

Abstract
Although fructooligosaccharides (FOS) can selectively stimulate the growth and activity of probiotics and beneficially modulate the balance of intestinal microbiota, knowledge of the molecular mechanism for FOS metabolism by probiotics is still limited. Here a combined transcriptomic and physiological approach was used to survey the global alterations that occurred during the logarithmic growth of Lactobacillus plantarum ST-III using FOS or glucose as the sole carbon source. A total of 363 genes were differentially transcribed; in particular, two gene clusters were induced by FOS. Gene inactivation revealed that both of the clusters participated in the metabolism of FOS, which were transported across the membrane by two phosphotransferase systems (PTSs) and were subsequently hydrolyzed by a β-fructofuranosidase (SacA) in the cytoplasm. Combining the measurements of the transcriptome- and membrane-related features, we discovered that the genes involved in the biosynthesis of fatty acids (FAs) were repressed in cells grown on FOS; as a result, the FA profiles were altered by shortening of the carbon chains, after which membrane fluidity increased in response to FOS transport and utilization. Furthermore, incremental production of acetate was observed in both the transcriptomic and the metabolic experiments. Our results provided new insights into gene transcription, the production of metabolites, and membrane alterations that could explain FOS metabolism in L. plantarum.
INTRODUCTION
Prebiotics are defined as nondigestible food ingredients that selectively stimulate the growth and activity of beneficial microbial strains residing in the host gastrointestinal tract (GIT) (1). Among the sugars that are qualified as prebiotics, fructooligosaccharides (FOS) are fructose polymers of diverse lengths that can be either derivatives of simple fructose polymers or fructose moieties attached to a sucrose molecule (2). Because of the linkage configuration, FOS are not digested in the upper GIT and have been shown in vivo to beneficially modulate the composition of the intestinal microbiota by preferentially increasing the numbers of bifidobacteria and lactobacilli (3, 4).
Despite considerable commercial and research interest in the beneficial effects of FOS, the molecular basis of FOS metabolism by specific members of the intestinal microbiota has only recently been examined. In order to understand the influence of environmental conditions on genome-wide gene expression levels, whole-genome DNA microarrays have often been used to survey the gene expression patterns of strains in the presence and absence of oligosaccharides (2, 5–8). On the basis of in silico analysis of the Lactobacillus acidophilus NCFM genome sequence, Barrangou et al. (2) identified a multiple-sugar metabolism (msm) operon that was involved in the metabolism of FOS. The msm operon encodes an ATP-dependent binding cassette-type transport system and a cytoplasmic β-fructosidase, which mediates FOS uptake and intracellular hydrolysis. Moreover, expression of the operon was induced by sucrose and FOS but was repressed by glucose. Similarly, a microarray analysis of Lactobacillus paracasei 1195 grown on FOS revealed that the main FOS metabolic pathway was located in a fosABCDXE operon, which encoded a fructose/mannose phosphotransferase system (PTS) and a cell wall-associated β-fructosidase (8). The cell surface localization of the β-fructosidase encoded by fosE suggested that FOS are hydrolyzed extracellularly, followed by uptake of the hydrolysis products via the fructose/mannose-specific PTS (9).
In general, the cell membrane is recognized as the first barrier against environmental changes. Its biophysical characteristics can be modified by bacteria in response to changes in growth conditions such as growth temperature, the source of carbon and energy, or various environmental stresses (10–12). The most important adaptive microbial response to stress exposure, in addition to the synthesis of specific proteins, is related to changes in the fatty acid (FA) composition of the membrane (13). The adaptive strategies include alterations in the degree of unsaturation, the length of the carbon chains, the branching position, and cis-trans isomerization (14, 15). These changes alter the dynamic structure, integrity, and fluidity of the cytoplasmic membrane, which are key factors in maintaining the viability of cells and their metabolic activities. Lactic acid bacteria (LAB), a group of bacteria widely employed in the food and pharmaceutical industries, may encounter various stress conditions during industrial processes or survival in the GIT. Several studies have suggested that the regulation of membrane status influences the survival of LAB cells under stress conditions, such as oxidative, heat, cold, acid, and osmotic stresses (10, 16, 17). However, the responses of the cellular membrane to alterations in the carbon source have not been described for LAB.
Lactobacillus plantarum is encountered in many food products and is a natural inhabitant of the human GIT. The ecological flexibility of L. plantarum is reflected by the high number of genes involved in sugar uptake and utilization, which allow the organism to grow on numerous types of carbohydrates. Previous reports have shown that various strains of L. plantarum can utilize FOS as efficiently as glucose (18, 19). In particular, microarray analysis suggested that a group of five genes (lp_0184 to lp_0189) was probably involved in the transport and degradation of FOS in L. plantarum WCFS1 (7). However, whether this cluster was solely responsible for the metabolism of FOS in L. plantarum and the adaptive responses of the cells to such oligosaccharides was not stated. In the present study, a global analysis of the physiological process of FOS metabolism in L. plantarum strain ST-III was carried out. We first used high-throughput RNA sequencing (RNA-seq) to identify and compare the transcriptome profiles of the cells during growth on FOS and those during growth on glucose. The determination of specific metabolic and membrane adaptations for the utilization of FOS complemented the transcriptome-based results. In addition, gene inactivation and the prediction of regulatory elements provided new insights into the pathways and regulation of FOS metabolism in L. plantarum ST-III.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All bacterial strains and plasmids used throughout this study are listed in Table 1. L. plantarum ST-III was originally isolated from kimchi and has many probiotic properties, such as cholesterol removal and strong adhesion to Caco-2 cells. Its complete genome (3.25 Mb) has been compared with two published L. plantarum genomes, those of WCFS1 and JDM1 (20). L. plantarum ST-III was routinely propagated in de Man-Rogosa-Sharpe (MRS) broth (Merck, Darmstadt, Germany) at 37°C without agitation. For phenotypic analysis, and for measurement of the growth of the parent and mutant strains, cells were grown in modified MRS (mMRS) basal medium or chemically defined medium (CDM) (21) supplemented with different carbon sources. FOS and glucose were used as the carbon sources, sterilized separately through a sterile 0.2-mm filter, and aseptically added to the fermentation medium. The FOS used in this study was a commercial mixture supplied by Meiji Seika Kaisha (Tokyo, Japan), composed of 37.3% (wt/wt) 1-kestose, 49.1% (wt/wt) nystose, 9.8% (wt/wt) fructosyl-nystose, 2.3% (wt/wt) sucrose, and 1.3% (wt/wt) glucose and fructose. To verify that the levels of residual sugars such as glucose, fructose, and sucrose in the commercial FOS mixture were negligible when cells were grown on this matrix, equivalent amounts of these sugars were added to the CDM as a control. Escherichia coli DH5α, used as a host for routine cloning procedures, was grown in Luria-Bertani (LB) medium at 37°C with aeration at 200 rpm. When appropriate, antibiotics were added to the media. For L. plantarum, 10 μg ml−1 chloramphenicol and 10 μg ml−1 or 30 μg ml−1 (for replica plating) erythromycin were used. For E. coli, 10 μg ml−1 chloramphenicol and 250 μg ml−1 erythromycin were used.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant feature(s)a | Source or reference |
|---|---|---|
| Strains | ||
| L. plantarum | CGMCC 0847b | |
| ST-III | Wild type | |
| BD1101CM | Derivative of ST-III containing a lox66-P32-cat-lox71 replacement of sacA (ΔsacA::cat) | This work |
| BD1102CM | Derivative of ST-III containing a lox66-P32-cat-lox71 replacement of pts1 (Δpts1::cat) | This work |
| BD1102 | Derivative of ST-III containing a lox72 replacement of pts1 (Δpts1) | This work |
| BD1103CM | Derivative of ST-III containing a lox66-P32-cat-lox71 replacement of pts26 (Δpts26::cat) | This work |
| BD1104CM | Derivative of BD1102 containing a lox66-P32-cat-lox71 replacement of pts26 (Δpts1 Δpts26::cat) | This work |
| E. coli DH5α | Cloning host; F− ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 endA1 recA1 hsdR17 (rK− mK+) phoA supE44 thi-1 gyrA96 relA1 λ− | Invitrogen |
| Plasmids | ||
| pNZ5319 | Cmr Emr; for multiple gene replacements in Gram-positive bacteria | 23 |
| pBD1101 | Cmr Emr; pNZ5319 derivative containing homologous regions up- and downstream of ST-III sacA | This work |
| pBD1102 | Cmr Emr; pNZ5319 derivative containing homologous regions up- and downstream of ST-III pts1 | This work |
| pBD1103 | Cmr Emr; pNZ5319 derivative containing homologous regions up- and downstream of ST-III pts26 | This work |
| pNZ5348 | Emr; contains cre under the control of the pcrA (lp_1144) promoter | 23 |
Cmr, chloramphenicol resistant; Emr, erythromycin resistant.
CGMCC, China General Microbiological Culture Collection Center.
Fermentation and sampling.
L. plantarum ST-III cultures were propagated in parallel for 2 passages in CDM with 1% (wt/vol) FOS or glucose as the sole carbon source. Cultures with each sugar were then transferred with a 2% (vol/vol) inoculum into 500 ml of CDM containing the same sugar. The cells were grown under aerobic conditions without agitation at 37°C, and cell density was monitored by the optical density at 600 nm (OD600) of the culture, measured by a UV-1601 spectrophotometer (Shimadzu, Kyoto, Japan). When the OD600 reached 0.65 or 1.5, cultures grown on FOS or glucose were harvested by centrifugation (8,000 × g, 10 min, 4°C), and the cell pellets and supernatants were collected for further RNA isolation, lipid extraction, detection of membrane fluidity, and analysis of metabolites.
RNA isolation and transcriptome analysis.
Cells grown on FOS or glucose were harvested at an OD600 of 0.65 for transcript profiling, and the pellets were flash frozen for storage at −80°C. Total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA) and was then purified with an RNeasy MinElute cleanup kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. The bacterial rRNA was then removed by a mixed treatment with the MICROBExpress kit (Ambion, Austin, TX, USA) and the mRNA-ONLY prokaryotic mRNA isolation kit (Epicentre, Madison, WI, USA). The quality of the RNA was determined by using an Agilent 2100 Bioanalyzer (Agilent, Palo Alto, CA, USA) and by visualization following 1% (wt/vol) agarose gel electrophoresis. RNA was quantified using a NanoDrop 2000 spectrophotometer after every step.
For the transcriptomic analysis, mRNA was fragmented, and a library was constructed with an mRNA Sample Prep Master Mix Set 1 kit (Illumina, San Diego, CA, USA). The library was sequenced using the Illumina/Solexa Genome Analyzer IIx system (Illumina) with samples loaded at a concentration of 10 pM. To obtain estimates of the expression levels, TopHat (http://ccb.jhu.edu/software/tophat/index.shtml) was used to align the trimmed sequencing reads to the L. plantarum ST-III genome. The reference genome and gene model annotation (GenBank accession number CP002222.1) used in this study were retrieved from GenBank. Cufflinks (http://cole-trapnell-lab.github.io/cufflinks/) was then used to estimate the gene expression levels based on the same gene model annotations (22). RPKM (reads per kilobase per million mapped reads) were used as normalized metrics to present the gene expression levels (23). Genes were considered differentially expressed when the ratio of transcription for cells grown in the presence of FOS to transcription for cells grown in the presence of glucose was ±2-fold, with a P value of ≤0.05.
Mutant construction.
Gene deletion mutants were constructed using the Cre-lox-based mutagenesis system according to the method of Lambert et al. (24). The primers used to construct the L. plantarum ST-III mutants are described in Table S1 in the supplemental material. In brief, upstream and downstream flanking regions of sacA, pts1, and pts26 were amplified by PCR using primers A, B, C, and D for sacA and primers E, F, G, and H for pts1 and pts26. The amplicons were cloned into the PmeI and Ecl136II restriction sites of the suicide vector pNZ5319, respectively, and the recombinant mutagenesis vectors, pBD1101, pBD1102, and pBD1103, were introduced into L. plantarum ST-III by electroporation. Chloramphenicol-resistant transformants were selected, and replicas were plated to check for an erythromycin-sensitive phenotype. Correct integration of the lox66-P32-cat-lox71 cassette into the genome was confirmed by PCR amplification of the flanking regions of the integrated cassette, using primers annealing uniquely to the genomic sequences combined with the mutagenesis vector-specific primers 85 and 87, which annealed to the P32-cat region. A single colony for each mutant confirmed by PCR was selected and was designated BD1101CM (ΔsacA::cat), BD1102CM (Δpts1::cat), or BD1103CM (Δpts26::cat). The pts1 pts26 mutant strain was constructed in the BD1102CM background in two steps. First, strain BD1102 (Δpts1) was constructed by expression of the Cre resolvase enzyme from pNZ5348 to remove the lox66-P32-cat-lox71 cassette. Then pBD1103 was introduced into BD1102, and the double mutant strain was selected using the approach described above, resulting in strain BD1104CM (Δpts1 Δpts26::cat). Mutants that had lost the ability to ferment FOS were selected on mMRS–1% FOS agar medium containing 30 mg liter−1 bromcresol purple. The growth of the parent and mutant strains was monitored using mMRS broth either alone (no added sugar) or supplemented with a 1% (wt/vol) commercial FOS mixture or glucose.
Analysis of metabolites.
The production of lactate, acetate, and formate as a result of the fermentation of sugars by L. plantarum ST-III was determined by high-performance liquid chromatography. Each culture sample was centrifuged (16,000 × g, 15 min), and the supernatant was filtered through a 0.45-μm nylon filter. Then 10 μl of supernatant was injected into an Agilent 1260 series chromatograph (Agilent) with a UV detector at a wavelength of 210 nm for analysis. Chromatographic analysis was performed on a Zorbax Eclipse XDB-C18 column (length, 250 mm; inside diameter [i.d.], 4.6 mm; particle size, 5 μm; Agilent). The organic acids were eluted with 10% (vol/vol) acetonitrile–0.05% (vol/vol) trifluoroacetic acid as the mobile phase at a flow rate of 1 ml min−1. The column temperature was kept at 30°C.
The amount of ethanol in the culture samples was analyzed by an HP6890 (Agilent) gas chromatograph with a flame ionization detector as described previously (25), with some modifications. The gas chromatograph was equipped with a DB-23 capillary column (length, 60 m; i.d., 0.32 mm; film thickness, 0.25 μm; Agilent), with helium as a carrier gas at 1.5 ml min−1. Samples (1 μl) were injected using a split ratio of 1:1 at 200°C. The temperature program was initially set at 40°C for 5 min, followed by an increase of 10°C min−1 to 180°C, and was then maintained at 180°C isothermally for 5 min.
FA extraction and analysis.
Since the FAs of LAB are derived mainly from phospholipids in the membrane bilayer, their analysis in the membrane was carried out directly on the cell pellets. Total bacterial lipids were extracted with chloroform-methanol according to a method described previously (26). The internal standard, heptadecanoic acid (C17:0) (99% pure; Sigma, St. Louis, MO, USA), was added to each sample to give a final concentration of 200 μg ml−1. FAs were converted to the corresponding methyl esters with 10% (vol/vol) methanol-HCl. The fatty acid methyl esters (FAMEs) were separated using a GC-2010 Plus gas chromatograph (Shimadzu) fitted with a QP2010 Ultra mass spectrometer on an Rtx-Wax column (length, 30 m; i.d., 0.25 mm; film thickness, 0.25 μm; Shimadzu). Injections of 1 μl were performed automatically at a split ratio of 10:1. Helium was used as the carrier gas. The temperature program was initially set at 170°C for 3 min, followed by increases of 10°C min−1 to 190°C, 5°C min−1 to 210°C, and 2°C min−1 to 190°C, and was then maintained at 190°C isothermally for 5 min. The injection and detection temperatures were 230°C. The electron energy was set at 70 eV, and the ion source temperature was kept at 220°C. FAMEs were identified by their retention times relative to those of the standards (C9 through C20; Supelco, Bellefonte, PA, USA) and by their mass spectra relative to a spectrum database. The relative amounts of FAMEs were calculated from peak areas. The degree of unsaturation (the ratio of unsaturated to saturated FAs, obtained without considering cyclopropane FAs) and the mean chain length were calculated.
Measurement of membrane fluidity.
The membrane fluidity characteristics of L. plantarum ST-III were investigated by fluorescence anisotropy according to a protocol described previously (27), with some modifications. Briefly, fresh cells grown in CDM containing FOS or glucose were harvested at an OD600 of 0.65 or 1.5, fixed in formaldehyde at a final concentration of 0.25% (vol/vol), and washed twice with a phosphate buffer solution (pH 7.4) containing 0.25% (vol/vol) formaldehyde. The samples were then incubated for 1 h at 37°C with 1,6-diphenyl-1,3,5-hexatriene (DPH) at a final concentration of 5 μM. The unlabeled probe was removed by centrifugation, the cells were resuspended in the phosphate buffer solution, and the OD600 was adjusted to 0.65 in all measurements. Fluorescence anisotropy was measured at 37°C using an F-7000 spectrofluorometer (Hitachi, Tokyo, Japan) with excitation at 360 nm and emission at 430 nm (5- and 5-nm slits, respectively). Anisotropy values (r) were calculated as , where I is the corrected fluorescence intensity and subscripts V and H indicate the values obtained with vertical or horizontal orientations, respectively, of the excitation polarizer and emission analyzer (in that order). In these experiments, decreases in the degree of fluorescence anisotropy reflected increases in the fluidity of the lipid bilayer, which controls or alters the mobility of DPH in the membrane.
Confirmation of the transcriptomic results by RT-qPCR.
To validate the RNA-seq data, 40 genes with altered expression levels (24 upregulated and 16 downregulated) were examined individually in cells grown on FOS or glucose at an OD600 of 0.65 or 1.5, using real-time quantitative PCR (RT-qPCR). Total RNA was isolated as described above and was reverse transcribed with a PrimeScript RT reagent kit (TaKaRa, Dalian, China) according to the manufacturer's instructions. The amplifications were performed using specific primers (see Table S2 in the supplemental material) and a Power SYBR green PCR master mix (Applied Biosystems, Foster City, CA, USA) with a 7300 Fast real-time PCR system (Applied Biosystems). All of the samples were measured in triplicate. Gene expression was normalized by the 2−ΔΔCT method, and the 16S rRNA gene was used as the normalized standard.
Statistical analysis.
The data shown in the tables and figures represent the results of at least three independent experiments. Student's t test was used to determine statistical differences. Differences between samples with a P value of ≤0.05 were considered to be statistically significant.
Microarray data accession number.
The transcriptomic data determined in this study have been deposited in the BioProject database at the National Center for Biotechnology Information (NCBI) under accession number PRJNA230898.
RESULTS
Comparison of growth on FOS with growth on glucose.
L. plantarum ST-III exhibited similar growth patterns in CDM supplemented with FOS and CDM supplemented with glucose, although the growth rate was slightly lower for the FOS-grown cultures (Fig. 1). As expected, growth in CDM with amounts of monosaccharides and disaccharides equivalent to those in 1% FOS was negligible, which indicated that L. plantarum ST-III mainly used the fructans in the commercial FOS mixture for growth. Based on the growth profiles of the cells on FOS and on glucose, the early-logarithmic phase was chosen for transcriptomic analysis; given the time lag from gene transcription to alterations in the phenotype, the early-logarithmic and mid-logarithmic phases were chosen for the measurement of metabolites and membrane-related features. RT-qPCR was used for the conformation of the expression levels of the key genes at the two sampling points (see Fig. S1 in the supplemental material).
FIG 1.
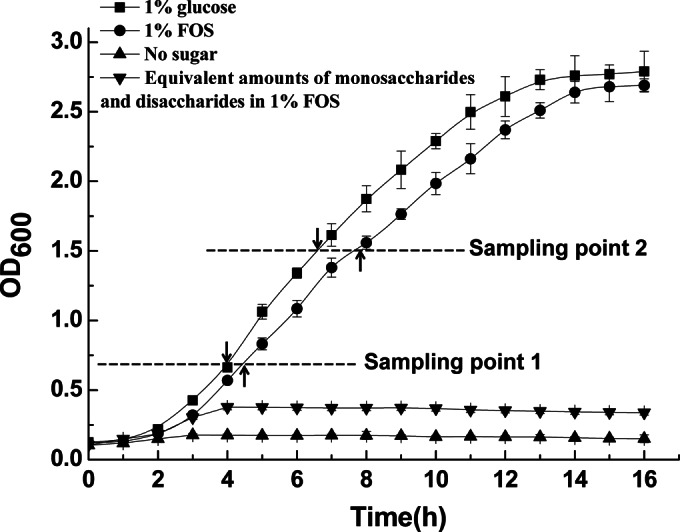
Growth of L. plantarum ST-III in CDM either alone (no added sugar) or supplemented with 1% commercial FOS or 1% glucose. To account for the glucose, fructose, and sucrose in the commercial FOS, cells were also grown in CDM containing equivalent amounts of these sugars. The two sampling points are indicated by dashed lines. Sampling point 1 was chosen for transcriptomic analysis; both of the sampling points were chosen for lipid extraction, the detection of membrane fluidity, and the analysis of metabolites. RT-qPCR was used for the conformation of the expression levels of the key genes at the two sampling points. Data are mean values based on at least three replicates. Error bars indicate standard deviations.
Global transcriptome profiles during growth on FOS compared with those during growth on glucose.
The differential global transcriptome of L. plantarum ST-III using FOS or glucose as the sole carbon source was studied by RNA-seq in the early-logarithmic phase of the bacterium. The numbers of cDNA reads obtained for L. plantarum ST-III grown on FOS and glucose were 27,107,812 and 17,145,874, with mapping rates of 97.7% and 98.8%, respectively. The average length was 101 bp (see Table S3 in the supplemental material). A total of 363 genes were found to be differentially transcribed in response to FOS compared with glucose; of these, 324 genes were upregulated (see Table S4 in the supplemental material) and 39 were downregulated (see Table S5 in the supplemental material). The genes that were differentially transcribed were classified into functional categories based on the designations of clusters of orthologous groups (COGs) (www.ncbi.nlm.nih.gov/COG), and the percentage of genes in each COGs category that were significantly altered was calculated (Fig. 2). The upregulated genes fell within almost all of the COGs except for those involved in extracellular structures. The COGs category with the largest proportion of upregulated genes was the carbohydrate transport and metabolism class, followed by the energy production and conversion category, in which approximately 40% and 20% of genes were activated by FOS, respectively. Among the 22 COGs categories, only 14 contained genes that were downregulated in response to FOS compared with glucose. Surprisingly, the COGs category with the largest proportion of FOS-repressed genes was the lipid transport and metabolism class, in which 22% of genes were downregulated.
FIG 2.

Functional classification according to COGs of genes differentially expressed by L. plantarum ST-III grown in the presence of FOS versus glucose. The profile of each functional classes is shown as the percentage of all genes in the class whose expression was significantly upregulated (open bars) or downregulated (filled bars).
In accordance with previous observations (7), significant upregulation (4- to 35-fold) was observed with a cluster (LPST_C0151 to LPST_C0154) composed of five genes that encoded a sucrose phosphoenolpyruvate transport system (PTS1), a β-fructofuranosidase (SacA), a fructokinase (SacK), an α-glucosidase (Agl2), and a repressor (SacR1). It was found that this cluster (designated the sacPTS1 cluster) was probably involved in the transport and degradation of FOS in L. plantarum (7). In addition, another gene cluster that showed 2.1- to 4.8-fold increases in the expression of its components was found in our study. This cluster (LPST_C2650 to LPST_C2652; designated the sacPTS26 cluster) was predicted to encode a sucrose PTS (PTS26), an α-glucosidase (Agl4), and a transcriptional regulator (SacR2). Within the sacPTS26 cluster, pts26 was upregulated 4.8-fold in the presence of FOS. This result differed from those of Saulnier et al., in whose microarray experiments two sucrose PTSs (lp_3219 and lp_3522) were not differentially expressed (7).
Another set of genes induced by FOS metabolism was related to mixed-acid fermentation. L. plantarum is a facultatively heterofermentative bacterium that can produce acetate and formate at the expense of lactate under certain conditions (28, 29). Some genes related to the proposed pathways for the conversion of lactate to acetate and formate were upregulated in the presence of FOS. These genes were predicted to encode a lactate dehydrogenase (LDH) (LPST_C1774), 3 pyruvate oxidases (POX) (LPST_C0667, LPST_C2161, and LPST_C2933), an acetate kinase (ACK) (LPST_C0255), a set of pyruvate formate lyase (PFL) complexes (LPST_C2728 to LPST_C2729), a set of pyruvate dehydrogenase (PDH) complexes (LPST_C1775 to LPST_C1778), and a phosphotransacetylase (PTA) (LPST_C0630) (Fig. 3). Previous physiological studies of L. plantarum indicated that acetate production was maximal under aerobic conditions with limited glucose (28), growth conditions similar to those used in this study with FOS as the sole carbon source.
FIG 3.

Genes differentially expressed in the carbohydrate utilization and metabolite production pathways in L. plantarum ST-III during growth on FOS compared with growth on glucose. Genes that were upregulated are shown in red. PTS, phosphotransferase system; SacA, β-fructofuranosidase; SacK, fructokinase; LDH, lactate dehydrogenase; POX, pyruvate oxidase; ACK, acetate kinase; PDH, pyruvate dehydrogenase; PFL, pyruvate formate lyase; PTA, phosphotransacetylase; ACDH, acetaldehyde dehydrogenase; ADHE, alcohol dehydrogenase; CoA, coenzyme A.
As stated above, several of the downregulated genes were related to membrane lipid transport and metabolism. In particular, 16 genes in a 10-kbp-region cluster (LPST_C1327 to LPST_C1342) involved in FA biosynthesis were repressed at least 6-fold. The cluster included genes that encoded FA initiation proteins (acetyl coenzyme A carboxylases [Acc]), elongation proteins (Fab) (see Fig. S2 in the supplemental material), an acyl carrier protein (ACP), and proteins that regulate the concentrations of intracellular cofactors (coenzyme A and ACP). This FA synthase system was structurally unique among all sequenced LAB and played a key role in the profiles of lipid composition and hence in the biophysical properties of the membrane bilayer (11).
To confirm the transcriptomic results and estimate the gene expression levels at an OD600 of 1.5 for L. plantarum ST-III grown on FOS and glucose, 40 key genes found in the transcriptomic results were selected and were tested for differences in their expression using RT-qPCR (see Table S2 in the supplemental material). At an OD600 of 0.65, the transcription levels of all these genes exhibited the same trends as the values obtained by RNA-seq. At an OD600 of 1.5, although the magnitude of the changes differed, the results for most genes corresponded well with the transcriptomic data at an OD600 of 0.65. For example, the upregulation of the two pox genes (LPST_C2933 and LPST_C2161) in response to FOS compared with glucose at an OD600 of 1.5 was more significant than that at an OD600 of 0.65. Overall, the direction of the changes and the approximate magnitudes confirmed the RNA-seq results at an OD600 of 0.65 and extended the observations of gene expression to an OD600 of 1.5.
Inactivation of the sacA and pts genes affected growth on FOS.
To investigate the potential involvement of the sacPTS1 cluster in FOS uptake and metabolism, pts1 and sacA deletion mutants were constructed using the Cre-lox-based mutagenesis system. The gene deletion had no detectable impact on cell morphology or growth in MRS medium or CDM when glucose, fructose, or galactose was used as the sole carbon source for L. plantarum ST-III (data not shown), suggesting that the functionality of these genes is not essential for the normal survival of these cells or their use of these monosaccharides. The growth of the sacA deletion mutant was significantly impaired on FOS (Fig. 4A). In contrast, the pts1 deletion mutant was able to grow on FOS, but at a lower growth rate than that of the wild type, reaching the stationary phase 5 h later (Fig. 4B). This phenomenon indicated that other, similar systems—possibly the sacPTS26 cluster—exist which could function after the deletion of pts1. The transcription of pts26 was evaluated by RT-qPCR in the wild type and a Δpts1 mutant (BD1102). A 2.3-fold-higher transcription level was observed for the BD1102 mutant grown on FOS than for the wild type grown on FOS, and the increase in the transcription level for BD1102 in response to FOS compared with glucose was more significant (3.2-fold) (see Fig. S3 in the supplemental material). To investigate whether pts26 is involved in the FOS metabolism of L. plantarum ST-III, a pts26 deletion mutant (BD1103CM) and a pts1 pts26 deletion mutant (BD1104CM) were constructed. The growth of BD1103CM was similar to that of BD1102 on FOS, reaching the stationary phase 2 h later than the wild type (Fig. 4C). In contrast, BD1104CM did not generate a distinct yellow zone against a purple background in mMRS-FOS agar medium (data not shown) and halted its growth prematurely in MRS broth with FOS as the sole carbon source (Fig. 4D). Overall, the results of the transcriptome and gene deletion analyses provided evidence that both PTS1 and PTS26 participate in the transport of FOS across the membrane of L. plantarum ST-III.
FIG 4.

Growth of mutant strains of L. plantarum ST-III in CDM either alone (no added sugar) or supplemented with 1% commercial FOS or glucose. (A) BD1101CM, a ΔsacA mutant; (B) BD1102, a Δpts1 mutant; (C) BD1103CM, a Δpts26 mutant; (D) BD1104CM, a Δpts1 Δpts26 mutant. Data are mean values based on at least three replicates. Error bars indicate standard deviations. CHO, carbohydrate.
FOS-induced alterations in the FA composition and fluidity of the membrane.
The FA compositions of the membranes of L. plantarum ST-III cells fermented with FOS were compared with those of cells fermented with glucose at OD600 values of 0.65 and 1.5. The FA composition of cells grown on FOS was dramatically altered from that of cells grown on glucose. An increase in the proportion of short-chain FAs, such as myristic acid (C14:0), palmitic acid (C16:0), and palmitoleic acid (C16:1), was observed, while the molar percentage of long-chain FAs, except for cyclopropane FA (C19:0-cyc), decreased (Fig. 5). As a result, the mean chain length of the FAs decreased for cells grown on FOS from that for cells grown on glucose, and this phenomenon was more significant at an OD600 of 1.5 than at an OD600 of 0.65 (Table 2). Furthermore, although the FA profiles changed significantly, the degree of unsaturation did not change. The adjustment of the degree of unsaturation of lipids is a well-known response of bacteria to a drastic modification of the environment (14), but changes in the source of carbon in our study did not alter the level of unsaturation of the membrane FAs in L. plantarum ST-III.
FIG 5.
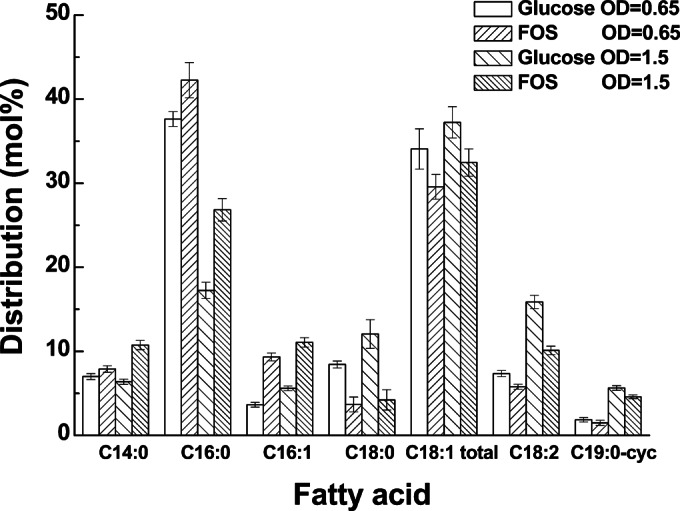
Differences in the distribution of FAs in L. plantarum ST-III cells grown in the presence of FOS from that in cells grown in the presence of glucose. The proportions of total-membrane FAs were determined in early-logarithmic (OD600, 0.65) and mid-logarithmic (OD600, 1.5) cultures of L. plantarum ST-III grown in CDM containing either 1% commercial FOS or glucose. The data are mean values based on at least three replicates. Error bars indicate standard deviations.
TABLE 2.
Differences in the degree of unsaturation and mean chain length between L. plantarum ST-III cultures using FOS or glucose as the sole carbon sourcea
| Conditions | Degree of unsaturationb | Mean chain length (no. of carbon atoms) |
|---|---|---|
| OD, 0.65 | ||
| Glucose | 0.88 ± 0.03 | 16.97 ± 0.11 |
| FOS | 0.86 ± 0.04 | 16.71 ± 0.07 |
| OD, 1.5 | ||
| Glucose | 0.94 ± 0.04 | 17.45 ± 0.08 |
| FOS | 0.93 ± 0.02 | 16.91 ± 0.06c |
Data are mean values based on at least three replicates. Error bars indicate standard deviations.
Ratio of unsaturated to saturated FAs (obtained without considering cyclopropane FAs).
The difference between cells grown on FOS and those grown on glucose was statistically significant (P ≤ 0.05).
To elucidate the possible membrane-related alterations in L. plantarum ST-III in response to FOS, membrane fluidity was evaluated by fluorescence anisotropy using DPH as a probe. At the two sampling points, cells grown on FOS maintained lower fluorescence anisotropy values than those grown on glucose (Fig. 6). Because the fluorescence anisotropy of DPH is directly related to membrane fluidity, higher membrane fluidity in the strain grown on FOS could be inferred.
FIG 6.
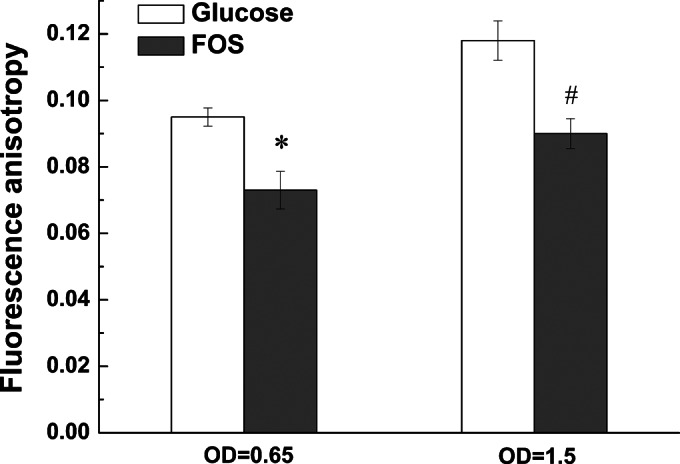
Differences in membrane fluidity between L. plantarum ST-III cells grown on FOS and those grown on glucose. Fluorescence anisotropy values were determined in early-logarithmic (OD600, 0.65) and mid-logarithmic (OD600, 1.5) cultures of L. plantarum ST-III grown in CDM containing either 1% commercial FOS (shaded bars) or glucose (open bars). Data are mean values based on at least three replicates. Error bars indicate standard deviations. Values that indicated statistically significant differences (P ≤ 0.05) between cells grown on FOS and those grown on glucose are marked with an asterisk or an octothorpe at an OD600 of 0.65 or 1.5, respectively.
Effects of FOS fermentation on the production of metabolites.
Lactate and acetate were the main end products of FOS and glucose fermentation in L. plantarum ST-III (Table 3). Cells grown on FOS produced more acetate and less lactate than cells grown on glucose at both sampling points. In addition, small amounts of formate and ethanol and traces of oxalate and malate were detected but did not differ between cells grown on FOS and those grown on glucose.
TABLE 3.
Comparison of metabolites resulting from the fermentation of FOS or glucose by L. plantarum ST-IIIa
| Conditions | Metabolite formation (mM) |
|||
|---|---|---|---|---|
| Lactate | Acetate | Formate | Ethanol | |
| OD, 0.65 | ||||
| Glucose | 15.4 ± 0.34 | 2.7 ± 0.05 | 1.32 ± 0.03 | 0.3 ± 0.10 |
| FOS | 13.1 ± 0.21* | 6.3 ± 0.10* | 1.35 ± 0.04 | 0.4 ± 0.08 |
| OD, 1.5 | ||||
| Glucose | 34.5 ± 0.89 | 4.4 ± 0.21 | 1.48 ± 0.03 | 0.5 ± 0.09 |
| FOS | 27.4 ± 0.79# | 10.4 ± 0.15# | 1.56 ± 0.08 | 0.4 ± 0.12 |
Data are mean values based on at least three replicates ± standard deviations. Values that indicate statistically significant differences (P ≤ 0.05) between cells grown on FOS and those grown on glucose are labeled with asterisks for an OD600 of 0.65 and with octothorpes for an OD600 of 1.5.
Prediction of the regulatory elements that mediate FOS metabolism.
The utilization of carbohydrates by lactobacilli is always subject to carbon catabolite regulation, which is achieved by the combined effects of global and operon-specific (local) regulatory mechanisms (2, 9). Global regulation results from the binding of the catabolite control protein (CcpA) to catabolite repression element (Cre) sites located within or downstream from the promoter in the presence of a preferred substrate (30, 31), while local regulation is achieved by the interaction of local regulators with specific operator motifs in the operon in the absence of a related substrate (32, 33). In the present study, two genes (sacR1 and sacR2) encoding putative repressor proteins, which display significant similarities to the GalR-LacI family of bacterial transcriptional regulators, were identified within the gene clusters related to FOS metabolism. Although the two regulators exhibited low sequence identity (28%) to each other, each contained a helix-turn-helix region (IPR000843) that is a DNA-binding region of the GalR-LacI family in the N-terminal end and a sugar-binding domain (IPR028082) in the C-terminal end. Correspondingly, potential Cre sites for CcpA binding and the specific operators controlled by the local regulators were predicted (Fig. 7) according to an analysis described previously (34). For the sacPTS1 cluster, two putative specific operators were found in the regions of putative promoters of sacA and pts1, respectively, and a Cre site was predicted within the promoter of sacK. For the sacPTS26 cluster, the genes were probably cotranscribed as a single polycistronic mRNA, because they were predicted to have only one promoter and one terminator. A specific operator was predicted in the region between the sacR2 and agl4 genes, and a potential Cre site lay downstream from the putative promoter of the operon. The potential existence of these regulators and operators in FOS-related gene clusters suggested that FOS metabolism in L. plantarum ST-III was under the negative control of both global and local regulation. A detailed study of the regulatory mechanism for the use of FOS by L. plantarum is currently being carried out.
FIG 7.

Prediction of the cre and operator sites in the sacPTS1 (A) and sacPTS26 (B) clusters of L. plantarum ST-III. Putative functions are indicated by color as follows: green, PTS; black, transcriptional regulators; red, glycoside hydrolases; yellow, enzymes involved in the glycolytic pathway. Predicted transcription terminators are shown as hairpin loops. The potential cre sites are underlined, and the specific operators are shaded. The presumed start codon of each gene is shown in capital letters, and the putative −10 and −35 promoter regions and possible ribosome-binding sites (RBS) are marked.
DISCUSSION
Although the prebiotic effects of FOS on enteric populations have been demonstrated both in vivo and in vitro (35–37), our knowledge of the mechanism of FOS metabolism by probiotic bacteria, particularly the regulation of transcription and the corresponding adaptive responses of the cells, is still limited. In the present study, the global alterations in L. plantarum ST-III using FOS or glucose as the sole carbon source were determined by the transcriptome, analysis of the metabolites, and the measurement of membrane FA composition and fluidity. We aimed to identify the connection between gene transcription and the production of metabolites, and between gene transcription and membrane alterations, in order to explain the FOS metabolism of L. plantarum ST-III in the GIT.
RNA-seq was performed to distinguish the gene expression patterns of L. plantarum ST-III grown on different carbon sources. RNA-seq provides several advantages over hybridization-based approaches, such as microarrays, especially markedly higher sensitivity for the detection of low-abundance transcripts and accuracy in determining gene transcription levels (38, 39). A total of 363 genes were found to be differentially transcribed in our study, a much higher number than that found in L. plantarum WCFS1 under similar conditions using microarray techniques. In addition to the discovery that the sacPTS1 cluster was probably involved in the transport and degradation of FOS, the sacPTS26 cluster was also found by RNA-seq to be induced by FOS in L. plantarum ST-III.
Experimental validation of the involvement of these gene clusters in the metabolism of FOS in L. plantarum ST-III was carried out using gene deletion. The complete inhibition of growth by excision of the sacA gene confirmed the hypothesis that the SacA enzyme was solely responsible for the hydrolysis of FOS in L. plantarum ST-III and that no extracellular or intracellular fructosidases were involved in this process. Our previous study showed that the SacA protein is an intracellular enzyme (40), which suggested that the hydrolysis of FOS occurs in the cytoplasm of L. plantarum ST-III. In contrast to previous opinions, both PTS1 and PTS26 were involved in the uptake of FOS in L. plantarum ST-III. As with some other PTSs, the specificity of sucrose PTSs may not be absolute; they may also transport and phosphorylate FOS, though at a relatively low rate (7). Taken together, these results demonstrated that FOS metabolism in L. plantarum ST-III was controlled by two gene clusters. FOS were found to be transported across the membrane by PTS1 and PTS26 through phosphorylation and to be subsequently hydrolyzed by SacA in the cytoplasm. Moreover, the sacPTS26 cluster was predicted to be related to the metabolism of sucrose or trehalose (34) and was conserved among the L. plantarum strains sequenced. This discovery suggested that the mechanism of FOS metabolism in L. plantarum ST-III could be extrapolated to all L. plantarum strains.
One of the core responses to the utilization of FOS in L. plantarum ST-III was the modification of FA metabolism and membrane-related features. The transcript profiles suggested that a reduction in FA biosynthesis was induced by FOS metabolism. Furthermore, FA analysis showed that L. plantarum ST-III cells grown on FOS had membrane FA profiles different from those of cells grown on glucose; in particular, the FAs had shorter mean chain lengths. Shorter chains have been reported to be unable to span the bilayer or form hydrophobic interactions with other lipids and proteins. As a result, the fluidity of the bilayer was increased due to the motion of the free acyl chain ends (16, 41). Therefore, the decreased carbon chain length contributed to the increase in cell membrane fluidity, which was in agreement with our results for fluorescence anisotropy detected by the DPH probe. The effects of different carbohydrates on the FA composition and fluidity of cellular membranes have been reported for Saccharomyces cerevisiae and Acinetobacter spp. (42, 43), but the mechanisms are not yet well understood. A possible explanation for the phenomenon observed in this study is as follows. FOS is an oligosaccharide molecule that is much larger than glucose; compared with the use of glucose, the use of FOS by L. plantarum ST-III can be regarded as nutrient stress, requiring high energy levels or the synthesis of specific proteins for FOS transport. Under these conditions, L. plantarum ST-III changes the FA composition and then increases the fluidity of the membrane to enable FOS transport and ensure growth and other physiological activities. Collectively, these results suggest that shortening of the carbon chains is the principal strategy by which L. plantarum ST-III modulates membrane fluidity for the transport and utilization of FOS, but the reasons for this strategy need further investigation.
The modification of metabolite profiles was another response of L. plantarum ST-III to FOS. Genes related to the proposed pathways for the production of acetate and formate were upregulated in the presence of FOS. The measurement of metabolites showed that cells grown on FOS altered their metabolism to produce more acetate and less lactate, although the formate contents did not differ between strains grown on FOS and those grown on glucose. These results were similar to those of previous in vitro studies with bifidobacteria and lactobacilli in which growth on FOS switched the metabolism toward the production of more acetate, formate, or ethanol at the expense of lactate (18, 44). Various pathways for the production of acetate have been identified in this species (28, 45). Pyruvate can be metabolized anaerobically into acetate via PFL, PTA, and ACK. A second possible pathway for the production of acetate is via the PDH complex, PTA, and ACK. A third proposed pathway for the conversion of lactate to acetate is via two stereospecific lactate dehydrogenases (LdhD and LdhL), POX, and ACK. Although most of the genes related to the three pathways for acetate production were upregulated in the presence of FOS, some of them may not function. For example, PFL cannot be involved, because this enzyme is oxygen sensitive (28), which could also explain the constant formate production in the metabolites. Although pdh genes have been identified in the genome sequences of sequenced L. plantarum strains, no detectable PDH activity has been reported under various growth conditions to date (45).
As stated before, a number of microarray or RNA-seq experiments have been performed to elucidate the interactions of probiotics with prebiotics, such as those of Bifidobacterium animalis subsp. lactis BB-12 with xylo-oligosaccharides (6), L. plantarum WCFS1 with FOS (7), L. paracasei 1195 with FOS (8), and Lactobacillus ruminis L5 with cellobiose (46). Although the results are different, some commonalities could be found. For example, genes of specific glycoside hydrolases and transporters were upregulated in the presence of the oligosaccharides and were repressed by glucose. In most cases, the genes encoding the transporter components and the associated glycoside hydrolases are clustered in conserved modules and are coregulated as single operons. In some cases, genes involved in membrane proteins (46) or fatty acid biosynthesis (7) were also downregulated, but the reasons were not investigated. These results are in accordance with our study and bolster the generalizability of the current findings.
The human GIT harbors a complex microbial ecosystem that comprises at least 400 to 500 different bacterial species based on competition and symbiosis. Although the main targets for prebiotic FOS are bifidobacteria and lactobacilli, other members of the GIT microbiota have also been demonstrated to consume FOS-type fructans and even to be stimulated by their ingestion (47, 48). Basically, two metabolic routes of FOS metabolism exist in bacteria in the GIT. Either the substrate is transported intact and is hydrolyzed in the cytoplasm, as is the case for most bifidobacteria, L. plantarum, and L. acidophilus, or it is hydrolyzed by extracellular enzymes, followed by accumulation of the hydrolysis products, as occurs in L. paracasei and some Bacteroides spp. In the GIT, extracellular hydrolysis of FOS may make free monosaccharides available to opportunistic competitors in the ecosystem (49). Strains capable of intracellular degradation, such as L. plantarum, would not suffer from that drawback and would experience the greater prebiotic effects of FOS. Moreover, the increased production of acetate by L. plantarum ST-III in the presence of FOS may be of interest for the inhibition of intestinal pathogens such as Escherichia coli and Salmonella spp., as stated previously (50, 51). Thus, the combination of L. plantarum ST-III and FOS may be a good choice for the modulation of microbiota in the GIT but needs to be validated further in in vivo experiments.
In conclusion, the transcriptome and mutagenesis analyses demonstrated that a β-fructofuranosidase and two sucrose PTSs participated in the metabolism of FOS in L. plantarum ST-III and that the strain modified its membrane-related features and metabolites in response to FOS, suggesting the possible adaptation of probiotics to complex sugars. Although we are still far from fully understanding the precise mechanisms by which prebiotics are metabolized by beneficial microbes and modulate microbiota composition in vivo, our current study on the mechanistic interactions of L. plantarum ST-III and FOS in vitro could reveal the adaptive strategies of the strain for FOS metabolism and could lay the basis for further study of the mechanisms of FOS utilization by probiotics in the gut.
Supplementary Material
ACKNOWLEDGMENTS
RNA-seq was performed at the Tianjin Research Center for Functional Genomics and Biochip.
This work was supported by the National Natural Science Foundation of China (award 31501451), the National Science Fund for Distinguished Young Scholars (award 31125021), the National High Technology Research and Development Program of China (863 Program, award 2011AA100901) and the Key Projects in the National Science & Technology Pillar Program during the Twelfth Five-Year Plan Period (award 2013BAD18B01).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02426-15.
REFERENCES
- 1.Gibson GR, Roberfroid MB. 1995. Dietary modulation of the human colonic microbiota: introducing the concept of prebiotics. J Nutr 125:1401–1412. [DOI] [PubMed] [Google Scholar]
- 2.Barrangou R, Altermann E, Hutkins R, Cano R, Klaenhammer TR. 2003. Functional and comparative genomic analyses of an operon involved in fructooligosaccharide utilization by Lactobacillus acidophilus. Proc Natl Acad Sci U S A 100:8957–8962. doi: 10.1073/pnas.1332765100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sivieri K, Morales ML, Saad SM, Adorno MA, Sakamoto IK, Rossi EA. 2014. Prebiotic effect of fructooligosaccharide in the simulator of the human intestinal microbial ecosystem (SHIME model). J Med Food 17:894–901. doi: 10.1089/jmf.2013.0092. [DOI] [PubMed] [Google Scholar]
- 4.Takemura N, Hagio M, Ishizuka S, Ito H, Morita T, Sonoyama K. 2010. Inulin prolongs survival of intragastrically administered Lactobacillus plantarum No. 14 in the gut of mice fed a high-fat diet. J Nutr 140:1963–1969. doi: 10.3945/jn.110.128082. [DOI] [PubMed] [Google Scholar]
- 5.Andersen JM, Barrangou R, Hachem MA, Lahtinen SJ, Goh YJ, Svensson B, Klaenhammer TR. 2012. Transcriptional analysis of prebiotic uptake and catabolism by Lactobacillus acidophilus NCFM. PLoS One 7:e44409. doi: 10.1371/journal.pone.0044409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilad O, Jacobsen S, Stuer-Lauridsen B, Pedersen MB, Garrigues C, Svensson B. 2010. Combined transcriptome and proteome analysis of Bifidobacterium animalis subsp. lactis BB-12 grown on xylo-oligosaccharides and a model of their utilization. Appl Environ Microbiol 76:7285–7291. doi: 10.1128/AEM.00738-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saulnier DM, Molenaar D, de Vos WM, Gibson GR, Kolida S. 2007. Identification of prebiotic fructooligosaccharide metabolism in Lactobacillus plantarum WCFS1 through microarrays. Appl Environ Microbiol 73:1753–1765. doi: 10.1128/AEM.01151-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goh YJ, Zhang C, Benson AK, Schlegel V, Lee JH, Hutkins RW. 2006. Identification of a putative operon involved in fructooligosaccharide utilization by Lactobacillus paracasei. Appl Environ Microbiol 72:7518–7530. doi: 10.1128/AEM.00877-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goh YJ, Lee JH, Hutkins RW. 2007. Functional analysis of the fructooligosaccharide utilization operon in Lactobacillus paracasei 1195. Appl Environ Microbiol 73:5716–5724. doi: 10.1128/AEM.00805-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montanari C, Sado Kamdem SL, Serrazanetti DI, Etoa FX, Guerzoni ME. 2010. Synthesis of cyclopropane fatty acids in Lactobacillus helveticus and Lactobacillus sanfranciscensis and their cellular fatty acids changes following short term acid and cold stresses. Food Microbiol 27:493–502. doi: 10.1016/j.fm.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 11.Zhang YM, Rock CO. 2008. Membrane lipid homeostasis in bacteria. Nat Rev Microbiol 6:222–233. doi: 10.1038/nrmicro1839. [DOI] [PubMed] [Google Scholar]
- 12.Ma DK, Li Z, Lu AY, Sun F, Chen S, Rothe M, Menzel R, Horvitz HR. 2015. Acyl-CoA dehydrogenase drives heat adaptation by sequestering fatty acids. Cell 161:1152–1163. doi: 10.1016/j.cell.2015.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murínová S, Dercova K. 2014. Response mechanisms of bacterial degraders to environmental contaminants on the level of cell walls and cytoplasmic membrane. Int J Microbiol 2014:873081. doi: 10.1155/2014/873081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Denich TJ, Beaudette LA, Lee H, Trevors JT. 2003. Effect of selected environmental and physico-chemical factors on bacterial cytoplasmic membranes. J Microbiol Methods 52:149–182. doi: 10.1016/S0167-7012(02)00155-0. [DOI] [PubMed] [Google Scholar]
- 15.Los DA, Mironov KS, Allakhverdiev SI. 2013. Regulatory role of membrane fluidity in gene expression and physiological functions. Photosynth Res 116:489–509. doi: 10.1007/s11120-013-9823-4. [DOI] [PubMed] [Google Scholar]
- 16.Wu C, Zhang J, Wang M, Du G, Chen J. 2012. Lactobacillus casei combats acid stress by maintaining cell membrane functionality. J Ind Microbiol Biotechnol 39:1031–1039. doi: 10.1007/s10295-012-1104-2. [DOI] [PubMed] [Google Scholar]
- 17.Hosseini Nezhad M, Hussain MA, Britz ML. 2015. Stress responses in probiotic Lactobacillus casei. Crit Rev Food Sci Nutr 55:740–749. doi: 10.1080/10408398.2012.675601. [DOI] [PubMed] [Google Scholar]
- 18.Arenahalli Ningegowda M, Siddalingaiya Gurudutt P. 2012. In vitro fermentation of prebiotics by Lactobacillus plantarum CFR 2194: selectivity, viability and effect of metabolites on beta-glucuronidase activity. World J Microbiol Biotechnol 28:901–908. doi: 10.1007/s11274-011-0887-z. [DOI] [PubMed] [Google Scholar]
- 19.Kaplan H, Hutkins RW. 2000. Fermentation of fructooligosaccharides by lactic acid bacteria and bifidobacteria. Appl Environ Microbiol 66:2682–2684. doi: 10.1128/AEM.66.6.2682-2684.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Chen C, Ai L, Zhou F, Zhou Z, Wang L, Zhang H, Chen W, Guo B. 2011. Complete genome sequence of the probiotic Lactobacillus plantarum ST-III. J Bacteriol 193:313–314. doi: 10.1128/JB.01159-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teusink B, van Enckevort FH, Francke C, Wiersma A, Wegkamp A, Smid EJ, Siezen RJ. 2005. In silico reconstruction of the metabolic pathways of Lactobacillus plantarum: comparing predictions of nutrient requirements with those from growth experiments. Appl Environ Microbiol 71:7253–7262. doi: 10.1128/AEM.71.11.7253-7262.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen G, Wang C, Shi T. 2011. Overview of available methods for diverse RNA-Seq data analyses. Sci China Life Sci 54:1121–1128. doi: 10.1007/s11427-011-4255-x. [DOI] [PubMed] [Google Scholar]
- 24.Lambert JM, Bongers RS, Kleerebezem M. 2007. Cre-lox-based system for multiple gene deletions and selectable-marker removal in Lactobacillus plantarum. Appl Environ Microbiol 73:1126–1135. doi: 10.1128/AEM.01473-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jo JH, Jeon CO, Lee DS, Park JM. 2007. Process stability and microbial community structure in anaerobic hydrogen-producing microflora from food waste containing kimchi. J Biotechnol 131:300–308. doi: 10.1016/j.jbiotec.2007.07.492. [DOI] [PubMed] [Google Scholar]
- 26.Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 27.Aricha B, Fishov I, Cohen Z, Sikron N, Pesakhov S, Khozin-Goldberg I, Dagan R, Porat N. 2004. Differences in membrane fluidity and fatty acid composition between phenotypic variants of Streptococcus pneumoniae. J Bacteriol 186:4638–4644. doi: 10.1128/JB.186.14.4638-4644.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lorquet F, Goffin P, Muscariello L, Baudry JB, Ladero V, Sacco M, Kleerebezem M, Hols P. 2004. Characterization and functional analysis of the poxB gene, which encodes pyruvate oxidase in Lactobacillus plantarum. J Bacteriol 186:3749–3759. doi: 10.1128/JB.186.12.3749-3759.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goffin P, Lorquet F, Kleerebezem M, Hols P. 2004. Major role of NAD-dependent lactate dehydrogenases in aerobic lactate utilization in Lactobacillus plantarum during early stationary phase. J Bacteriol 186:6661–6666. doi: 10.1128/JB.186.19.6661-6666.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zotta T, Ricciardi A, Guidone A, Sacco M, Muscariello L, Mazzeo MF, Cacace G, Parente E. 2012. Inactivation of ccpA and aeration affect growth, metabolite production and stress tolerance in Lactobacillus plantarum WCFS1. Int J Food Microbiol 155:51–59. doi: 10.1016/j.ijfoodmicro.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 31.Kremling A, Geiselmann J, Ropers D, de Jong H. 2015. Understanding carbon catabolite repression in Escherichia coli using quantitative models. Trends Microbiol 23:99–109. doi: 10.1016/j.tim.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 32.O'Connell KJ, Motherway MO, Liedtke A, Fitzgerald GF, Ross RP, Stanton C, Zomer A, van Sinderen D. 2014. Transcription of two adjacent carbohydrate utilization gene clusters in Bifidobacterium breve UCC2003 is controlled by LacI- and repressor open reading frame kinase (ROK)-type regulators. Appl Environ Microbiol 80:3604–3614. doi: 10.1128/AEM.00130-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teixeira JS, Abdi R, Su MS, Schwab C, Ganzle MG. 2013. Functional characterization of sucrose phosphorylase and scrR, a regulator of sucrose metabolism in Lactobacillus reuteri. Food Microbiol 36:432–439. doi: 10.1016/j.fm.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 34.Francke C, Kerkhoven R, Wels M, Siezen RJ. 2008. A generic approach to identify transcription factor-specific operator motifs; inferences for LacI-family mediated regulation in Lactobacillus plantarum WCFS1. BMC Genomics 9:145. doi: 10.1186/1471-2164-9-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sims IM, Ryan JL, Kim SH. 2014. In vitro fermentation of prebiotic oligosaccharides by Bifidobacterium lactis HN019 and Lactobacillus spp. Anaerobe 25:11–17. doi: 10.1016/j.anaerobe.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 36.Arboleya S, Salazar N, Solis G, Fernandez N, Gueimonde M, de los Reyes-Gavilan CG. 2013. In vitro evaluation of the impact of human background microbiota on the response to Bifidobacterium strains and fructo-oligosaccharides. Br J Nutr 110:2030–2036. doi: 10.1017/S0007114513001487. [DOI] [PubMed] [Google Scholar]
- 37.Rajkumar H, Kumar M, Das N, Kumar SN, Challa HR, Nagpal R. 2015. Effect of probiotic Lactobacillus salivarius UBL S22 and prebiotic fructo-oligosaccharide on serum lipids, inflammatory markers, insulin sensitivity, and gut bacteria in healthy young volunteers: a randomized controlled single-blind pilot study. J Cardiovasc Pharmacol Ther 20:289–298. doi: 10.1177/1074248414555004. [DOI] [PubMed] [Google Scholar]
- 38.Haas BJ, Chin M, Nusbaum C, Birren BW, Livny J. 2012. How deep is deep enough for RNA-Seq profiling of bacterial transcriptomes? BMC Genomics 13:734. doi: 10.1186/1471-2164-13-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gagarinova A, Emili A. 2012. Genome-scale genetic manipulation methods for exploring bacterial molecular biology. Mol Biosyst 8:1626–1638. doi: 10.1039/c2mb25040c. [DOI] [PubMed] [Google Scholar]
- 40.Chen C, Zhou F, Ren J, Ai L, Dong Y, Wu Z, Liu Z, Chen W, Guo B. 2014. Cloning, expression and functional validation of a β-fructofuranosidase from Lactobacillus plantarum. Process Biochem 49:758–767. doi: 10.1016/j.procbio.2014.02.013. [DOI] [Google Scholar]
- 41.Cao-Hoang L, Marechal PA, Le-Thanh M, Gervais P, Wache Y. 2008. Fluorescent probes to evaluate the physiological state and activity of microbial biocatalysts: a guide for prokaryotic and eukaryotic investigation. Biotechnol J 3:890–903. doi: 10.1002/biot.200700206. [DOI] [PubMed] [Google Scholar]
- 42.Tuller G, Nemec T, Hrastnik C, Daum G. 1999. Lipid composition of subcellular membranes of an FY1679-derived haploid yeast wild-type strain grown on different carbon sources. Yeast 15:1555–1564. doi:. [DOI] [PubMed] [Google Scholar]
- 43.Barbaro SE, Trevors JT, Inniss WE. 1999. Effect of different carbon sources on membrane permeability, membrane fluidity, and fatty acid composition of a psychrotrophic Acinetobacter sp. HH1-1 during growth at low temperatures and after cold shock. World J Microbiol Biotechnol 15:683–692. [Google Scholar]
- 44.Van der Meulen R, Avonts L, De Vuyst L. 2004. Short fractions of oligofructose are preferentially metabolized by Bifidobacterium animalis DN-173 010. Appl Environ Microbiol 70:1923–1930. doi: 10.1128/AEM.70.4.1923-1930.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goffin P, Muscariello L, Lorquet F, Stukkens A, Prozzi D, Sacco M, Kleerebezem M, Hols P. 2006. Involvement of pyruvate oxidase activity and acetate production in the survival of Lactobacillus plantarum during the stationary phase of aerobic growth. Appl Environ Microbiol 72:7933–7940. doi: 10.1128/AEM.00659-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lawley B, Sims IM, Tannock GW. 2013. Whole-transcriptome shotgun sequencing (RNA-seq) screen reveals upregulation of cellobiose and motility operons of Lactobacillus ruminis L5 during growth on tetrasaccharides derived from barley beta-glucan. Appl Environ Microbiol 79:5661–5669. doi: 10.1128/AEM.01887-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Porcheron G, Chanteloup NK, Trotereau A, Bree A, Schouler C. 2012. Effect of fructooligosaccharide metabolism on chicken colonization by an extra-intestinal pathogenic Escherichia coli strain. PLoS One 7:e35475. doi: 10.1371/journal.pone.0035475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adamberg S, Tomson K, Vija H, Puurand M, Kabanova N, Visnapuu T, Jogi E, Alamae T, Adamberg K. 2014. Degradation of fructans and production of propionic acid by Bacteroides thetaiotaomicron are enhanced by the shortage of amino acids. Front Nutr 1:21. doi: 10.3389/fnut.2014.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Falony G, Lazidou K, Verschaeren A, Weckx S, Maes D, De Vuyst L. 2009. In vitro kinetic analysis of fermentation of prebiotic inulin-type fructans by Bifidobacterium species reveals four different phenotypes. Appl Environ Microbiol 75:454–461. doi: 10.1128/AEM.01488-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tejero-Sariñena S, Barlow J, Costabile A, Gibson GR, Rowland I. 2013. Antipathogenic activity of probiotics against Salmonella Typhimurium and Clostridium difficile in anaerobic batch culture systems: is it due to synergies in probiotic mixtures or the specificity of single strains? Anaerobe 24:60–65. doi: 10.1016/j.anaerobe.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 51.Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, Tobe T, Clarke JM, Topping DL, Suzuki T, Taylor TD, Itoh K, Kikuchi J, Morita H, Hattori M, Ohno H. 2011. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 469:543–547. doi: 10.1038/nature09646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.