

Supplemental Digital Content is available in the text
Abstract
Proteolipid protein 2 (PLP2) has been shown to be upregulated in several cancers, including breast cancer, hepatocellular carcinoma, osteosarcoma, and melanoma. PLP2 specifically binds to phosphatidylinositol 3 kinase to activate the protein kinase B pathway to enhance cell proliferation, adhesion, and invasion in melanoma cells. Therefore, we speculated that PLP2 exhibits oncogenic potential. However, the regulatory mechanisms of PLP2 in cancer cells remain unclear.
Herein, we found that microRNA (miR)-664 expression was significantly downregulated in cutaneous malignant melanoma (CMM) cells and tissues compared with normal human melanocytes and benign melanocytic naevi. MiR-664 expression level was significantly correlated with patient survival. Ectopic expression of miR-664 reduced CMM cell proliferation and anchorage-independent growth, whereas the inhibition of miR-664 induced these effects. Furthermore, inhibition of miR-664 in CMM cells resulted in modulation of their entry into the G1/S transitional phase, which was caused by downregulation of the cyclin-dependent kinase inhibitor P21 and upregulation of the cell-cycle regulator cyclin D1. Moreover, we demonstrated that miR-664 downregulated PLP2 expression by directly targeting the PLP2 untranslated region.
Taken together, our results suggest that miR-664 may play an important role in suppressing proliferation of CMM cells and present a novel mechanism of miR-mediated direct suppression of PLP2 expression in cancer cells.
INTRODUCTION
Cutaneous malignant melanoma (CMM) is a highly aggressive skin cancer with rapidly increasing incidence worldwide; the 5-year survival of metastatic melanoma patients does not exceed 10% to 15%.1–8 As the curative effects of conventional treatments, including ionizing radiation, systemic chemotherapy, and immunotherapy, are quite limited for the advanced stages of melanoma, a wide surgical excision in the primary stages remains the main therapeutic option for the disease.9–11 Rapid progression of melanoma tumors is a major obstacle; therefore, early effective diagnosis and curative therapeutic intervention would be of great clinical interest for melanoma therapy.8,9,12 Recent discoveries in cell signaling led to the rapid development of targeted drugs and new therapeutic approaches for melanoma treatment, but the effects have been limited.13,14 Therefore, new indicators of prognosis and therapeutic targets are demanded.
Growing evidence indicates that an aberrant phosphatidylinositol 3 kinase (PI3K)/protein kinase B (AKT) pathway is frequently activated in melanomas.15–21 Dai et al22 demonstrated that activated AKT expression was significantly associated with the progression of melanoma and poorer patient survival. Stahl et al17 reported that the loss or haploinsufficiency of the phosphatase and tensin homolog (PTEN) gene caused the deregulation of AKT in 43% to 60% of melanomas.17 Activation of the PI3K/AKT pathway can downregulate the cyclin-dependent kinase (CDK) inhibitors p21 and p27, and stabilize the cell-cycle regulator cyclin D1/2, consequently leading to increased cell proliferation.23–29 In the recent decades, a 4-transmembrane protein, proteolipid protein 2 (PLP2), has been reported to be upregulated in various cancers, such as breast cancer, hepatocellular carcinoma, and osteosarcoma.30–32 Additionally, PLP2 was found to induce cell proliferation, adhesion, and invasion by specifically binding to PI3K to activate the AKT pathway in melanoma cells, but the knowledge of the PLP2 regulatory mechanism remains unclear.33,34
MicroRNAs (miRs) are a class of short (approximately 22 nucleotides) noncoding single-stranded ribonucleic acids that posttranscriptionally regulate gene expression by binding to the untranslated region (3′UTR) of a target messenger RNA (mRNA) molecule.35,36 Numerous recent studies have proved that miRs are involved in the initiation, development, and progression of melanoma.37–40 MiR-664 has been implicated in several cancers, including hepatocellular carcinoma, prolactinoma, and papillary thyroid carcinoma.41–43
In the present work, we observed that miR-664 suppressed CMM by directly targeting the 3′UTR of PLP2 RNA, which consequently led to inhibition of the PI3K/AKT pathway and decreased proliferation. Therefore, our results suggest that downregulated expression of miR-664 plays an important role in the development and progression of CMM.
MATERIALS AND METHODS
Ethics Statement
This study was approved by the Institutional Review Board of Nanfang Hospital, Guangzhou, China. The 9 melanoma and 2 BMN tissues used in this study were obtained from patients who underwent radical operations at the Affiliated Hospital of Southern Medical University.
Cell Culture
Melanoma cell lines, including A375.S2, A7, MeWo, RPMI-7951, SK-MEL-5, SK-MEL-24, and SK-MEL-28, were maintained in Dulbecco's modification of Eagle's medium Dulbecco medium (Invitrogen, Carlsbad, CA, USA, 12430054) supplemented with 10% fetal bovine serum (HyClone, Logan, UT, USA, SH30370.03) and 1% penicillin/streptomycin (Invitrogen, 15140163). In addition, the above culture conditions were used to culture the primary epidermal melanocyte (PEM) cell line (ATCC, American Type Culture Collection, USA PCS-200-013).
Plasmids and Transfection
The full-length sequence of the PLP2 3′UTR is 420 base pairs (bp) and contains 2 conserved miR-664-binding sites (REs), including RE #1 from 368 to 375 bp, and RE #2 from 400 to 407 bp. The human PLP2 3′UTR was amplified by polymerase chain reaction (PCR) from A375 cells and cloned into the XbaI sites of the pGL3-control luciferase reporter plasmid (Promega, Madison, WI, USA, E1741) and the pGFP-C3 plasmid (Clontech, Mountain View CA, USA, 632370). The primers selected are as follows: Homo-pGL3-PLP2-3′UTR-F-XbaI: 5′-AGATCGCCGTGTAATTCTAGACAATAGGAGACACCAGTTCTGACTG-3′; Homo-pGL3-PLP2-3′UTR-R-XbaI: 5′-GCCGGCCGCCCCGACTCTAGATTTGATGAAAGGATTACTTTATTCATTATC-3’; PLP2-3′UTR-F-mut1: 5′-TATAGAGGGATAAATAGCCAATAAACATTGTTAAAA-3′; PLP2-3′UTR-R-mut1: 5′-TTTTAACAATGTTTATTGGCTATTTATCCCTCTATA-3′; PLP2-3′UTR-F-mut2: 5′-AAAATATACGATAATAGCCAAAGTAATCCTTTCA-3′; PLP2-3′UTR-R-mut2: 5′-TGAAAGGATTACTTTGGCTATTATCGTATATTTT-3′; Homo-pGFP-C3-PLP2-3′UTR-F-250-EcoRI: 5′-CCGGAATTCTGTGCCTAGGTCCTCCTTCT-3′; Homo-pGFP-C3-PLP2-3′UTR-R-250-BamHI: 5′-CGCGGATCCGAAAGGATTACTTTATTCATTATCG-3’. The miR-664 mimics, negative control (NC), and anti-miR-664 inhibitor were purchased from RiboBio Co., Ltd, Guangzhou, GD, CN Transfection of the miR and miR inhibitor was performed using the Lipofectamine 2000 reagent (Invitrogen, 11668019) according to the manufacturer's instructions.
Western Blot
Cells were lysed in sample buffer [62.5 mmol/L Tris-HCl pH 6.8, 10% glycerol, 2% sodium dodecyl sulfate (SDS)] and boiled for 5 minutes. Equal amounts of lysate (50 μg total protein) were electrophoretically separated on 10% SDS/polyacrylamide gels and transferred onto nitrocellulose filter membranes (PALL, Boston, MA, USA, 66485) followed by incubation with a 1:1000-diluted anti-PLP2 antibody (Abcam, ab180131), anti-AKT phosphorylation (p-AKT) (CST, Danvers, MA, USA, 13038), anti-AKT (CST, 4685), anti-p21 (CST, 2947), anti-cyclin D (CST, 2978), and anti-GFP antibodies (CST, 2956). Proteins were detected by incubation with horseradish peroxidase-conjugated donkey anti-rabbit immunoglobulin G (1:3000) and enhanced by chemiluminescence (Pierce, Rockford, IL, USA, 32106). The membranes were stripped and reprobed with an anti-β-actin mouse monoclonal antibody (1:1000; Boster, Wuhan, CN, BM0627), which was used as a loading control.
MiRNA Extraction and Real-Time Quantitative PCR
Total miRNA from cultured cells and fresh surgical melanoma cancer tissues was extracted using the mirVana miRNA Isolation Kit (Ambion, Austin, TX, USA, AM1560) according to the manufacturer's instructions. The expression levels of miR-664 were quantified using miRNA-specific TaqMan MiRNA Assay Kits (Applied Biosystems, Foster, CA, USA, 4364031). Real-time PCR was performed using the Applied Biosystems 7500 Sequence Detection system. The expression of miRNA was defined based on the threshold cycle (Ct), wherein Ct represents the threshold cycle for each transcript. Relative expression levels were calculated as 2-(Ct of miR-664)—(Ct of U6) after normalization with reference to expression of U6 small nuclear RNA.
3-(4, 5-Dimethyl-2-Thiazolyl)-2, 5-Diphenyl-2H-Tetrazolium Bromide (MTT) Assay
Cells were stained with sterile MTT dye (0.5 mg/mL; Sigma, Saint Louis, MO, USA, 03285) for 4 hours at 37°C at each indicated time point, followed by addition of dimethyl sulfoxide (Sigma, D2650). Absorbance was measured at 570 nm, with 655 nm as the reference wavelength. All experiments were performed in triplicates.
Anchorage-Independent Growth Ability Assay
Mix the cells (5 × 102) and 2 mL complete medium along with 0.3% agar (Sigma, A1296) to an agar-cell mixture, seed this agar-cell mixture on the top of a bottom layer of 1% complete medium agar, and culture for 10 days. Viable colonies that contained >50 cells or were >0.1 mm were counted. Colony size was measured with an ocular micrometer. All experiments were performed in triplicates.
Colony Formation Assays
Cells were trypsinized and seeded on 6-well plates (500 cells per well) and cultured for 10 days. Colonies were fixed with formaldehyde (10%) for 5 minutes followed by staining with crystal violet (1.0%) for 30 seconds.
Bromodeoxyuridine Labeling and Immunofluorescence
Cells grown on coverslips (Fisher, Waltham, MA, USA, 12-545-87) were incubated with bromodeoxyuridine (BrdU) for 1 hour and stained with an anti-BrdU antibody (Abcam, ab1893) according to the manufacturer's instructions. Gray-level images were acquired under a laser-scanning microscope (Axioskop 2 plus; Carl Zeiss Co. Ltd. Oberkochen, BW, DE).
Tumor Xenografts
Nude mice were randomly divided into 2 groups (n = 5 per group), and indicated CMM cells were inoculated into the inguinal folds of individual mice. Tumor volume was determined using an external caliper and calculated using the equation (L × W2)/2. The mice were sacrificed 31 days after inoculation and the tumors were excised.
Luciferase Assays
A total of 100 ng of pGL3-PLP2-3′UTR (wt/mu), or the control luciferase plasmid, along with 1 ng of pRL-TK renilla plasmid (Promega, E2241) were transfected into melanoma cells for 48 hours. Cells were lysed and luciferase activity was measured using the Dual Luciferase Reporter Assay Kit (Promega, E1960) according to the manufacturer's protocol. Three independent experiments were performed. The data are presented as mean ± standard deviation.
Flow Cytometry Analysis
Cells in ice-cold phosphate buffer saline were fixed by 100% ice-cold ethanol followed by treatment with RNAase (Sigma-Aldrich, 83931) at a final concentration of 2 mg/mL, and subsequently incubated at 37°C for 30 minutes followed by incubation in 20 mg/mL of propidium iodide (Sigma-Aldrich, P4170) for 20 minutes at room temperature. Then, 50,000 cells were analyzed using a flow cytometer (FACSCalibur; BD Biosciences, San Diego, CA, USA).
Statistical Analysis
Student t test was used to evaluate the significant difference of 2 groups of data in all pertinent experiments. P value <0.05 (using a 2-tailed paired t test) was considered statistically significant.
RESULTS
Downregulation of MiR-664 Correlates With CMM Progression
We observed that miR-664 expression was significantly decreased in CMM tissues compared with normal human melanocytes (NHMs) and benign melanocytic naevi (BMN) (Figure 1A). To confirm this observation, real-time PCR analyses was performed that showed decreased expression of miR-664 in CMM cell lines, including A375.S2, A7, MeWo, RPMI-7951, SK-MEL-5, SK-MEL-24, and SK-MEL-28 cells, compared with NHM tissues and a PEMs cell line (Figure 1B). Furthermore, the downregulation of miR-664 has been identified in a previously published microarray-based high-throughput assessment of CMM cells (NCBI/GEO/GSE34460; Figure 1C). Additionally, the expression of miR-664 significantly correlated with CMM patient survival (P < 0.05; Figure 1D). We determined that the downregulated expression of miR-664 was closely associated with a shorter overall survival time (P < 0.05; Supplemental Table 1, http://links.lww.com/MD/A380), which suggests that low levels of miR-664 may contribute to the progression of CMM, and highlights the potential value of miR-664 as a predictive biomarker of disease outcome.
FIGURE 1.
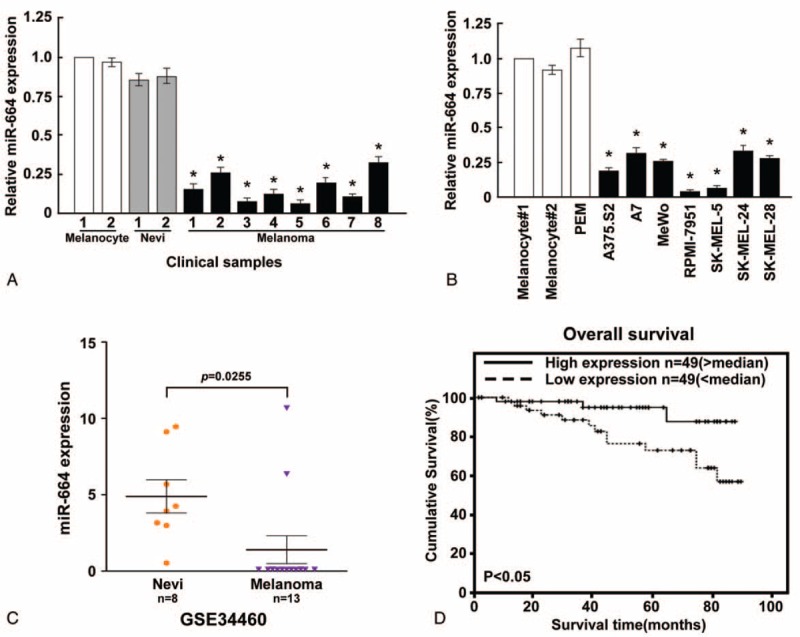
Expression analysis of microRNA (miR)-664 in CMM cell lines and tissues. (A) MiR-664 expression was examined in CMM, NHM, and BMN tissues. (B) Real-time PCR analysis of miR-664 expression in NHM tissues and CMM cell lines, including A375.S2, A7, MeWo, RPMI-7951, SK-MEL-5, SK-MEL-24, and SK-MEL-28. (C) Analysis of microarray data (accession number GSE334460) revealed miR-664 downregulation in 13 patients. (D) Correlation between miR-664 levels and survival by Kaplan–Meier analysis of patients with high (greater than the median; n = 49) or low miR-664 (less than the median; n = 49) expression. The average miR-664 expression was normalized by U6 expression. Each bar represents the mean of 3 independent experiments. ∗P < 0.05. BMN = benign melanocytic naevi, CMM = cutaneous malignant melanoma, NHM = normal human melanocytes, PCR = polymerase chain reaction.
MiR-664 Upregulation Reduced Proliferation and Tumorigenicity in CMM Cells
Next, we aimed to investigate the biological role of miR-664 in CMM development and progression; therefore, we transfected A375.S2 and SK-MEL-28 CMM cell lines with an hsa-miR-664 mimic oligonucleotide and assessed its effect on cell proliferation. We observed that miR-664 overexpression dramatically decreased the growth rate of A375.S2 and SK-MEL-28 cells compared with NC-transfected cells by using MTT and colony formation assays (Figure 2A and B). Furthermore, as indicated by a decrease in colony number and size, ectopically expressing miR-664 in both the CMM cell lines significantly reduced anchorage-independent growth ability (Figure 2C), suggesting that miR-664 upregulation could decrease the tumorigenicity of CMM cells in vitro. A BrdU incorporation assay was utilized to further explore the ability of miR-664 to reduce cell proliferation, and the results showed that the percentage of cells in S phase was dramatically decreased in A375.S2 (22.36%) and SK-MEL-28 (21.01%) cells overexpressing miR-664 compared with control cells A375.S2 cells (36.17%), SK-MEL-28 (34.61%); Figure 2D. Likewise, we further analyzed miR-664 overexpressing A375.S2 and SK-MEL-28 cells by flow cytometry. Our results showed a significant decrease in the percentage of cells in the S peak and an increase in the percentage of cells in the G1/G0 peak, which indicates that miR-664 induced G1/S arrest of CMM cells (Figure 2E). Together, our results suggest that a decline of CMM cell growth by miR-664 overexpression may be mediated through the regulation of cellular entry into the G1/S transitional phase.
FIGURE 2.
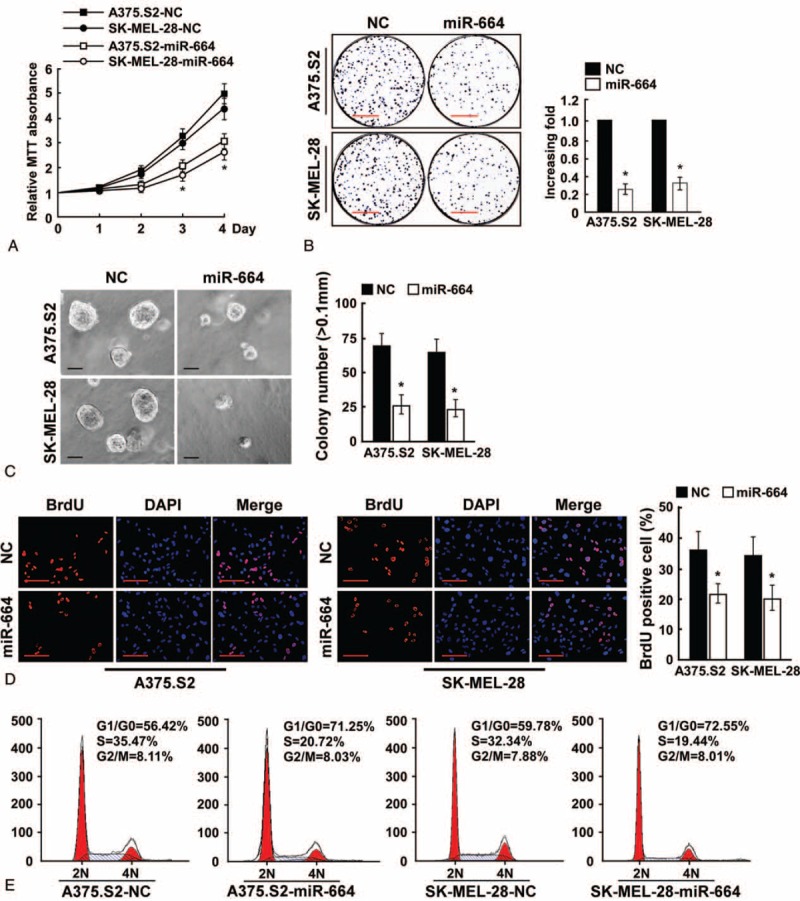
Upregulation of microRNA (miR)-664 suppresses the proliferation of CMM cells. (A) MTT assays revealed that upregulation of miR-664 reduced cell growth in CMM cell lines A375.S2 and SK-MEL-28. (B) Representative micrographs (left) and quantification (right) of crystal violet-stained cell colonies. Scale bars: 10 mm. (C) Upregulation of miR-664 reduced CMM cell tumorigenicity as determined by anchorage-independent growth assay. Representative micrographs (left) and quantification of colonies that were >0.1 mm (right) were scored. Scale bars: 100 μm. (D) Representative micrographs (left) and quantification of BrdU-incorporated cells after transfection with miR-664 or NC. Scale bars: 50 μm. (E) Flow cytometric analysis of indicated CMM cells after miR-664 overexpression. Each bar represents the mean of 3 independent experiments. ∗P < 0.05. BrdU = bromodeoxyuridine, CMM = cutaneous malignant melanoma, MTT = 3-(4, 5-dimethyl-2-thiazolyl)-2, 5-diphenyl-2H-tetrazolium bromide, NC = negative control.
Inhibition of MiR-664 Enhances the Growth Rate of CMM Cells
We further examined CMM cell proliferation after the downregulation of miR-664. As shown in Figure 3A and B, the growth rates of A375.S2 and SK-MEL-28 CMM cells following transfection with the hsa-miR-664 inhibitor increased compared with NC-transfected cells. In addition, inhibition of miR-664 in A375.S2 and SK-MEL-28 cells significantly induced anchorage-independent growth, which resulted in an increased number and size of colonies (Figure 3C). Moreover, we found that ectopic expression of the miR-664 inhibitor drastically decreased the percentage of CMM cells in the G0/G1 peak but increased the amount of cells in the S peak (Figure 3D and E).
FIGURE 3.
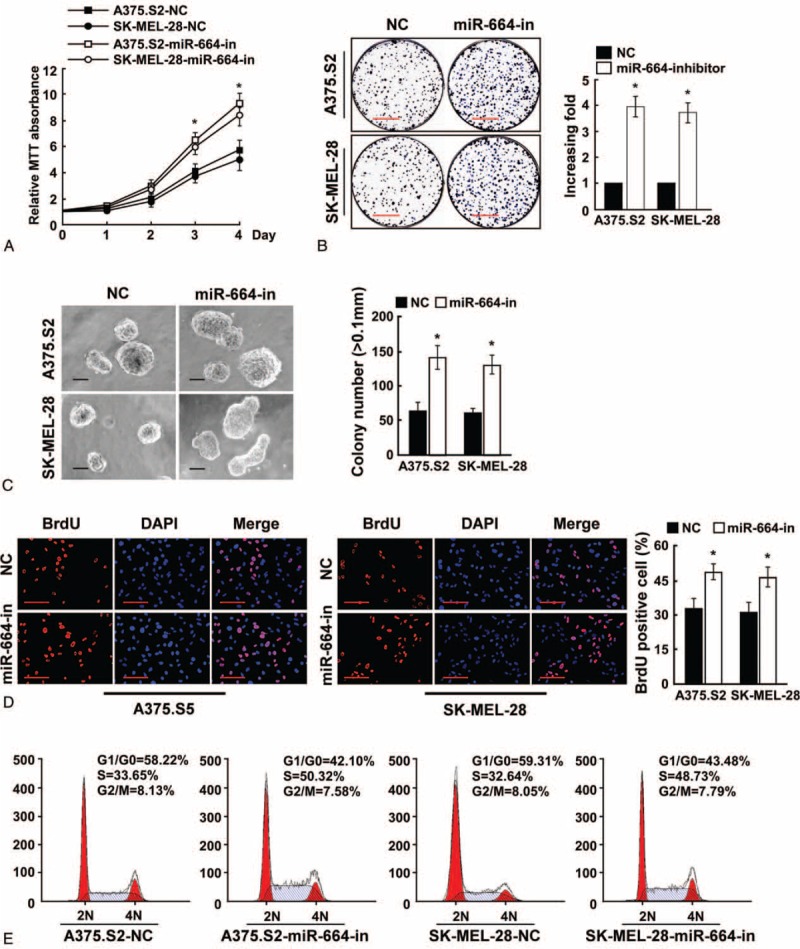
Inhibition of microRNA (miR)-664 promotes CMM cell proliferation. (A) MTT assays revealed that inhibition of miR-664 promoted cell growth of CMM cell lines A375.S2 and SK-MEL-28. (B) Representative micrographs (left) and quantification (right) of crystal violet-stained cell colonies. Scale bars: 10 mm. (C) Inhibition of miR-664 promoted the anchorage-independent growth of CMM cells. Representative micrographs (left) and quantification of colonies that were larger than 0.1 mm (right) were scored. Scale bars: 100 μm. (D) Representative micrographs (left) and quantification of BrdU-incorporated cells after transfection with miR-664 inhibitor or NC. Scale bars: 50 μm. (E) Flow cytometric analysis of indicated CMM cells after miR-664 inhibition. Each bar represents the mean of 3 independent experiments. ∗P < 0.05. BrdU = bromodeoxyuridine, CMM = cutaneous malignant melanoma, MTT = 3-(4, 5-dimethyl-2-thiazolyl)-2, 5-diphenyl-2H-tetrazolium bromide, NC = negative control.
MiR-664 Upregulation Suppresses Tumorigenicity of CMM Cells In Vivo
To begin to understand whether miR-664 is involved in the tumorigenesis of CMM cells in vivo, we subcutaneously inoculated CMM cells into the inguinal folds of nude mice. Tumors formed in nude mice by miR-664-transduced CMM cells were noticeably smaller than the vector control tumors (Figure 4A and B). Therefore, these results validate that reintroduction of miR-664 into CMM cells could suppress tumorigenic behavior in vivo.
FIGURE 4.
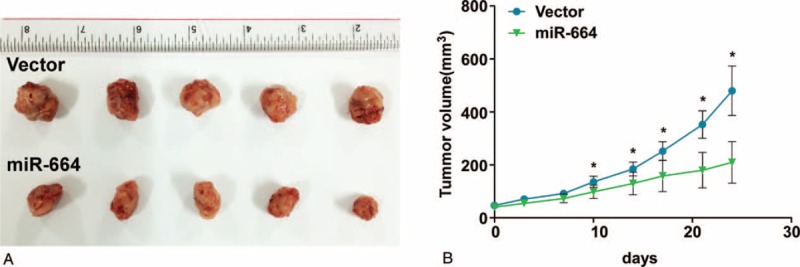
Upregulation of microRNA (miR)-664 augments tumorigenicity of CMM cells in vivo. Mice were injected subcutaneously with 106 cells/mouse. (A) Representative images of tumor growth in nude mice injected with A375.S2-Vector (control) and A375.S2-miR-664 cells. (B) Tumor volumes were measured and plotted as the mean ± standard error of the mean. Each bar represents the mean ± standard deviation of 3 independent experiments. ∗P < 0.05. CMM = cutaneous malignant melanoma.
PLP2 Is a Direct Target of MiR-664 in CMM Cells
Western blot analysis revealed that the proliferative marker, proliferating cell nuclear antigen (PCNA), decreased in miR-664-transfected CMM cells and increased in miR-664-inhibited cells, further demonstrating that miR-664 plays an important role in the proliferation of CMM cells (Figure 5B). Importantly, analysis using publicly available algorithms (TargetScan, Pictar, and miRANDA) indicated that the expression of miR-664 was inversely correlated with the expression of PLP2, a proliferative protein in CMM. Furthermore, the ectopic expression of miR-664 in CMM cells decreased PLP2 protein expression and p-AKT, a downstream target of PLP2; however, the inhibition of miR-664 showed the contradictory results (Figure 5B). Additionally, we subcloned the 3′UTR of PLP2 that contained 2 miR-664-binding sites (REs; Figure 5A) into the pEGFP-C3 and pGL3 dual luciferase reporter vectors. As shown in Figure 5C, ectopic expression of miR-664 in both A375.S2 and SK-MEL-28 CMM cells dramatically inhibited green fluorescent protein (GFP) protein expression, but not GFP-γ-tubulin expression, which was used as a control for transfection efficiency, suggesting that miR-664 specifically affects the PLP2 3′UTR. Meanwhile, we observed a consistent and dose-dependent reduction of luciferase activity in both post-transfected miR-664 melanoma cell lines, which was abolished by the addition of the miR-664 inhibitor (Figure 5D). Moreover, point mutations in the tentative miR-664-binding sites abrogated the suppressive effect of PLP2 mediated by miR-664 (Figure 5E). In addition, statistical analysis demonstrated that miR-664 expression was inversely correlated with the expression of PLP2 (r = −0.889, P < 0.001; Figure 5F). Therefore, our results determine that PLP2 is a bona fide target of miR-664.
FIGURE 5.
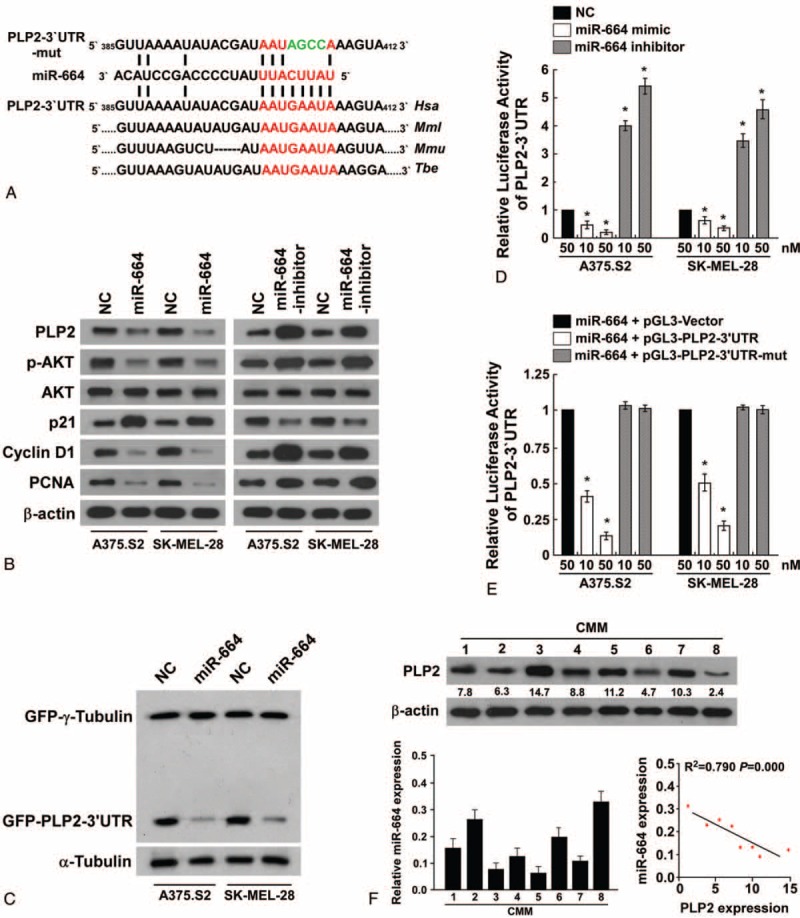
MicroRNA (MiR)-664 suppresses PLP2 expression via direct targeting of the 3′UTR. (A) Predicted miR-664 target sequence in the 3′UTR of PLP2 (PLP2-3′UTR) and PLP2-3′UTR mutant containing 4 altered nucleotides in the seed sequence (PLP2-3′UTR-mut). (B) Western blot analysis of PLP2, p-AKT, AKT, p21, cyclin D1, and PCNA expression by miR-664 overexpression or miR-664 inhibition. β-actin served as the loading control. (C) Western blot analysis of GFP expression in indicated cells. (D) Luciferase assay of indicated cells transfected with the pGL3-PLP2-3′UTR reporter with increasing amounts (10, 50 nM) of miR-664 mimic or miR-664 inhibitor. (E) Luciferase assay of indicated cells transfected with pGL3-PLP2-3′UTR or pGL3-PLP2-3′UTR-mut reporter with increasing amounts (10, 50 nM) of miR-664 oligonucleotides. (F) Analysis of expression (up) and correlation (down) of miR-664 and PLP2 expression in 8 freshly collected human CMM tissues. Each bar represents the mean ± standard deviation of 3 independent experiments. ∗P < 0.05. 3′UTR = untranslated region, AKT = protein kinase B, CMM = cutaneous malignant melanoma, p-AKT = AKT phosphorylation, PLP2 = proteolipid protein 2.
PLP2 Plays an Important Role in MiR-664-Suppressed Proliferation of Melanoma Cells
In the attempt to understand the role of PLP2 in miR-664-suppressed proliferation, we subcloned PLP2 with its 3′UTR (PLP2 w/3′UTR) or without its 3′UTR (PLP2 w/out 3′UTR) into the pSin vector. Our results showed that cotransfection of miR-664 and PLP2 w/out 3′UTR significantly enhanced PLP2 protein expression and the growth rate of A375.S2 and SK-MEL-28 melanoma cell lines (Figure 6A and B). However, the combination of PLP2 w/3′UTR and miR-664 in melanoma cells exhibited no obvious effect on PLP2 protein expression and the growth rate over that of CMM cells transfected only with miR-664 (Figure 6A and B). Furthermore, flow cytometry showed that the overexpression of miR-664 and PLP2 w/out 3′UTR increased the percentage of cells in S phase and significantly decreased the percentage of cells in G1/G0. However, the overexpression of miR-664 and PLP2 w/3′UTR did not increase the percentage of cells in S phase compared to CMM cells only transfected with miR-664 alone (Figure 6C). Taken together, these results demonstrate that miR-664 inhibits proliferation of melanoma cells through suppression of PLP2.
FIGURE 6.
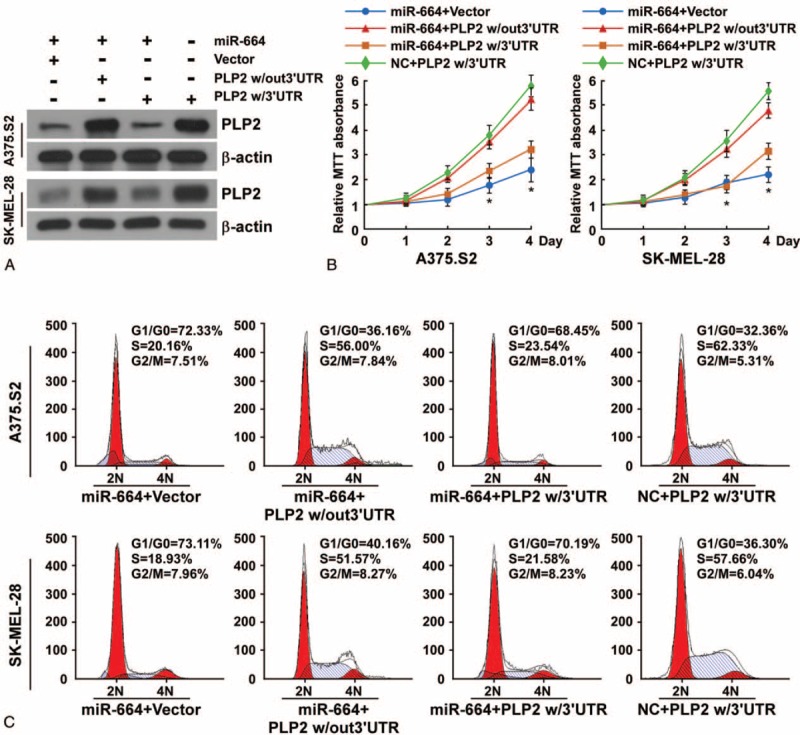
MicroRNA (MiR)-664 inhibits CMM cell proliferation through direct targeting of PLP2. (A) Western blot analysis of PLP2 expression in indicated cells. (B) MTT assays of indicated melanoma cells. (C) Flow cytometric analysis of indicated melanoma cells. CMM = cutaneous malignant melanoma, MTT = 3-(4, 5-dimethyl-2-thiazolyl)-2, 5-diphenyl-2H-tetrazolium bromide, PLP2 = proteolipid protein 2.
DISCUSSION
The current study has shown that the expression of miR-664 was significantly downregulated in CMM cells and tissues compared with BMN tissues. Furthermore, the progression of CMM was correlated with miR-664 expression. Moreover, we observed that the ectopic expression of miR-664 could diminish melanoma cell proliferation and anchorage-independent growth, whereas miR-664 inhibition enhanced proliferation and anchorage-independent growth in vitro and in vivo. Furthermore, we determined that miR-664 overexpression led to the increase of CDK inhibitor p21 in melanoma cells. Additionally, miR-664 overexpression resulted in a decrease of the cell-cycle regulator cyclin D1 in melanoma cells. These events occurred because of the downregulation of PLP2 via miR-664 directly targeting the 3′UTR. These findings suggest that miR-664 deregulation might play an important role in promoting carcinogenesis and progression of melanoma.
Epidemiological studies show that the incidence of CMM has increased at an alarming rate. From 1970 to 2009, the incidence of melanoma increased by 400% among young men and 800% among young women.44 Furthermore, melanoma is 1 of only 3 cancers, along with liver and esophageal cancers, with an increasing mortality rate for men.6 Although both environmental and genetic factors are considered to be principal causes of melanoma cancer, the molecular pathogenesis of its development and progression remain unclear.45 Previous studies have demonstrated that the PI3K/AKT pathway is aberrantly regulated in approximately 70% of melanomas in vitro and in vivo.46 Furthermore, previous studies have shown that the loss of PTEN or loss-of-function mutations in PTEN result in PI3K/AKT pathway activation in melanoma cells.15,19 In addition, a previous study has shown that mutant B-Raf Proto-Oncogene, Serine/Threonine Kinase cooperates with the activation of the PI3K/AKT signaling pathway in melanomagenesis.47 Recently, Ye et al48 reported that decreased expression of PIB5PA contributed to increased activation of the PI3K/AKT pathway in melanoma cells. PLP2 is an integral membrane protein that can multimerize and may function as an ion channel. PLP2 has been reported to contribute to melanoma cell proliferation, adhesion, and invasion by binding specifically to PI3K to activate the AKT pathway.33,34
MiRNAs are a group of small noncoding RNAs that negatively regulate gene expression by binding to 3′UTR regions of target mRNAs to repress protein translation and induce mRNA degradation. MiRNAs play pivotal roles in multiple biological processes, such as cellular differentiation, proliferation, oncogenesis, angiogenesis, invasion, and metastasis and can function as either tumor suppressors or oncogenes.35–40 In the current study, we found that the downregulation of miR-664 may correlate with clinical CMM progression and could possibly function as a tumor-suppressor miRNA. Interestingly, expression level of miR-664 is frequently deregulated in various human tumor types, including hepatocellular carcinoma, prolactinoma, and papillary thyroid carcinoma.41–43
We have indicated that the PLP2 gene is a theoretical target gene of miR-664 through bioinformatics analysis, and we determined that PLP2 was a bona fide target of miR-664 by triplicate experiments. We showed through Western blot analysis that overexpression of miR-664 resulted in downregulation of PLP2 protein. We also determined that downstream targets of PLP2, including p21, were significantly upregulated, whereas cyclin D1 was downregulated in miR-664-transfected CMM cells. Furthermore, we demonstrated that the downregulation of PLP2 was mediated by miR-664 through binding of the PLP2 3′UTR. Moreover, transfection with PLP2 w/out the 3′UTR significantly abrogated the miR-664-suppressed proliferation, but ectopic expression of PLP2 w/3′UTR only partially attenuated the decrease in proliferation by miR-664 overexpression, suggesting that the effect of miR-664 on proliferation of CMM cells may be through downregulation of PLP2 via direct targeting of the PLP2 3′UTR. Therefore, the biological function of miR-664 in triggering the suppression of melanoma cell proliferation, by way of PLP2 function, is currently under investigation in our laboratory.
Sustaining proliferative signaling is one of the hallmarks of cancer.49 A growing number of miRNAs are reported to facilitate melanoma proliferation through regulation of the PI3K/AKT pathway. Restoration of miR-205 expression decreases AKT phosphorylation leading to decreased melanoma proliferation in vitro and in vivo.50 Furthermore, ectopic expression of miR-221/222 in melanoma cells yielded an increased proliferation rate by binding to the 3′UTR of PTEN.51 Our study enriched these findings.
In summary, the current study provides, for the first time, an important link between miR-664-mediated proliferation of CMM cells and downregulation of PLP2. Our findings suggest an essential role for miR-664 in the regulation of CMM cell growth. Understanding the precise role played by miR-664 in CMM progression will not only increase our knowledge of tumor biology, but its inhibition may also allow development of novel therapeutic strategies.
Footnotes
Abbreviations: 3′UTR = untranslated region, AKT = protein kinase B, BMN = benign melanocytic naevi, BrdU = bromodeoxyuridine, CDK = cyclin-dependent kinase, CMM = cutaneous malignant melanoma, miR-664 = microRNA-664, MTT = 3-(4, 5-dimethyl-2-thiazolyl)-2, 5-diphenyl-2H-tetrazolium bromide, NC = negative control, NHMs = normal human melanocytes, PI3K = phosphatidylinositol 3 kinase, PLP2 = proteolipid protein 2, PLP2 w/3′UTR = PLP2 with 3′UTR, PLP2 w/out 3′UTR = PLP2 without 3′UTR, 3′UTR = untranslated region.
ZD, SJ, and XP have equally contributed to this work.
This work was supported by grants from the National Natural Science Foundation of China (81172634, 81202152, and 81472922), the School of Public Health and Tropical Medicine of Southern Medical University, China (GW201314, GW201418), the Guangdong Natural Science Foundation of China (S2013040015308), and the Science and Technology Projects Foundation of Guangzhou City (2013J4200014).
The authors have no conflicts of interest to disclose.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Website (www.md-journal.com).
REFERENCES
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA Cancer J Clin 2009; 59:225–249. [DOI] [PubMed] [Google Scholar]
- 2.Trotter SC, Sroa N, Winkelmann RR, et al. A global review of melanoma follow-up guidelines. J Clin Aesthet Dermatol 2013; 6:18–26. [PMC free article] [PubMed] [Google Scholar]
- 3.Tsao H, Atkins MB, Sober AJ. Management of cutaneous melanoma. N Engl J Med 2004; 351:998–1012. [DOI] [PubMed] [Google Scholar]
- 4.Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature 2007; 445:851–857. [DOI] [PubMed] [Google Scholar]
- 5.Garbe C, Leiter U. Melanoma epidemiology and trends. Clin Dermatol 2009; 27:3–9. [DOI] [PubMed] [Google Scholar]
- 6.Jemal A, Siegel R, Xu J, et al. Cancer statistics, 2010. CA Cancer J Clin 2010; 60:277–300. [DOI] [PubMed] [Google Scholar]
- 7.Balch CM, Gershenwald JE, Soong SJ, et al. Final version of the American Joint Committee on Cancer staging system for cutaneous melanoma. J Clin Oncol 2001; 19:3635–3648. [DOI] [PubMed] [Google Scholar]
- 8.Francken AB, Bastiaannet E, Hoekstra HJ. Follow-up in patients with localised primary cutaneous melanoma. Lancet Oncol 2005; 6:608–621. [DOI] [PubMed] [Google Scholar]
- 9.Dunki-Jacobs EM, Callender GG, McMasters KM. Current management of melanoma. Curr Probl Surg 2013; 50:351–382. [DOI] [PubMed] [Google Scholar]
- 10.Batus M1, Waheed S, Ruby C, et al. Optimal management of metastatic melanoma: current strategies and future directions. Am J Clin Dermatol 2013; 14:179–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bastiaannet E, Beukema JC, Hoekstra HJ. Radiation therapy following lymph node dissection in melanoma patients: treatment, outcome and complications. Cancer Treat Rev 2005; 31:18–26. [DOI] [PubMed] [Google Scholar]
- 12.Comis RL. DTIC (NSC-45388) in malignant melanoma: a perspective. Cancer Treat Rep 1976; 60:165–176. [PubMed] [Google Scholar]
- 13.Mimeault M, Batra SK. Novel biomarkers and therapeutic targets for optimizing the therapeutic management of melanomas. World J Clin Oncol 2012; 3:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scott AM, Allison JP, Wolchok JD. Monoclonal antibodies in cancer therapy. Cancer Immun 2012; 12:14. [PMC free article] [PubMed] [Google Scholar]
- 15.Madhunapantula SV, Robertson GP. The PTEN-AKT3 signaling cascade as a therapeutic target in melanoma. Pigment Cell Melanoma Res 2009; 22:400–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inamdar GS, Madhunapantula SV, Robertson GP. Targeting the MAPK pathway in melanoma: why some approaches succeed and other fail. Biochem Pharmacol 2010; 80:624–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stahl JM, Sharma A, Cheung M, et al. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res 2004; 64:7002–7010. [DOI] [PubMed] [Google Scholar]
- 18.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007; 129:1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stahl JM, Cheung M, Sharma A, et al. Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res 2003; 63:2881–2890. [PubMed] [Google Scholar]
- 20.Robertson GP. Functional and therapeutic significance of Akt deregulation in malignant melanoma. Cancer Metastasis Rev 2005; 24:273–285. [DOI] [PubMed] [Google Scholar]
- 21.Madhunapantula SV, Robertson GP. Is B-Raf a good therapeutic target for melanoma and other malignancies? Cancer Res 2008; 68:5–8. [DOI] [PubMed] [Google Scholar]
- 22.Dai DL, Martinka M, Li G. Prognostic significance of activated Akt expression in melanoma: a clinicopathologic study of 292 cases. J Clin Oncol 2005; 23:1473–1482. [DOI] [PubMed] [Google Scholar]
- 23.Lee JO, Yang H, Georgescu MM, et al. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 1999; 99:323–334. [DOI] [PubMed] [Google Scholar]
- 24.Maehama T, Dixon JE. PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol 1999; 9:125–128. [DOI] [PubMed] [Google Scholar]
- 25.Mikhail M, Velazquez E, Shapiro R, et al. PTEN expression in melanoma: relationship with patient survival, Bcl-2 expression, and proliferation. Clin Cancer Res 2005; 11:5153–5157. [DOI] [PubMed] [Google Scholar]
- 26.Mounir Z, Krishnamoorthy JL, Robertson GP, et al. Tumor suppression by PTEN requires the activation of the PKR-eIF2alpha phosphorylation pathway. Sci Signal 2009; 2:ra85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramaswamy S, Nakamura N, Vazquez F, et al. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci USA 1999; 96:2110–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu H, Goel V, Haluska FG. PTEN signaling pathways in melanoma. Oncogene 2003; 22:3113–3122. [DOI] [PubMed] [Google Scholar]
- 29.Huang W, Chang HY, Fei T, et al. GSK3 beta mediates suppression of cyclin D2 expression by tumor suppressor PTEN. Oncogene 2007; 26:2471–2482. [DOI] [PubMed] [Google Scholar]
- 30.Longo A, Librizzi M, Luparello C. Effect of transfection with PLP2 antisense oligonucleotides on gene expression of cadmium-treated MDA-MB231 breast cancer cells. Anal Bioanal Chem 2013; 405:1893–1901. [DOI] [PubMed] [Google Scholar]
- 31.Zimmerman JW, Pennison MJ, Brezovich I, et al. Cancer cell proliferation is inhibited by specific modulation frequencies. Br J Cancer 2012; 106:307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee SM, Shin H, Jang SW, et al. PLP2/A4 interacts with CCR1 and stimulates migration of CCR1-expressing HOS cells. Biochem Biophys Res Commun 2004; 324:768–772. [DOI] [PubMed] [Google Scholar]
- 33.Sonoda Y, Warita M, Suzuki T, et al. Proteolipid protein 2 is associated with melanoma metastasis. Oncol Rep 2010; 23:371–376. [PubMed] [Google Scholar]
- 34.Ozawa H, Sonoda Y, Suzuki T, et al. Knockdown of proteolipid protein 2 or focal adhesion kinase with an artificial microRNA reduces growth and metastasis of B16BL6 melanoma cells. Oncol Lett 2012; 3:19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116:281–297. [DOI] [PubMed] [Google Scholar]
- 36.Ambros V. The functions of animal microRNAs. Nature 2004; 431:350–355. [DOI] [PubMed] [Google Scholar]
- 37.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer 2006; 6:857–866. [DOI] [PubMed] [Google Scholar]
- 38.Gregory RI, Shiekhattar R. MicroRNA biogenesis and cancer. Cancer Res 2005; 65:3509–3512. [DOI] [PubMed] [Google Scholar]
- 39.Esquela-Kerscher A, Slack FJ. Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer 2006; 6:259–269. [DOI] [PubMed] [Google Scholar]
- 40.Zhou M, Liu W, Ma S, et al. A novel onco-miR-365 induces cutaneous squamous cell carcinoma. Carcinogenesis 2013; 34:1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Z, Zhang H, Zhang P, et al. Upregulation of miR-2861 and miR-451 expression in papillary thyroid carcinoma with lymph node metastasis. Med Oncol 2013; 30:577. [DOI] [PubMed] [Google Scholar]
- 42.Yang H, Cho ME, Li TW, et al. MicroRNAs regulate methionine adenosyltransferase 1A expression in hepatocellular carcinoma. J Clin Invest 2013; 123:285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen YX, Li Q, Wang CD, et al. Differential expression analysis of prolactinoma-related microRNAs. Zhonghua Yi Xue Za Zhi 2012; 92:320–323. [PubMed] [Google Scholar]
- 44.Reed KB, Brewer JD, Lohse CM, et al. Increasing incidence of melanoma among young adults: an epidemiological study in Olmsted County, Minnesota. Mayo Clin Proc 2012; 87:328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller AJ, Mihm MC., Jr Melanoma. N Engl J Med 2006; 355:51–65. [DOI] [PubMed] [Google Scholar]
- 46.Madhunapantula SV, Mosca PJ, Robertson GP. The Akt signaling pathway: an emerging therapeutic target in malignant melanoma. Cancer Biol Ther 2011; 12:1032–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen B, Tardell C, Higgins B, et al. BRAFV600E negatively regulates the AKT pathway in melanoma cell lines. PLoS One 2012; 7:e42598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ye Y, Li Q, Hu WL, et al. Loss of PI(4,5)P2 5-phosphatase A contributes to resistance of human melanoma cells to RAF/MEK inhibitors. Transl Oncol 2013; 6:470–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646–674. [DOI] [PubMed] [Google Scholar]
- 50.Dar AA, Majid S, de Semir D, et al. miRNA-205 suppresses melanoma cell proliferation and induces senescence via regulation of E2F1 protein. J Biol Chem 2011; 286:16606–16614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Felicetti F, Errico MC, Bottero L, et al. The promyelocytic leukemia zinc finger-microRNA-221/-222 pathway controls melanoma progression through multiple oncogenic mechanisms. Cancer Res 2008; 68:2745–2754. [DOI] [PubMed] [Google Scholar]