), 148 (eGFR: 32 mL/min), 150 (eGFR: 44 mL/min), and 160 μmol/L (eGFR: 38 mL/min). These patients were, however, included in the analysis, as all experienced a rapid decline in eGFR (>25% of initial SCr, mean decrease of eGFR of 10 mL/min) in the 2 years following the diagnosis of myositis, along with the development of significant proteinuria (>1.5 g/d) in 2 of them.
Twenty patients (64.5%) had CKD grade III-a (eGFR: 45–60 mL/min), 4 (12.9%) had CKD grade III-b (eGFR: 30–45 mL/min), 5 (16.1%) had CKD grade IV (eGFR: 15–30 mL/min), and 2 (6.5%) had CKD grade V. Two patients underwent kidney biopsy in the setting of proteinuria (2 and 7 g/d) and a decrease in eGFR >30%, at 2 and 8 years, respectively, after the onset of myositis. Both had normal renal function at the diagnosis of myositis. Kidney biopsies disclosed severe fibrotic lesions with 30% and 60% of sclerotic glomeruli and focal segmental glomerulosclerosis lesions.
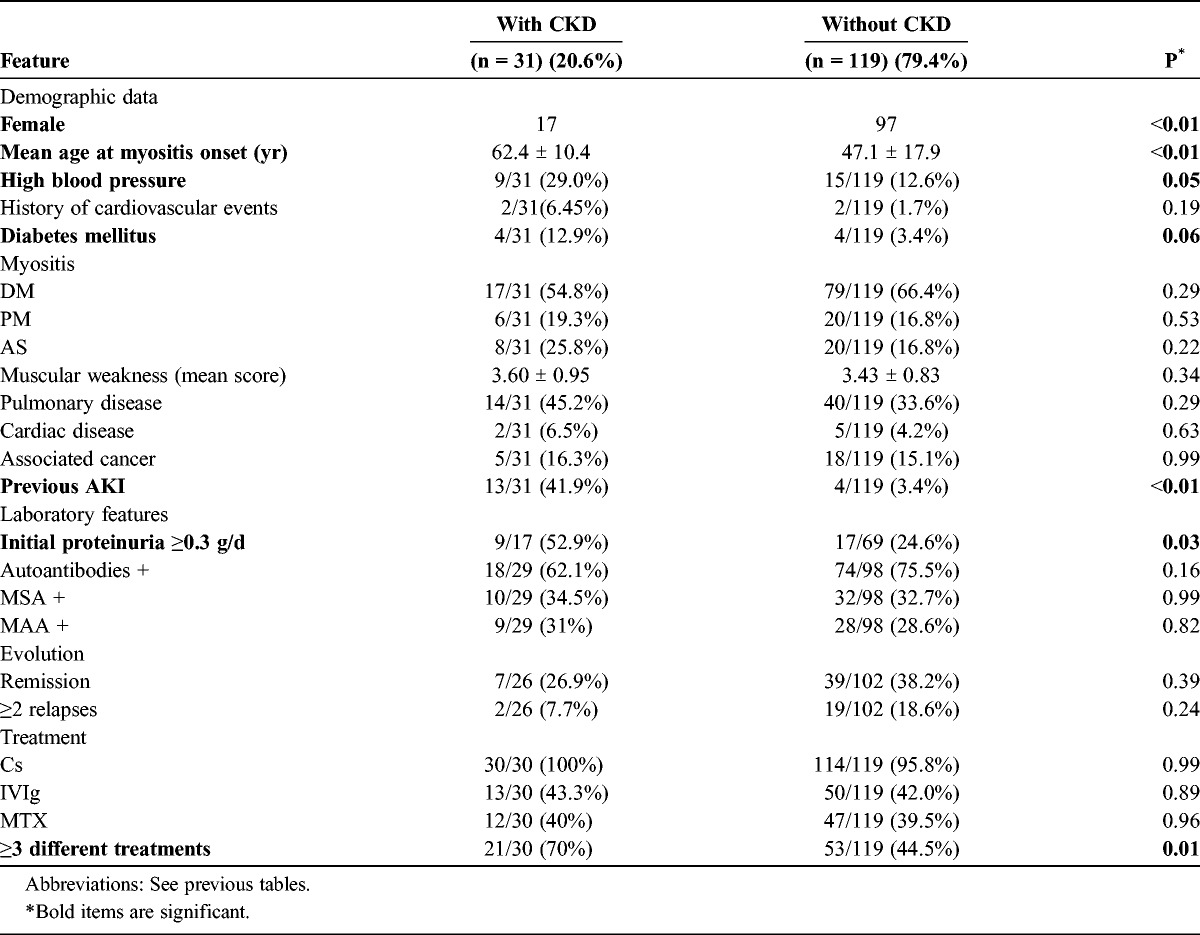
Male sex, cardiovascular risk factors, previous episode of AKI, initial proteinuria (≥0.3 g/d), and the severity of IM as exemplified by the number of different treatments required to induce remission were associated with the development of CKD (Table 3).
TABLE 3.
Clinical and Laboratory Features of 150 IM Patients With or Without Chronic Kidney Disease (Univariate Analysis)
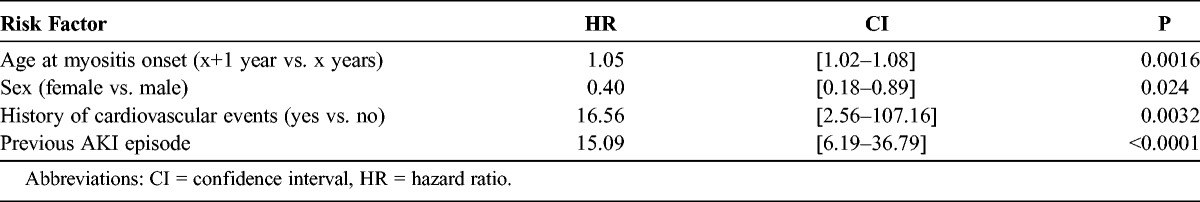
In multivariate survival analysis (Table 4), only age at IM onset, male sex, history of cardiovascular events, and a previous episode of AKI were identified as risks factors of CKD. History of cardiovascular events affected too few patients (only 4 patients were included in the cohort) to be considered a significant and representative risk factor.
TABLE 4.
Independent Risk Factors for the Occurrence of CKD in IM Patients, Identified by Multivariate Regression Analysis
A prognostic score of the occurrence of CKD within 5 years after the onset of IM was established based on the baseline covariates significantly associated with CKD in the multivariate Cox model. With an area under the ROC curve equal to 0.83 (confidence interval [CI], 0.73–0.89), this score accurately identified patients with low (score <1.80) or high (score ≥1.80) risk for CKD with 90% sensitivity and 60% specificity (Figure 1).
FIGURE 1.
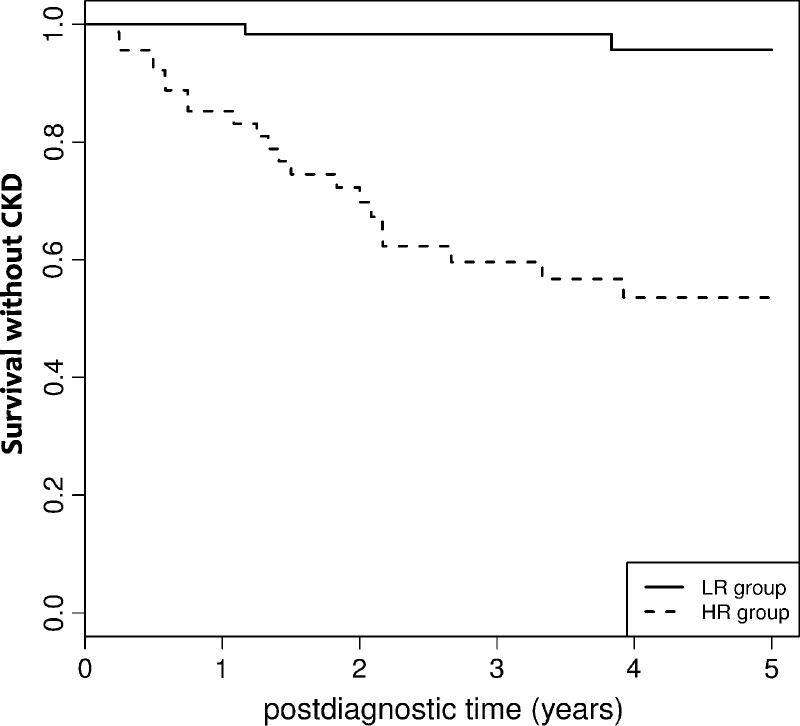
Survival without CKD in patients with IM with low risk (LR) or high risk (HR) based on the prognostic score of CKD. For a given patient, the prognostic score is calculated using the following formulas: Score = 0.068 * Patient age at IM diagnosis − -0.88 * Sex + 2.4 * History of cardiovascular event (with Sex = 1 if the patient is a women and = 0 if a man, and History of cardiovascular event = 1 if the patient has history of cardiovascular event and = 0 if not). Low risk was defined by a score <1.80, and high risk by a score ≥1.80.
The Spectrum of Nephropathies Documented by Kidney Biopsy in Myositis Patients
We identified 14 patients (7 male and 7 female patients) with myositis (6 PM, 5 DM, and 3 AS) who underwent kidney biopsy in 10 French nephrology centers (Table 5). All these patients had no clinical or laboratory features of any other autoimmune diseases (including sarcoidosis, systemic sclerosis, and systemic lupus erythematosus) or IM-associated cancer. Kidney biopsy was performed based on the presence of proteinuria (1.2–7.0 g/d) (n = 13), and/or AKI (SCr: 200–737 μmol/L) (n = 5), and/or CKD (n = 3) (eGFR: 34–46 mL/min) (see Table 4). Microscopic hematuria was present in 8 of 11 cases (72.73%). In 8 cases (57%), renal abnormalities were detected within the first 6 months after the diagnosis of myositis.
TABLE 5.
Clinical and Laboratory Features of 14 Patients With IM With Nephropathy Documented by a Kidney Biopsy, Present Report
Pathologic patterns disclosed by kidney biopsies included the following (see Table 5):
Immune complex-related glomerulonephritis (n = 4), with IgA nephropathy (IgAN) in 2 patients and MN in 2 cases, 1 of whom had crescentic MN (described above) (Figure 2 A).
FIGURE 2.
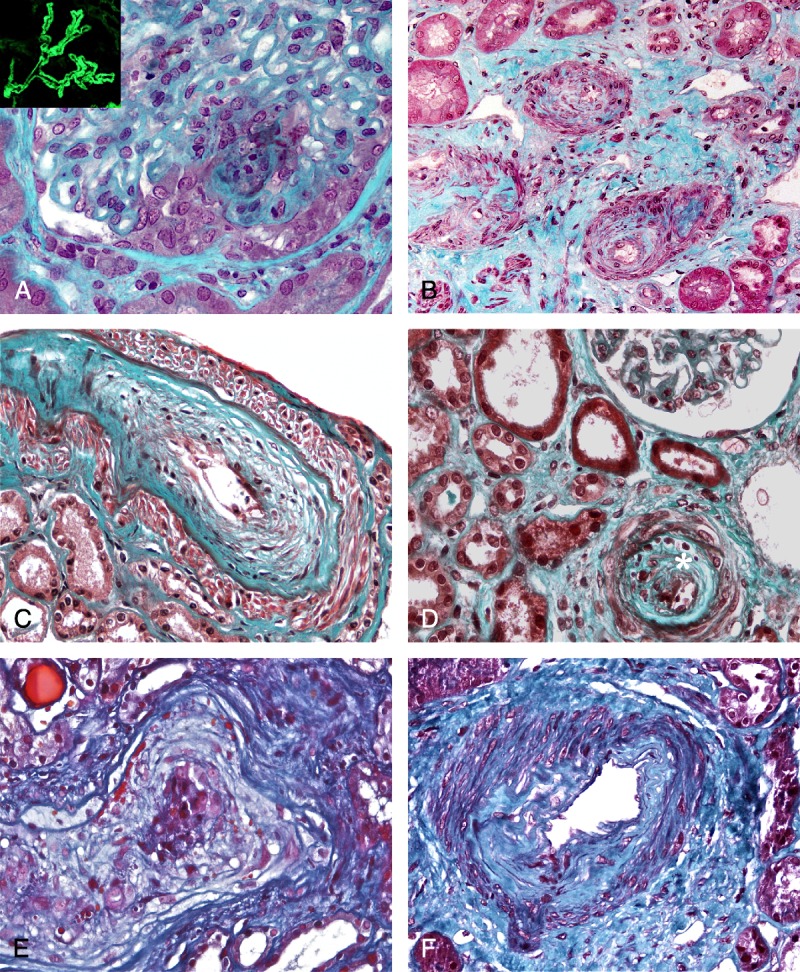
A, Kidney biopsy (light microscopy, Masson trichrome, ×400) performed in a 45-year-old woman (Table 5, Patient 4) with a history of antisynthetase syndrome and MN diagnosed 10 years earlier. She presented with an exacerbation of the antisynthetase syndrome and an AKI. Kidney biopsy disclosed a crescentic MN with extracapillary proliferation noted in 11/20 glomeruli. Test for ANCA was negative. (Insert shows IgG staining.) B, Kidney biopsy (light microscopy, Masson trichrome, ×200) performed in a 68-year-old normotensive man (Table 5, Patient 6) with a history of DM who presented with AKI. Edematous thickening of the intima of interlobular arteries associated with interstitial fibrosis and ischemic glomerular lesions was noted. C and D, Kidney biopsy (light microscopy, Masson trichrome, ×400) performed in a 67-year-old normotensive woman (Table 5, Patient 7) who presented with an exacerbation of PM (rhabdomyolysis, lung involvement) and AKI. Kidney biopsy disclosed edematous thickening of the intima of an interlobular artery superimposed on chronic arteriosclerosis. Mild lymphocytic infiltration (*) was detected in 2 interlobular arteries within the marked fibrous intimal thickening (2D). E, Kidney biopsy (light microscopy, Masson trichrome, ×400) performed in a 55-year-old normotensive man (Table 5, Patient 5) who presented with PM (rhabdomyolysis, interstitial pneumopathy) and AKI. Pathologic analysis disclosed the presence of marked edematous thickening of the intima of the interlobular arteries leading to an obstruction of the vessel lumen. F, Kidney biopsy (light microscopy, Masson trichrome, ×400) performed in a 34-year-old normotensive man (Table 5, Patient 8) who presented with DM (rhabdomyolysis, interstitial pneumopathy), mild proteinuria (0.8 g/d and subsequently 1.2 g/d), and hematuria. On pathologic examination, a marked fibrous thickening of the intima of an arcuate artery was present.
Predominant vascular lesions, noted in 5 patients with DM (n = 2) or PM (n = 3). Four patients (aged 34–70 yr) with no history of high blood pressure presented with severe AKI (SCr: 200–570 μmol/L) due to severe acute vascular lesions. The first patient (Table 5, Patient 6) was a 68-year-old normotensive man with a history of DM who presented with AKI (SCr: 420 μmol/L). Kidney biopsy disclosed edematous thickening of the intima of interlobular arteries associated with an interstitial fibrosis and ischemic glomerular lesions (Figure 2B). This patient eventually progressed to ESRD. The second patient with vascular lesions (Table 5, Patient 7) was a 67-year-old normotensive woman who presented with exacerbated PM (rhabdomyolysis, lung involvement) and AKI (SCr: 570 μmol/L). Edematous thickening of the intima of an interlobular artery superimposed on chronic arteriosclerosis was present in a kidney biopsy (Figure 2C). A mild lymphocytic infiltration was detected in 2 interlobular arteries (Figure 2D). The patient required hemodialysis and died 2 years after the onset of AKI. The third patient (Table 5, Patient 5) was a 55-year-old normotensive man who presented with PM (rhabdomyolysis, interstitial pneumopathy) and AKI (SCr: 200 μmol/L). Renal pathologic analysis disclosed the presence of marked edematous thickening of the intima of the interlobular arteries leading to an obstruction of the vessel lumen (Figure 2E). This patient reached ESRD. The fourth patient (Table 5, Patient 8) was a 34-year-old normotensive man who presented with DM (rhabdomyolysis, interstitial pneumopathy), mild proteinuria (0.8 g/d and subsequently 1.2 g/d), and hematuria. On pathologic examination, a marked fibrous thickening of the intima of an arcuate artery was present (Figure 2 F). He progressed to mild CKD (eGFR: 54 mL/min). The last patient with predominant vascular lesions was a 70-year-old man with PM (Table 5, Patient 9). Three months after introduction of high-dose steroids, he developed AKI (SCr: 737 μmol/L), and the only remarkable feature in kidney biopsy was extensive arteriolosclerosis lesions.
The remaining cases included focal segmental glomerulosclerosis lesions (n = 2), minimal change disease (n = 2), and chronic tubulointerstitial nephritis (n = 1).
Among these 14 patients, 11 (78.6%) progressed to CKD. At last follow-up, among the 4 patients with immune complex-related glomerulonephritis, 2 developed CKD (median eGFR: 39.5 mL/min). Among the 5 patients with predominant vascular lesions, 4 reached ESRD and 1 had CKD (eGFR: 50 mL/min). In the last group (n = 5), 4 patients progressed to CKD with a mean eGFR of 33.8 mL/min (27–44 mL/min).
In the literature search we retrieved 15 published cases1,3,4,6,9,14,15,21,22,25–29,31 of nephropathies documented by kidney biopsy in IM patients (Table 6). These cases include IgAN (n = 7), MN (n = 1), membranoproliferative glomerulonephritis (n = 1), pauci-immune crescentic glomerulonephritis (n = 2), minimal change disease (n = 2), focal segmental glomerulosclerosis lesions (n = 1), and acute tubulointerstitial nephritis (n = 1). Renal disease was diagnosed early in the course of myositis, during the first 6 months in 10 cases (66%). Renal outcome of these published cases was not totally reported, but at least 3 of the cases (20%) progressed to CKD.
TABLE 6.
Clinical and Laboratory Features of Patients With IM With Nephropathy Documented by a Kidney Biopsy, Previous Reports
DISCUSSION
Data regarding renal involvement in the setting of IM are relatively scarce. To our knowledge, a single retrospective study32 has assessed the occurrence of acute or chronic kidney damage in a relatively small cohort of 65 patients with IM. AKI was reported in 14 patients (21%), CKD in 1 (1.5%), and 4 (6%) additional patients had isolated proteinuria. However, some patients included in the study had SLE, a clear bias in the analysis of renal involvement in that cohort.
The current study included a large number of myositis patients, and patients with other autoimmune systemic disorders were carefully excluded. Our data clearly indicate that AKI and CKD are frequent in patients with IM. Renal damage was noted in 23% of all included patients: AKI occurred in 11% of cases and CKD developed in 21% of the patients. Indeed, ATN is the leading cause of AKI in myositis patients. This is due to the combination of 2 factors. On the one hand, well-identified causes of toxic ATN are frequently encountered in IM patients, mainly myoglobinuria and the use of nephrotoxic drugs such as intravenous immunoglobulins (IVIg). Myoglobin and IVIg cause tubular damage through renal medullar vasoconstriction and ischemia, a direct tubular cytotoxic effect and intratubular myoglobin cast formation.23,33 On the other hand, IM patients also present with several well-identified risk factors for toxic ATN. In our cohort, advanced age, diabetes, and cardiac disease were associated with the occurrence of AKI in IM patients. These findings highlight the need for 1) a careful monitoring of renal function in IM patients who require potentially nephrotoxic drugs during the acute phases of the disease and 2) the implementation of specific nephroprotective measures such as hydration with saline or bicarbonates or a slow infusion rate of IVIg. Markers that may predict the occurrence of AKI, including the neutrophil gelatinase-associated lipocalin (NGAL),20 warrant assessment in IM patients, and the prevention of AKI is crucial in these patients. In at least 40% of cases, AKI was not a direct manifestation of IM but rather was due to drug toxicity. Nevertheless, AKI is a frequent event in IM patients, may limit optimal treatment options, and contributes to the relatively high incidence of CKD (21%) in these patients. Eighty-one percent of patients who presented with an episode of AKI progressed to CKD, and 12.5% to ESRD.
One-fifth of the patients included in this relatively large and unselected cohort of IM patients had CKD during follow-up. This incidence may even be underestimated. Some patients with severe IM may present with significant amyotrophy, and SCr may not be the optimal biomarker of renal function, as previously underlined in similar populations of patients with amyotrophy.18,30 CKD developed after the onset of IM in virtually all patients, and was clearly precipitated by an episode of IM-related AKI in 42% of cases, which strongly suggests a link between IM and the occurrence of CKD. We established a predictive score of CKD in IM patients, a score based on simple, easily available clinical and laboratory parameters. This score may help clinicians identify IM patients at high risk of CKD and refer these patients to the nephrologist for long-term renal monitoring.
Renal pathology data available in our cohort are relatively in keeping with those previously published in cases reports and suggest that IM are associated with a wide range of renal disorders. IM-associated nephropathies consisted mainly of immune complex glomerulopathies, which represent roughly half of all available cases. The predominance of IgAN in previously published cases may be biased by the fact that several previous reports are from the Far East. These glomerulopathies were not paraneoplastic manifestations of IM-related cancers, as only 1 patient (IgAN) out of 19 had a history of cancer. IM are associated with the occurrence of various autoantibodies that were detected in three-fourths of patients included in the present study. The mechanisms underlying this dysregulated generation of autoantibodies remain unclear. Nevertheless, the occurrence of immune complex glomerulopathies, including IgAN 24 or ANCA-associated renal disease, in IM patients may arise from this dysregulated generation of autoantibodies.
We identified a new peculiar pattern of, mostly acute, renal vascular damage in 4 IM patients with no clinical or laboratory features of systemic sclerosis. One patient presented will full-blown thrombotic microangiopathy 2 weeks after the start of steroids, a clinical feature reminiscent of the triggering of renal scleroderma crisis by steroids.10 Three additional patients with no history of high blood pressure presented with severe AKI coincidental with an exacerbation of IM (rhabdomyolysis, NSIP). Their only notable renal pathologic feature was severe acute vascular damage consisting of edematous thickening of the intima of arterioles, a feature reminiscent of vascular lesions seen in the setting of accelerated hypertension or renal scleroderma crisis. Finally, in a 34-year-old normotensive man with isolated proteinuria, kidney biopsy disclosed surprising lesions suggestive of chronic arteriolosclerosis. We believe that this pattern of vascular damage is part of the spectrum of renal diseases associated with IM. It is noteworthy that DM is a complement-driven microangiopathy in the muscle. Our findings should be put in perspective with the accumulating data linking complement activation to another acute vascular renal disease, atypical hemolytic uremic syndrome (HUS).7
In all, our data indicate that AKI and CKD are frequent in IM patients. However, AKI and CKD are probably not a direct manifestation of IM, but rather result from drug and myoglobin-induced renal damage. Nevertheless, AKI and CKD contribute to an increased morbidity in these patients. Thus, careful monitoring of renal function is required, and adequate measures are crucial to prevent AKI in these patients, as AKI is a major contributor to the relatively high risk of CKD in IM patients. In contrast, the peculiar pattern of acute vascular damage we identified is most probably directly related to IM and is part of the spectrum of renal diseases associated with these disorders.
Abbreviations
- ANCA
antineutrophil cytoplasmic antibodies
- AKI
acute kidney injury
- AS
antisynthetase syndrome
- ATN
acute tubular necrosis
- CKD
chronic kidney disease
- CPK
creatine phosphokinase
- DM
dermatomyositis
- eGFR
estimated glomerular filtration rate
- ESRD
end-stage renal disease
- IgAN
IgA nephropathy
- IM
inflammatory myopathies
- IVIg
intravenous immunoglobulins
- MAC
membrane attack complex
- MN
membranous nephropathy
- NGAL
neutrophil gelatinase-associated lipocalin
- NSIP
nonspecific interstitial pneumonia
- PM
polymyositis
- ROC
receiver operating characteristic
- SCr
serum creatinine
Footnotes
Financial support and conflicts of interest: F. Fakhouri is a member of Alexion Pharmaceutical’s national advisory board. The remaining authors have no declared funding or conflict of interest.
REFERENCES
- 1.Akashi Y, Inoh M, Gamo N, Kinashi M, Ohbayashi S, Miyake H, Kurata N. Dermatomyositis associated with membranous nephropathy in a 43-year-old female. Am J Nephrol. 2002;22:385–388. [DOI] [PubMed] [Google Scholar]
- 2.Benveniste O, Dubourg O, Herson S. [New classifications and pathophysiology of the inflammatory myopathies]. Rev Med Interne. 2007;28:603–612. [DOI] [PubMed] [Google Scholar]
- 3.Chiu KC, Tsai TC, Lin WT, Lee WJ, Su CC, Chen CY. Paraneoplastic polymyositis associated with crescentic glomerulonephritis. Ren Fail. 2008;30:939–942. [DOI] [PubMed] [Google Scholar]
- 4.Civilibal M, Selcuk Duru N, Ozagari A, Durali K, Elevli M. Immunoglobulin A nephropathy associated with juvenile dermatomyositis. Pediatr Nephrol. 2009;24:2073–2075. [DOI] [PubMed] [Google Scholar]
- 5.Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971–982. [DOI] [PubMed] [Google Scholar]
- 6.Das J, George P, Pawar B, Calton N. Acute interstitial nephritis in association with polymyositis. J Postgrad Med. 2008;54:170–171. [DOI] [PubMed] [Google Scholar]
- 7.Fakhouri F, Fremeaux-Bacchi V. Does hemolytic uremic syndrome differ from thrombotic thrombocytopenic purpura? Nat Clin Pract Nephrol. 2007;3:679–687. [DOI] [PubMed] [Google Scholar]
- 8.Fathi M, Lundberg IE, Tornling G. Pulmonary complications of polymyositis and dermatomyositis. Semin Respir Crit Care Med. 2007;28:451–458. [DOI] [PubMed] [Google Scholar]
- 9.Frost NA, Morand EF, Hall CL, Maddison PJ, Bhalla AK. Idiopathic polymyositis complicated by arthritis and mesangial proliferative glomerulonephritis: case report and review of the literature. Br J Rheumatol. 1993;32:929–931. [DOI] [PubMed] [Google Scholar]
- 10.Guillevin L, Berezne A, Seror R, Teixeira L, Pourrat J, Mahr A, Hachulla E, Agard C, Cabane J, Vanhille P, Harle JR, Deleveaux I, Mouthon L. Scleroderma renal crisis: a retrospective multicentre study on 91 patients and 427 controls. Rheumatology (Oxford). 2012;51:460–467. [DOI] [PubMed] [Google Scholar]
- 11.Heagerty PJ, Lumley T, Pepe MS. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics. 2000;56:337–344. [DOI] [PubMed] [Google Scholar]
- 12.Hoogendijk JE, Amato AA, Lecky BR, Choy EH, Lundberg IE, Rose MR, Vencovsky J, de Visser M, Hughes RA. 119th ENMC International Workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10–12 October 2003, Naarden, The Netherlands. Neuromuscul Disord. 2004;14:337–345. [DOI] [PubMed] [Google Scholar]
- 13.Imbert-Masseau A, Hamidou M, Agard C, Grolleau JY, Cherin P. Antisynthetase syndrome. Joint Bone Spine. 2003;70:161–168. [DOI] [PubMed] [Google Scholar]
- 14.Kamata K, Kobayashi Y, Shigematsu H, Saito T. Childhood type polymyositis and rapidly progressive glomerulonephritis. Acta Pathol Jpn. 1982;32:801–806. [DOI] [PubMed] [Google Scholar]
- 15.Kaneoka H, Sasatomi Y, Miyagi K, Kiyoshi Y, Takeda S, Takebayashi S, Naito S, Saito T. A rare case of dermatomyositis associated with immune-complex type glomerulonephritis, idiopathic thrombopenic purpura, pulmonary fibrosis and lung cancer. Clin Exp Rheumatol. 2003;21:801–802. [PubMed] [Google Scholar]
- 16.Kissel JT, Mendell JR, Rammohan KW. Microvascular deposition of complement membrane attack complex in dermatomyositis. N Engl J Med. 1986;314:329–334. [DOI] [PubMed] [Google Scholar]
- 17.Lundberg IE. Cardiac involvement in autoimmune myositis and mixed connective tissue disease. Lupus. 2005;14:708–712. [DOI] [PubMed] [Google Scholar]
- 18.MacDiarmid SA, McIntyre WJ, Anthony A, Bailey RR, Turner JG, Arnold EP. Monitoring of renal function in patients with spinal cord injury. BJU Int. 2000;85:1014–1018. [DOI] [PubMed] [Google Scholar]
- 19.Mammen AL. Dermatomyositis and polymyositis: clinical presentation, autoantibodies, and pathogenesis. Ann N Y Acad Sci. 2010;1184:134–153. [DOI] [PubMed] [Google Scholar]
- 20.Mishra J, Ma Q, Prada A, Mitsnefes M, Zahedi K, Yang J, Barasch J, Devarajan P. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol. 2003;14:2534–2543. [DOI] [PubMed] [Google Scholar]
- 21.Moutsopoulos H, Fye KH. Letter: lipoid nephrosis and focal glomerulosclerosis in a patient with polymyositis. Lancet. 1975;1:1039. [DOI] [PubMed] [Google Scholar]
- 22.Nickavar A, Mehr Azma M. Nephrotic syndrome and juvenile dermatomyositis. Rheumatol Int. 2012;32:2933–2935. [DOI] [PubMed] [Google Scholar]
- 23.Orbach H, Katz U, Sherer Y, Shoenfeld Y. Intravenous immunoglobulin: adverse effects and safe administration. Clin Rev Allergy Immunol. 2005;29:173–184. [DOI] [PubMed] [Google Scholar]
- 24.Sigdel TK, Woo SH, Dai H, Khatri P, Li L, Myers B, Sarwal MM, Lafayette RA. Profiling of autoantibodies in IgA nephropathy, an integrative antibiomics approach. Clin J Am Soc Nephrol. 2011;6:2775–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takizawa Y, Kanda H, Sato K, Kawahata K, Yamaguchi A, Uozaki H, Shimizu J, Tsuji S, Misaki Y, Yamamoto K. Polymyositis associated with focal mesangial proliferative glomerulonephritis with depositions of immune complexes. Clin Rheumatol. 2007;26:792–796. [DOI] [PubMed] [Google Scholar]
- 26.Tsunemi M, Ishimura E, Tsumura K, Shoji S, Sugimura T, Nishizawa Y, Morii H. A case of crescentic glomerulonephritis associated with polymyositis. Nephron. 1993;64:488–489. [DOI] [PubMed] [Google Scholar]
- 27.Valenzuela OF, Reiser IW, Porush JG. Idiopathic polymyositis and glomerulonephritis. J Nephrol. 2001;14:120–124. [PubMed] [Google Scholar]
- 28.Vilppula AH, Aine RA. Polymyositis associated with several immunological disorders. Clin Rheumatol. 1984;3:533–539. [DOI] [PubMed] [Google Scholar]
- 29.Xie Q, Liu Y, Liu G, Yang N, Yin G. Diffuse proliferative glomerulonephritis associated with dermatomyositis with nephrotic syndrome. Rheumatol Int. 2010;30:821–825. [DOI] [PubMed] [Google Scholar]
- 30.Yahiaoui Y, Jablonski M, Hubert D, Mosnier-Pudar H, Noel LH, Stern M, Grenet D, Grunfeld JP, Chauveau D, Fakhouri F. Renal involvement in cystic fibrosis: diseases spectrum and clinical relevance. Clin J Am Soc Nephrol. 2009;4:921–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yen TH, Huang JY, Chen CY. Unexpected IgA nephropathy during the treatment of a young woman with idiopathic dermatomyositis: case report and review of the literature. J Nephrol. 2003;16:148–153. [PubMed] [Google Scholar]
- 32.Yen TH, Lai PC, Chen CC, Hsueh S, Huang JY. Renal involvement in patients with polymyositis and dermatomyositis. Int J Clin Pract. 2005;59:188–193. [DOI] [PubMed] [Google Scholar]
- 33.Zager RA. Rhabdomyolysis and myohemoglobinuric acute renal failure. Kidney Int. 1996;49:314–326. [DOI] [PubMed] [Google Scholar]