

Abstract
Previously, we successfully cloned a d-cycloserine (d-CS) biosynthetic gene cluster consisting of 10 open reading frames (designated dcsA to dcsJ) from d-CS-producing Streptomyces lavendulae ATCC 11924. In this study, we put four d-CS biosynthetic genes (dcsC, dcsD, dcsE, and dcsG) in tandem under the control of the T7 promoter in an Escherichia coli host. SDS-PAGE analysis demonstrated that the 4 gene products were simultaneously expressed in host cells. When l-serine and hydroxyurea (HU), the precursors of d-CS, were incubated together with the E. coli resting cell suspension, the cells produced significant amounts of d-CS (350 ± 20 μM). To increase the productivity of d-CS, the dcsJ gene, which might be responsible for the d-CS excretion, was connected downstream of the four genes. The E. coli resting cells harboring the five genes produced d-CS at 660 ± 31 μM. The dcsD gene product, DcsD, forms O-ureido-l-serine from O-acetyl-l-serine (OAS) and HU, which are intermediates in d-CS biosynthesis. DcsD also catalyzes the formation of l-cysteine from OAS and H2S. To repress the side catalytic activity of DcsD, the E. coli chromosomal cysJ and cysK genes, encoding the sulfite reductase α subunit and OAS sulfhydrylase, respectively, were disrupted. When resting cells of the double-knockout mutant harboring the four d-CS biosynthetic genes, together with dcsJ, were incubated with l-serine and HU, the d-CS production was 980 ± 57 μM, which is comparable to that of d-CS-producing S. lavendulae ATCC 11924 (930 ± 36 μM).
INTRODUCTION
The cyclic structural analog of the amino acid d-cycloserine (d-CS) (Fig. 1) is a broad-spectrum antibiotic produced by Streptomyces lavendulae and Streptomyces garyphalus (1). The antibiotic inhibits both alanine racemase and d-alanyl–d-alanine ligase, which are necessary for the biosynthesis of peptidoglycan in the bacterial cell wall (2, 3). Rifampin and isoniazid have been clinically used for the treatment of tuberculosis caused by infection with Mycobacterium tuberculosis (4). However, M. tuberculosis that is resistant to these drugs has recently occurred. Presently, d-CS is clinically used as a second-line-of-defense drug against these antibiotic-resistant M. tuberculosis strains (4). In this connection, it has been shown that M. smegmatis overproducing alanine racemase is resistant to d-CS (5, 6). Recently, d-CS has been shown to function as a partial agonist for the N-methyl-d-aspartate receptor. As a result, the application of d-CS for the treatment of some psychological dysfunctions has been extensively studied (7–9).
FIG 1.

Biosynthetic pathway for d-CS.
Our group has successfully cloned a d-CS biosynthetic gene cluster from the chromosomal DNA of d-CS-producing S. lavendulae ATCC 11924, which is composed of 10 open reading frames, designated dcsA to dcsJ (10). The functions of dcsI and dcsJ had previously been analyzed using the corresponding genes cloned from other d-CS-producing strains, i.e., S. lavendulae ATCC 25233 (11) and S. garyphalus (CSH) 5-12 (12), demonstrating that both gene products are responsible for self-resistance in the d-CS producer. Gene disruption and recombinant protein analyses have demonstrated that the revised d-CS biosynthetic pathway is as follows. l-Serine is O-acetylated by DcsE to generate O-acetyl-l-serine (OAS) (10, 13). The resultant OAS reacts with hydroxyurea (HU) to yield O-ureido-l-serine by use of DcsD, which is a pyridoxal phosphate-dependent enzyme (14). O-Ureido-l-serine is racemized by DcsC (14, 15), followed by cyclization with DcsG, which is a member of the ATP-grasp fold family of proteins (10, 14) (Fig. 1).
We have previously hypothesized that l-arginine, as a precursor in the d-CS biosynthetic pathway, must be hydroxylated by nitric oxide synthase (NOS) expressed in d-CS-producing S. lavendulae (10). However, we have corrected the hypothesis as follows: DcsA as a heme protein, but not as an NOS protein, contributes to the formation of Nω-hydroxy-l-arginine (16). As shown in Fig. 1, HU is generated by the hydrolysis of Nω-hydroxy-l-arginine with DcsB (10).
In recent years, the heterologous expression of secondary metabolic pathways using a surrogate host, such as Escherichia coli, has emerged as an effective way of producing natural products. However, practical antibiotics have not yet been successfully produced using E. coli as a host. Our goal is to realize high production of d-CS by expressing its biosynthetic genes (dcsA to -dcsE and dcsG) in E. coli as a host cell. In this study, we tried to introduce the four d-CS biosynthetic genes (dcsC, dcsD, dcsE, and dcsG) into E. coli cells to express these gene products and to construct a d-CS production system by incubating resting cells with precursors of d-CS. We show that coexpression of the four d-CS biosynthetic genes and a self-resistance gene, dcsJ, which encodes a putative d-CS efflux protein from d-CS-producing S. lavendulae, in combination with metabolic engineering of the E. coli host, is effective for the high production of the antibiotic.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
S. lavendulae ATCC 11924 was maintained at 28°C on FB agar medium (17). To produce d-CS, the ATCC 11924 strain was cultivated at 28°C in liquid medium B (18). E. coli DH5α (19) and plasmids pUC19 (20), pBR322 (21), pTA2 (Toyobo, Japan), and pGEM-T (Promega, USA) were used for cloning, sequencing, and creating gene disruption constructs. Plasmid pRedET (Gene Bridges, Germany) was used for gene disruption by Red/ET recombination (22). pET-28a(+) (Merck, Germany) and pBR322 were used as the sources of kanamycin and tetracycline resistance genes, respectively. E. coli BL21(DE3) and plasmid pET-21a(+) (Merck) were used for protein expression. All E. coli strains were cultivated in Luria broth or on Luria agar supplemented with the appropriate antibiotics when necessary (23).
DNA manipulations.
Plasmid DNA was isolated from E. coli using the Wizard Plus Minipreps DNA purification system (Promega). DNA fragments separated in an agarose gel were extracted using a GeneClean III kit (MP Biomedicals, USA). Genomic DNA of Streptomyces was isolated as described previously (24).
Construction of expression vectors for bioconversion.
The primers used in this study are listed in Table 1. dcsE was amplified by PCR using KOD-Plus DNA polymerase (Toyobo) and genomic DNA of the ATCC 11924 strain as a template with primers dcsE_F and dcsE_R. The amplified dcsE was cloned into a pGEM-T vector to generate pGEM/dcsE. After confirmation of the nucleotide sequence, dcsE was cut off from pGEM/dcsE by digestion with NdeI and HindIII, followed by ligation to the same sites of pET-21a(+) to yield pET-21a(+)/dcsE. The dcsD gene was amplified by PCR with primers dcsD_F and dcsD_R and inserted into pGEM-T to generate pGEM/dcsD. The dcsD gene obtained from pGEM/dcsD after digestion with NdeI and HindIII was ligated to the same sites of pET-21a(+) to yield pET-21a(+)/dcsD. The dcsD fragment was cut off from pET-21a(+)/dcsD by digestion with XbaI and HindIII and ligated to pET-21a(+)/dcsE previously digested with SpeI and HindIII to generate pET-21a(+)/dcsED. Each dcsC or dcsG gene was amplified by PCR with the primer sets listed in Table 1 and subcloned into pET-21a(+) to yield pET-21a(+)/dcsC and pET-21a(+)/dcsG, respectively. Subsequently, dcsC and dcsG were successively connected to pET-21a(+)/dcsED using the same procedure as described for dcsD to generate pET-21a(+)/dcsEDCG. A d-CS self-resistance gene, designated dcsJ (10), was amplified by PCR with primers dcsJ_F and dcsJ_R; it was then inserted into pET-21a(+)/dcsEDCG in the same way to yield pET-21a(+)/dcsEDCGJ.
TABLE 1.
Primers used in this study
| Name | Sequence (5′→3′) |
|---|---|
| dcsE_F | CATATGAGGGAATTCATACCCCCGGCCTCC |
| dcsE_R | AAGCTTGGACTAGTTCAGGCGACGATCGCCAGGAACTTCG |
| dcsD_F | CATATGCCTCTGTTTAACAGCATCCTCGACACCATC |
| dcsD_R | AAGCTTGGACTAGTCTAGAGGCCGGAGCCGGTGTC |
| dcsC_F | CATATGATACGCATGAGAACGCCGAGCACG |
| dcsC_R | AAGCTTGGACTAGTTTAAACAGAGGCATGGAGGAAGGTTCCTTCG |
| dcsG_F | CATATGGGCATCCTCGCCTTGGTCACC |
| dcsG_R | AAGCTTGGACTAGTCTAGGGCTTGAGCCGTTCGGC |
| dcsJ_F | CATATGGATGCACGACACAGCACGATCACGTTGA |
| dcsJ_R | AAGCTTGGACTAGTCTACGCTCTCGCCCGCTGCGT |
| cysJ_U_F | GAATTCTTTACCCACCAGGCCCACTTCCGCCA |
| cysJ_U_R | TCTAGAGAGCGCCGTTATCAGCGAGATGTCTAC |
| cysJ_D_F | CTGCAGCGGAAGGTGGGACCTGTGTCGTCAT |
| cysJ_D_R | GCATGCCACCAGATGCGCACCATACTCCAGTGATTC |
| Tetr_F | TCTAGAAATAAGGGCGACACGGAAATGTTGAATACTCATACTCTTCCT |
| Tetr_R | CTGCAGGTCGCAGACGTTTTGCAGCAGCAGTC |
| cysJ_dis_F | ATGATCAGATTCTGGTTCGCCGTAATGCGGA |
| cysJ_dis_R | CGGAGAATATCGTCACCACAAATAACGCCACA |
| cysJ_check_F | CATCTCCTTGCATGCACCATTCCTTGC |
| cysJ_check_R | GCCGTGACGACGTAAATGATGCCAGAAT |
| cysK_U_F | GAATTCCGAACATATCCTAACTGTCCATTGCGCAATTACCC |
| cysK_U_R | AAGCTTCTCCGGACCGGTGGTTTTTTCGTGAATTTCAG |
| cysK_D_F | GCATGCGGGTGAGCGTTATTTAAGCACCGCATTGTTTGC |
| cysK_D_R | GTCGACCTCTGCACCTTCAACGTCACGAACCGTA |
| Kmr_F | AAGCTTTTGAAGTGGTGGCCTAACTACGGCTACACTAGAA |
| Kmr_R | GCATGCTTTTTCGCCCTTTGACGTTGGAGTCCACGTTCTTTAATA |
| cysK_dis_F | CTAACTGTCCATTGCGCAATTACCCGGT |
| cysK_dis_R | CGTACCAATGTTAGCGCATACTTCTACCTG |
| cysK_check_F | CCGATGCGCCAGAGTTGTTTCTGAAACA |
| cysK_check_R | CGTCGCGCAGTTCCTGTTTGTAGATTTC |
Protein analysis.
Whole-cell proteins of E. coli BL21(DE3) that expressed d-CS biosynthetic genes were analyzed by SDS-PAGE (25).
Disruption of cysJ and cysK located on the E. coli BL21(DE3) chromosome.
A 1.5-kb DNA fragment upstream of cysJ was amplified by PCR with primers cysJ_U_F and cysJ_U_R; it was then cloned into pGEM-T to yield pGEM/ΔcysJ-U. After confirmation of the nucleotide sequence, a 1.5-kb DNA fragment, which was cut off from pGEM/ΔcysJ-U by digestion with EcoRI and XbaI, was inserted into the same sites of pUC19 to generate pUC19/ΔcysJ-U. A 1.6-kb DNA fragment downstream of cysJ was amplified by PCR with primers cysJ_D_F and cysJ_D_R, inserted into pGEM-T, and subsequently cloned into the PstI/SphI sites of pUC19/ΔcysJ-U to yield pUC19/ΔcysJ-UD. A pBR322-derived tetracycline resistance gene that includes its promoter and terminator (1.8 kb) was amplified by PCR with primers Tetr_F and Tetr_R, cloned into pGEM-T, and inserted into the XbaI/PstI sites of pUC19/ΔcysJ-UD to construct pUC19/ΔcysJ. Using pUC19/ΔcysJ as a template DNA, a disruption cassette for cysJ (4.3 kb) was amplified by PCR with primers cysJ_dis_F and cysJ_dis_R. With the cysJ cassette, cysJ, which is located on the E. coli BL21(DE3) chromosome, was disrupted by Red/ET recombination using the Quick and Easy E. coli gene deletion kit (Gene Bridges). A cysJ disruption mutant (ΔcysJ) was selected on an LB agar plate containing tetracycline, and the authenticity of the mutant was confirmed by PCR with primers cysJ_check_F and cysJ_check_R.
DNA fragments upstream (1.5 kb) and downstream (1.6 kb) of cysK were amplified by PCR with the primer sets listed in Table 1 and cloned into the EcoRI/HindIII and SphI/SalI sites of pBR322, respectively, to generate pBR322/ΔcysK-UD. A kanamycin resistance gene that includes its promoter and terminator (1.4 kb) on pET-28a(+) was amplified by PCR with primers Kmr_F and Kmr_R and inserted into the HindIII/SphI sites of pBR322/ΔcysK-UD to construct pBR322/ΔcysK. A disruption cassette for cysK (4.5 kb) was amplified by PCR with primers cysK_dis_F and cysK_dis_R, and a cysK-disrupted mutant (ΔcysK) was obtained using the same procedure as described for ΔcysJ, with the exception of the selection for kanamycin. The validity of ΔcysK was checked by PCR with primers cysK_check_F and cysK_check_R. A cysJ cysK double mutant (ΔcysJ ΔcysK) was created by introducing the cysK disruption cassette into ΔcysJ, selected with tetracycline and kanamycin, and confirmed by PCR.
Generation of d-CS from l-serine supplemented with HU by bioconversion.
E. coli BL21(DE3) and its mutants harboring pET-21a(+)/dcsEDCG or pET-21a(+)/dcsEDCGJ were cultivated in 4 ml of LB supplemented with reagents of the Overnight Express autoinduction system 1 (GE Healthcare, United Kingdom) at 28°C for 24 h. The system allows the induction of T7 RNA polymerase under the control of the lac promoter automatically at the stationary phase of growth based on the medium components. Subsequently, the expression of desired proteins under the control of the T7 promoter is induced. The E. coli cells were collected by centrifugation, washed once with 10 mM potassium phosphate buffer (pH 7.2), and resuspended in the same buffer to an optical density at 600 nm (OD600) of 1.0. The cell suspension (4 ml), called resting cells here, was incubated with l-serine (2.5 mM) and HU (2.5 mM) (Sigma-Aldrich, USA) at 28°C for 4 h.
Assay of d-CS production.
After incubation of the resting cells with the addition of the precursors in the biosynthetic pathway, the cells were centrifuged, and the resulting supernatant fluid was filtered through a 0.2-μm-pore-size filter (Advantech, Japan). An aliquot of the fluid was analyzed by HPLC as described previously (26) to evaluate the amount of d-CS, except that the detection was carried out at 226 nm. Briefly, a solution of authentic d-CS (Wako, Japan) was mixed with l-tryptophan (1 mM) as an internal standard at a volume ratio of 3:1, followed by analysis with high-pressure liquid chromatography (HPLC). A standard curve for quantitation of d-CS was generated by plotting the ratio of peak areas (d-CS to l-tryptophan) versus the concentrations of d-CS. The sample solution was analyzed in the same way as the authentic d-CS. The concentration of d-CS was calculated from the ratio of peak areas and the standard curve. The d-CS titer was expressed as the mean ± standard error (SE) from three independent experiments.
Bioassay.
After bioconversion, the reaction mixture was centrifuged to remove the E. coli cells. The resulting supernatant fluid was sterilized by filtration. Fifty microliters of the solution was applied on a paper disk, which was put on a bioassay plate overlaid with spores of Bacillus subtilis IFO3134 as a test organism. Bioassay was carried out at 37°C for 16 h.
Mass spectrometry.
The eluate from the HPLC column was collected and freeze-dried. The resulting residue was dissolved in methanol-water (1:1) and analyzed by electrospray ionization-time of flight (ESI-TOF) mass spectrometry. ESI-TOF mass analysis was done using a TripleTOF 5600 instrument (AB Sciex, USA). Mass spectra were obtained in the negative-ion mode.
Statistical analysis.
Statistical analysis was carried out using Student's t test. P values of <0.05 were used to identify statistically significant differences.
RESULTS AND DISCUSSION
Expression of four d-CS biosynthetic genes in E. coli and formation of d-CS.
Four genes, dcsE, dcsD, dcsC, and dcsG (in order), were located polycistronically under the control of the T7 promoter using an E. coli expression vector, pET-21a(+), to create pET-21a(+)/dcsEDCG (Fig. 2a). In this case, a ribosome-binding sequence derived from pET-21a(+) was designed to be located in front of each gene. SDS-PAGE analysis of the whole-cell protein of E. coli BL21(DE3) harboring pET-21a(+)/dcsEDCG showed that the four proteins were obviously produced, but the expression of DcsG was relatively low (Fig. 2b).
FIG 2.

(a) Vectors used for bioconversion in this study. T7p, T7 promoter; RBS, ribosome-binding sequence; T7t, T7 terminator; amp, ampicillin resistance gene; ori, replication origin; H, HindIII; N, NdeI; S, SpeI; X, XbaI. (b) SDS-PAGE analysis of E. coli BL21(DE3) harboring various plasmids. Whole-cell proteins of E. coli BL21(DE3) harboring the indicate plasmids and grown at 28°C for 24 h were analyzed by SDS-PAGE (25). Lane 1, molecular mass standards; lane 2, pET-21a(+); lane 3, pET-21a(+)/dcsC; lane 4, pET-21a(+)/dcsD; lane 5, pET-21a(+)/dcsE; lane 6, pET-21a(+)/dcsG; lane 7, pET-21a(+)/dcsEDCG.
The resting cell suspension of E. coli harboring pET-21a(+)/dcsEDCG was incubated with l-serine and HU dissolved in potassium phosphate buffer at 28°C. HPLC analysis of the supernatant fluid from the incubation mixture showed that a peak corresponding to d-CS was obtained at the retention time of 17 min (Fig. 3d). When HU was eliminated from the reaction mixture, the peak did not appear (Fig. 3c). The peak also did not appear when l-serine and HU were incubated with cells of E. coli harboring pET-21a(+) without the insertion of dcsEDCG (Fig. 3b). In addition, the supernatant fluid from the incubation mixture, which is composed of both substrates (l-serine and HU) and the cells of E. coli harboring pET-21a(+)/dcsEDCG, displayed an antibiotic activity against Bacillus subtilis (Fig. 4). Furthermore, a negative-ion peak of m/z 101.0 (molecular weight [MW] of d-CS, 102.09) was observed by ESI-TOF mass analysis, demonstrating that d-CS is produced by the bioconversion system. To determine the suitable time for bioconversion, l-serine and HU were incubated with E. coli cells harboring pET-21a(+)/dcsEDCG at 28°C. The d-CS titer was determined every 1 h until 8 h (Fig. 5). Since the d-CS titer was saturated at 4 h, the d-CS titers were evaluated after 4 h of incubation in all of the experiments described below. When l-serine and HU (each at 2.5 mM) were incubated together with the cell suspension, the d-CS production was 350 ± 20 μM (Fig. 6), suggesting that the added substrates are not fully used for the synthesis of d-CS. Because the d-CS production by the mutant E. coli BL21(DE3) strain increased, as described below, the saturation of the antibiotic production may be due to the limitation of OAS rather than to the depletion of ATP, which is necessary for the catalytic activity of DcsG (14).
FIG 3.

Analysis of bioconverted product by HPLC. The supernatant fluid from the indicated mixtures incubated at 28°C for 4 h was analyzed by HPLC using a Senshu Pak SCX-1251-N column. (a) Authentic d-CS. (b) Cells of E. coli BL21(DE3) harboring pET-21a(+) with l-serine and HU (each at 2.5 mM). (c) Cells of E. coli BL21(DE3) harboring pET-21a(+)/dcsEDCG with l-serine (2.5 mM). (d) Cells of E. coli BL21(DE3) harboring pET-21a(+)/dcsEDCG with l-serine and HU (each at 2.5 mM).
FIG 4.
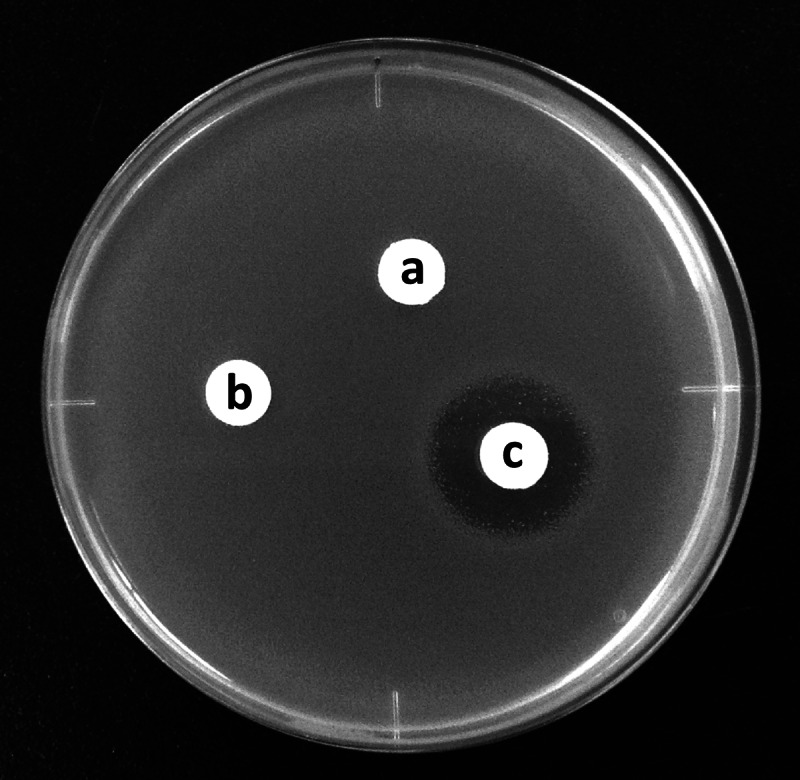
Antibiotic activity of bioconverted mixture. (a) l-Serine and HU (each at 2.5 mM) in 10 mM potassium phosphate (pH 7.2) as a control. (b) Incubation mixture containing both substrates and cells of E. coli BL21(DE3) harboring pET-21a(+). (c) Incubation mixture containing both substrates and cells of E. coli BL21(DE3) harboring pET-21a(+)/dcsEDCG. The supernatant fluid from each mixture was assayed for antibacterial activity using B. subtilis IFO3134 as a test organism.
FIG 5.

Time course of d-CS production. Cells of E. coli harboring pET-21a(+)/dcsEDCG were incubated with l-serine (2.5 mM) and HU (2.5 mM) at 28°C for 8 h. The amount of d-CS produced was determined by HPLC analysis every 1 h.
FIG 6.

Production of d-CS obtained by bioconversion using a resting cell suspension of E. coli BL21(DE3) or its mutants harboring the indicated plasmid. l-Serine (2.5 mM) and HU (2.5 mM) were bioconverted to d-CS by E. coli BL21(DE3) and its mutants carrying pET-21a(+)/dcsEDCG or pET-21a(+)/dcsEDCGJ at 28°C for 4 h; the amount of d-CS was determined by HPLC analysis. *, P < 0.05; **, P < 0.01.
Yield improvement by coexpression of dcsJ, which encodes a putative d-CS efflux protein.
We have previously cloned a self-resistance gene, designated orfB, from a d-CS-producing S. garyphalus (CSH) 5-12 (12). The orfB gene product deduced from the amino acid sequence was similar to several membrane proteins, suggesting that OrfB functions to efflux d-CS outside the cells (12). Since the dcsJ gene product displays a high sequence similarity (97%) with OrfB, the protein is deduced to have the same function as OrfB (10). To increase the production of d-CS, we examined the coexpression of dcsJ with dcsEDCG by the construction of pET-21a(+)/dcsEDCGJ (Fig. 2a). Resting cells of E. coli BL21(DE3) harboring pET-21a(+)/dcsEDCGJ which were incubated with l-serine and HU produced 650 ± 31 μM d-CS (Fig. 6), demonstrating that the self-resistance gene dcsJ increases d-CS production significantly. This increment seems to have occurred due to the high efflux of d-CS toward outside the cells. However, the added l-serine and HU, which were incubated with resting cells harboring pET-21a(+)/dcsEDCGJ as well as those harboring pET-21a(+)/dcsEDCG, were not completely utilized in the synthesis of d-CS.
High production of d-CS by metabolic engineering of the E. coli host.
We have previously shown that OAS, an intermediate in the d-CS biosynthetic pathway, is synthesized by the dcsE product (10). On the other hand, since E. coli cells possess cysE, which encodes l-serine-O-acetyltransferase, OAS can be supplied by the gene product. However, it is known that the enzymatic activity of CysE is strongly repressed in the presence of l-cysteine (27). In this study, since the E. coli cells were grown in LB broth, l-cysteine must be tightly bound to the CysE protein. Therefore, it is likely that OAS is supplied from l-serine and acetyl coenzyme A (acetyl-CoA) by the catalytic activity of DcsE. Because the l-serine-O-acetyltransferase activity of DcsE is relatively low (13), there may be a small OAS pool in the E. coli cells. Moreover, OAS is consumed by CysK, an OAS sulfhydrylase that forms l-cysteine from OAS and H2S as substrates. In addition, DcsD catalyzes the same reaction as does CysK, and the OAS sulfhydrylase activity of DcsD is superior to the enzymatic ability to synthesize O-ureido-l-serine (14).
One of the strategies for increasing OAS, which is necessary for d-CS synthesis, is to reduce the above reactions. For this purpose, we disrupted both cysJ and cysK on the E. coli BL21(DE3) chromosome. cysJ encodes the α subunit of sulfite reductase, which catalyzes the synthesis of H2S (28). Using the Red/ET recombination method (22), we made two single-knockout mutants, in which cysJ or cysK on the chromosome of a parental E. coli BL21(DE3) strain was knocked out (designated ΔcysJ or ΔcysK, respectively), and a cysJ cysK double-knockout mutant (designated ΔcysJ ΔcysK). The authenticity of each mutant was confirmed by PCR. Although the d-CS production by each ΔcysJ and ΔcysK mutant harboring pET-21a(+)/dcsEDCG scarcely increased compared with that by the parental BL21(DE3) strain, the d-CS titer was significantly increased to 820 ± 36 μM (Fig. 6) when using the ΔcysJ ΔcysK mutant as a host; this shows that the double mutation effectively increases the d-CS yield. This was also observed when using pET-21a(+)/dcsEDCGJ, and the d-CS titer was significantly increased to 980 ± 57 μM when the ΔcysJ ΔcysK mutant with pET-21a(+)/dcsEDCGJ was used as a biocatalyst (Fig. 6). The d-CS titer obtained by this conversion system is comparable to that for the d-CS-producing microorganism S. lavendulae ATCC 11924 (930 ± 36 μM).
In the present study, we established a bioconversion system for the production of d-CS that uses an E. coli resting cell suspension and l-serine supplemented with HU as precursors of the antibiotic biosynthesis. The d-CS produced by the system can be easily purified from the incubation mixture using a chromatographic technique. Therefore, the system will be a powerful tool for synthesizing a practical antibiotic. The establishment of a bioconversion system to synthesis HU is in progress. In the near future, a complete bioconversion system for d-CS will be established using the E. coli resting cell suspension supplemented with l-serine and l-arginine as substrates.
ACKNOWLEDGMENTS
Part of this work was supported by JSPS KAKENHI grant number 25460119.
DNA sequence determination was carried out at the Analysis Center of Life Science, Hiroshima University.
REFERENCES
- 1.Harris DA, Ruger M, Reagan MA, Wolf FJ, Peck RL, Wallick H, Woodruff HB. 1955. Discovery, development, and antimicrobial properties of d-4-amino-3-isoxazolidone (oxamycin), a new antibiotic produced by Streptomyces garyphalus n. sp. Antibiot Chemother 5:183–190. [PubMed] [Google Scholar]
- 2.Lambert MP, Neuhaus FC. 1972. Mechanism of d-cycloserine action: alanine racemase from Escherichia coli W. J Bacteriol 110:978–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neuhaus FC, Lynch JL. 1964. The enzymatic synthesis of d-alanyl-d-alanine. III. On the inhibition of d-alanyl-d-alanine synthetase by the antibiotic d-cycloserine. Biochemistry 3:471–480. [DOI] [PubMed] [Google Scholar]
- 4.Humma LM. 1996. Prevention and treatment of drug-resistant tuberculosis. Am J Health Syst Pharm 53:2291–2298. [DOI] [PubMed] [Google Scholar]
- 5.Cáceres NE, Harris NB, Wellehan JF, Feng Z, Kapur V, Barletta RG. 1997. Overexpression of the d-alanine racemase gene confers resistance to d-cycloserine in Mycobacterium smegmatis. J Bacteriol 179:5046–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feng Z, Barletta RG. 2003. Roles of Mycobacterium smegmatis d-alanine:d-alanine ligase and d-alanine racemase in the mechanisms of action of and resistance to the peptidoglycan inhibitor d-cycloserine. Antimicrob Agents Chemother 47:283–291. doi: 10.1128/AAC.47.1.283-291.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goff DC, Tsai G, Levitt J, Amico E, Manoach D, Schoenfeld DA, Hayden DL, McCarley R, Coyle JT. 1999. Placebo-controlled trial of d-cycloserine added to conventional neuroleptics in patients with schizophrenia. Arch Gen Psychiatry 56:21–27. doi: 10.1001/archpsyc.56.1.21. [DOI] [PubMed] [Google Scholar]
- 8.Herberg LJ, Rose IC. 1990. Effects of d-cycloserine and cycloleucine, ligands for the NMDA-associated strychnine-insensitive glycine site, on brain-stimulation reward and spontaneous locomotion. Pharmacol Biochem Behav 36:735–738. doi: 10.1016/0091-3057(90)90069-T. [DOI] [PubMed] [Google Scholar]
- 9.Leeson PD, Iversen LL. 1994. The glycine site on the NMDA receptor: structure-activity relationships and therapeutic potential. J Med Chem 37:4053–4067. doi: 10.1021/jm00050a001. [DOI] [PubMed] [Google Scholar]
- 10.Kumagai T, Koyama Y, Oda K, Noda M, Matoba Y, Sugiyama M. 2010. Molecular cloning and heterologous expression of a biosynthetic gene cluster for the antitubercular agent d-cycloserine produced by Streptomyces lavendulae. Antimicrob Agents Chemother 54:1132–1139. doi: 10.1128/AAC.01226-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noda M, Kawahara Y, Ichikawa A, Matoba Y, Matsuo H, Lee DG, Kumagai T, Sugiyama M. 2004. Self-protection mechanism in d-cycloserine-producing Streptomyces lavendulae: gene cloning, characterization, and kinetics of its alanine racemase and d-alanyl-d-alanine ligase, which are target enzymes of d-cycloserine. J Biol Chem 279:46143–46152. doi: 10.1074/jbc.M404603200. [DOI] [PubMed] [Google Scholar]
- 12.Matsuo H, Kumagai T, Mori K, Sugiyama M. 2003. Molecular cloning of a d-cycloserine resistance gene from d-cycloserine-producing Streptomyces garyphalus. J Antibiot 56:762–767. doi: 10.7164/antibiotics.56.762. [DOI] [PubMed] [Google Scholar]
- 13.Oda K, Matoba Y, Kumagai T, Noda M, Sugiyama M. 2013. Crystallographic study to determine the substrate specificity on an l-serine-acetylating enzyme found in the d-cycloserine biosynthetic pathway. J Bacteriol 195:1741–1749. doi: 10.1128/JB.02085-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uda N, Matoba Y, Kumagai T, Oda K, Noda M, Sugiyama M. 2013. Establishment of an in vitro d-cycloserine-synthesizing system by using O-ureido-l-serine synthase and d-cycloserine synthetase found in the biosynthetic pathway. Antimicrob Agents Chemother 57:2603–2612. doi: 10.1128/AAC.02291-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dietrich D, van Belkum MJ, Vederas JC. 2012. Characterization of DcsC, a PLP-independent racemase involved in the biosynthesis of d-cycloserine. Org Biomol Chem 10:2248–2254. doi: 10.1039/c2ob06864h. [DOI] [PubMed] [Google Scholar]
- 16.Kumagai T, Takagi K, Koyama Y, Matoba Y, Oda K, Noda M, Sugiyama M. 2012. Heme protein and hydroxyarginase necessary for biosynthesis of d-cycloserine. Antimicrob Agents Chemother 56:3682–3689. doi: 10.1128/AAC.00614-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ikeda K, Masujima T, Suzuki K, Sugiyama M. 1996. Cloning and sequence analysis of the highly expressed melanin-synthesizing gene operon from Streptomyces castaneoglobisporus. Appl Microbiol Biotechnol 45:80–85. doi: 10.1007/s002530050652. [DOI] [PubMed] [Google Scholar]
- 18.Yanagimoto M, Enatsu T. 1983. Regulation of a blue pigment production by γ-nonalactone in Streptomyces sp. J Ferment Technol 61:545–550. [Google Scholar]
- 19.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580. doi: 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 20.Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 21.Bolivar F, Rodriguez RL, Greene PJ, Betlach MC, Heyneker HL, Boyer HW, Crosa JH, Falkow S. 1977. Construction and characterization of new cloning vehicles. II. A multipurpose cloning system. Gene 2:95–113. [PubMed] [Google Scholar]
- 22.Zhang Y, Buchholz F, Muyrers JPP, Stewart AF. 1998. A new logic for DNA engineering using recombination in Escherichia coli. Nat Genet 20:123–128. doi: 10.1038/2417. [DOI] [PubMed] [Google Scholar]
- 23.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 24.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. 2000. Practical Streptomyces genetics. The John Innes Foundation, Norwich, United Kingdom. [Google Scholar]
- 25.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 26.Kitani S, Yamada Y, Nihira T. 2001. Gene replacement analysis of the butyrolactone autoregulator receptor (FarA) reveals that FarA acts as a novel regulator in secondary metabolism of Streptomyces lavendulae FRI-5. J Bacteriol 183:4357–4363. doi: 10.1128/JB.183.14.4357-4363.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hindson VJ. 2003. Serine acetyltransferase of Escherichia coli: substrate specificity and feedback control by cysteine. Biochem J 375:745–752. doi: 10.1042/bj20030429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blattner FR, GPlunkett 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]