

Abstract
Forest fires are a common natural disturbance in forested ecosystems and have a large impact on the microbial communities in forest soils. The response of soil fungal communities to forest fire is poorly documented. Here, we investigated fungal community structure and function across a 152-year boreal forest fire chronosequence using high-throughput sequencing of the internal transcribed spacer 2 (ITS2) region and a functional gene array (GeoChip). Our results demonstrate that the boreal forest soil fungal community was most diverse soon after a fire disturbance and declined over time. The differences in the fungal communities were explained by changes in the abundance of basidiomycetes and ascomycetes. Ectomycorrhizal (ECM) fungi contributed to the increase in basidiomycete abundance over time, with the operational taxonomic units (OTUs) representing the genera Cortinarius and Piloderma dominating in abundance. Hierarchical cluster analysis by using gene signal intensity revealed that the sites with different fire histories formed separate clusters, suggesting differences in the potential to maintain essential biogeochemical soil processes. The site with the greatest biological diversity had also the most diverse genes. The genes involved in organic matter degradation in the mature forest, in which ECM fungi were the most abundant, were as common in the youngest site, in which saprotrophic fungi had a relatively higher abundance. This study provides insight into the impact of fire disturbance on soil fungal community dynamics.
INTRODUCTION
Boreal forest soils play an important role in the global carbon cycle (1) and are a net sink for atmospheric CO2 (2, 3). Approximately 16% of the terrestrial carbon (C) stock is estimated to be stored in the boreal forest ecosystem (1). Fire is a natural disturbance in most forest ecosystems (4), and about 1% of the boreal forest burns annually (5). The frequency of forest fires in the northern boreal zone is expected to increase because of rising temperatures and more frequent dry periods resulting from ongoing climate change (6).
Soil microbes perform essential ecological functions in forested ecosystems via nutrient cycling and decomposition of organic matter. Microbial activities control the turnover of organic C in soil and thus contribute to global C cycling (7). Fungi are the predominant decomposers in boreal soils and play a central role in the turnover of carbon and nitrogen (8). Ectomycorrhizal fungi form symbioses with both trees and ground vegetation in boreal forests. Many species of ground vegetation can additionally form symbioses with ericoid mycorrhizal fungi. Boreal forest ecosystem productivity is linked to plant nutrient acquisition from the microbial community via soil organic matter (SOM) decomposition. Forest fires result in the loss of mycorrhizal host plants and may modify the SOM chemistry, leading to dramatic changes in soil microbial activity (9, 10). Fire disturbance often increases soil pH and causes changes in soil temperature due to loss of canopy cover, both of which likely have large direct and indirect effects on soil microbial communities (11–13). It is important to understand how microbial communities respond to the postfire environment and what are the most important environmental factors driving the fungal community structure and function.
The impact of fire on microbial communities has received less attention than the effects of fire on vegetation and soil properties. Detailed investigations of long-term changes in microbial community dynamics following fires are rare, especially for fungi. The recovery of fungal biomass has been reported to be related to a decrease in SOM turnover time in a boreal fire chronosequence (14). Previous studies have shown that the increase in soil pH after fire favors bacteria (15) and reduces the richness and diversity of mycorrhizal fungi (16, 17). Fire can also lead to significant losses in fungal biomass in organic horizons (18). The microbial community responses to reoccurring low-intensity prescribed burning have been reported to be minimal unless fires are implemented at high frequency (2- to 3-year intervals) (19, 20). However, only a few studies have investigated whether these changes in the microbial community persist over long periods of time. There are large numbers of studies using nonmolecular methods to investigate the effects of fire on fungal communities, from boreal to Mediterranean areas (21–25). Studies examining the recovery of fungal components of the soil microbial communities in boreal forests are lacking (26, 27). There is also an urgent need to determine how fire affects fungal communities and their ecosystem functions using high-throughput molecular biological tools.
The recent advances in next-generation sequencing (NGS) have provided powerful tools to address microbial diversity and community composition differences in complex environments (19, 28–30). Further, high-throughput functional gene arrays provide a useful tool to characterize the functional attributes of microbial communities for environmental microbial community analysis (31–34). The GeoChip 4.0 functional array contains approximately 82,000 probes covering 142,000 coding sequences from 410 functional gene families related to microbe-driven biogeochemical, ecological, and environmental processes (34). The GeoChip microarray provides, therefore, the ability to analyze targeted functional gene families originating from a wide range of different microbial groups (i.e., eukaryotes, prokaryotes, and viruses).
The main aims of the study were to evaluate the short- and long-term effects of forest fires and the postfire succession on soil fungal communities and to assess the functional potential of fungal communities after fire. We hypothesized that with increasing time since the last fire, the fungal community composition gradually shifts from being saprophytic to being more strongly dominated by mycorrhizal fungi. The community functions involved in different biochemical processes were hypothesized to shift correspondingly. We coupled a high-throughput metabarcoding approach with functional gene arrays to analyze the diversity and composition of soil fungal communities and to evaluate their functional potential across four different periods of time postfire in a 152-year fire chronosequence. The results are discussed in the context of dynamics and postfire responses of fungal community structure and function in boreal forest ecosystems.
MATERIALS AND METHODS
Study area and soil sampling.
The five sampling sites were located in the northern boreal subarctic coniferous forest of the Värriö Strict Nature Reserve in northeastern Finland (67°46′N, 29°35′E). The study area (18 km2) was dominated by Pinus sylvestris L., with scattered Betula pubescens Ehrh. and Picea abies (L.) Karst trees. The ground vegetation consists mostly of Vaccinium myrtillus L., Vaccinium vitis-idaea (Lodd.) Hulten, Empetrum nigrum L., and Cladina species. The study was conducted at five sampling sites across a fire chronosequence and included four different periods of time (five sites) since the last fire: 2 years (2y), 42 years (42y), 60 years (60y-A and 60y-B), and 152 years (152y) after fire. The fire disturbance occurring 60 years prior included two sites, 60y-A and 60-B, which were in two different areas about 1.5 km from each other. None of the fires detected on the sample plots were completely stand-replacing events, as some trees survived in areas where the fire occurred 2, 42, and 60 years ago (see Table S1 in the supplemental material), and we found some trees that were >152 years old in the sites where fire occurred 152 years ago (see Table S1). A thin humus layer was left after the fire at the 2y site. Non-stand-replacing fire disturbances are frequent in Eurasian boreal forests. Also, as in many boreal fire chronosequence studies, we were not able to find an unburnt control, since all the landscape has burned at some point in time (35). Detailed information on the study sites was previously described (14) (see Table S1).
Two plots were chosen at each site and separated by a distance of ≥100 m. This was considered to be sufficient to break any spatial correlation of soil characteristics or fire intensity (36). We took five consecutive soil samples at each plot (with a distance of 4 m from each other), resulting in 10 individual samples per site (5 samples/plot and 2 plots/site). All samples were collected in August 2011. The humus layer samples (0.5 to 1.0 cm thick) were collected from a 0.25 m by 0.25 m quadrat of soil at the interface of the humus and the mineral soil after removing the litter. The samples were mixed well and transferred to 1.5-ml Eppendorf vials. The samples were frozen at −180°C in liquid nitrogen within a few hours after collection and transported to the laboratory on dry ice for subsequent DNA isolation. The soil microbial biomass was estimated by measuring the ergosterol content from the samples collected from the humus layer (14).
Five soil cores, each 0.05 m in diameter and 0.15 m in length, were taken 4 months apart at each sampling plot in order to determine soil C and N content, root biomass, and soil pH. These samples were stored at 4°C until subsequent analysis. Soil C and N contents were measured with an elemental analyzer (vario MAX CN elemental analyzer; Elementar Analysensysteme GmbH, Germany) after the soil samples were dried and passed through a 2-mm-pore-size mesh sieve. The soil temperature and water content were also measured at each sampling site, as previously described (14).
DNA extraction, amplification of ITS2 region, and pyrosequencing.
Genomic DNA was extracted from 0.25 g (fresh weight) of each homogenized soil sample using the PowerSoil DNA isolation kit (Mo Bio Laboratories, Carlsbad, CA, USA), according to the manufacturer's instructions. The genomic DNA was purified using the GeneClean Turbo kit (MP Biomedicals, LLC, France), quantified with a Qubit 2.0 fluorometer (Life Technologies, Eugene, OR, USA), and adjusted to a final concentration of 10 ng μl−1.
The fungus-specific primers gITS7, containing a 454 pyrosequencing A-adapter, and ITS4, containing a 454 pyrosequencing B-adapter and a 6-bp barcode, were used to amplify the internal transcribed spacer 2 (ITS2) region (37). Thirty nanograms of template genomic DNA was used for a 25-μl-volume PCR using Phusion high-fidelity DNA polymerase (Thermo Scientific, Vantaa, Finland), according to the manufacturer's protocol. The PCRs were performed in triplicate for each sample to minimize PCR biases. The possible amplification of contaminants was tested through the use of PCR negative controls in which the template DNA was replaced with sterile H2O.
Five microliters of each PCR product was separated on a 1.5% agarose gel run at 130 V for 20 min. The mixtures from triplicate PCRs per sample were pooled and purified using Agencourt AMPure XP beads (Beckman Coulter) to remove nonincorporated primers and deoxynucleoside triphosphates (dNTPs). The DNA concentration of each sample was determined after purification using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). One hundred nanograms of the PCR products was sequenced using the ITS4 primer with the 454 GS-FLX Titanium protocol (454 Life Sciences/Roche Diagnostics, CT, USA).
Pyrosequencing data processing.
The sequence data were analyzed using the mothur pipeline (version 1.31.2) (38) using a modified operation procedure (19). Briefly, the raw reads were quality controlled, and each sequence was screened for a match to the sequencing primer (ITS4) and valid DNA tag. Sequences were removed if shown to contain ambiguous (N) bases, homopolymers longer than eight nucleotides, or a Phred quality score of <25. Sequences that passed the quality filtering were truncated to 200 bp in length after primer and tag removal. The remaining sequences were preclustered within a distance of 1 bp using a pseudo-single-linkage algorithm implemented in mothur to minimize sequences that contain pyrosequencing errors (39). All potentially chimeric sequences were identified using the mothur-embedded UCHIME program (40) and removed. Unique sequences were pairwise aligned using the Needleman method (41), and the aligned distance matrices were clustered into operational taxonomic units (OTUs) using the average neighbor algorithm and 97% sequence similarity. All global singletons (OTUs that contain only one sequence across all samples) were omitted due to their uncertain origin (19, 42). The OTUs were assigned to taxa using the naive Bayesian classifier (43) and the mothur-formatted UNITE taxonomy reference (UNITE+INSD, version 6 [http://unite.ut.ee/repository.php]) with an 80% confidence threshold (43). Nonfungal sequences were removed using the remove lineage command in mothur. The top 50 identified fungal genera were categorized into ectomycorrhizae (ECM), ericoid mycorrhizae (ERM), and saprotrophs (SAP) based on the ecological/functional role, in accordance with information in the published literature. The definition of ECM referred to genera for which most of the members formed an ectomycorrhizal association. ERM referred to genera for which most of the members formed an ericoid mycorrhizal association. SAP referred to genera that are not ECM or ERM and not known to be pathogenic fungi. All other functional forms were classified as unknown. Observed and estimated species richness (Chao1) (44), diversity (inverse Simpson's index [1/D]), and evenness (Simpson's equitability [ED]) were estimated in mothur. The commands used in mothur to process the data are listed in Table S2 in the supplemental material.
The sequence data were randomly subsampled to 2,400 reads per sample (45) to calculate richness and diversity estimators for a comparison between fungal communities in order to correct for differences in sampling effort and ensure comparable estimators across samples. The cumulative-sum scaling (CSS) method (46) without subsampling was used to validate the results obtained from rarified data (subsample normalization).
DNA labeling, GeoChip 4.0 microarray hybridization, data processing, and visualization.
DNA samples from the 2y, 60y-B, and 152y sites were used for GeoChip 4.0 microarray analyses with three replicates to assess the gene structure of the fungal communities. The genomic DNA of each replicate was obtained by randomly pooling three individual genomic DNA samples that were obtained from each site and used previously for sequence analysis. One hundred nanograms of genomic DNA was amplified by rolling circle amplification using the TempliPhi kit (GE Healthcare) and a modified protocol (47). Approximately 2 μg of amplified genomic DNA was mixed with random primers (3 μg/μl random hexamers; Life Technologies) and then labeled with 15 μl of labeling master mix (2.5 μl of dNTP [5 mM dATP, dGTP, and dCTP and 2.5 mM dTTP], 0.5 μl of Cy-3 dUTP [25 nM; GE Healthcare], 1 μl of Klenow fragment [40 U ml−1; Imer, San Diego, CA], 5 μl of Klenow buffer, 2.5 μl of water) (48). The labeled genomic DNA was purified using the QIAquick purification kit (Qiagen), according the manufacturer's instructions, and then dried in a SpeedVac at 45°C for 45 min (Thermo Savant). Labeled genomic DNA was hybridized on the GeoChip 4.0 microarray, as previously described (48, 49). Data normalization and quality filtering were performed in multiple steps (34, 48). Briefly, the probes in the GeoChip microarray were first organized by gene lineage (bacteria/fungi/archaea/viruses), and only fungal genes were included in the analysis. Spots were scored as positive and retained if the signal-to-noise ratio (SNR), calculated as (signal mean − background mean)/background standard deviation, was ≥2.0. Only the spots automatically scored as positive in the raw data were used for further data analysis. Spots with a signal intensity of <1,000 were discarded. The process and functions used for the complete analysis of the remaining data were performed in the R (50) gplots package (51) for hierarchical cluster analysis (HCA). The gene diversity in each sample was calculated by inverse Simpson's index (1/D).
Statistical analysis.
Linear mixed models were used to compare the differences in fungal diversity (1/D), richness (Chao1), and evenness between sites, assuming times after the last forest fire to be fixed effects. Plots and samples were considered nested random effects within each time period after the last fire. The mixed models break down the variance into inter- and intraplot components and thus improve the true structure of the randomness present in the data in spatial sampling (52). Analysis was done using the lme4 package in R (53). Multiple comparisons of means were done using the multcomp package of the R statistical environment (54).
The stepwise linear regression analysis method was performed to compare the relative abundances of a phylum or genus across sites. The relative abundance of a fungal group or OTU at each period of time was calculated by dividing the number of sequences in each group or OTU by the number of sequences in each period of time since fire. Nonmetric multidimensional scaling (NMDS) was used to visualize the difference in fungal community and function between fire ages (sites), including environmental variables as vectors contributing to fungal community composition. The environmental variable data (e.g., soil pH, water content, soil C and N contents, tree root biomass, and ergosterol content) were derived from a study by Köster et al. (14). The NMDS analysis was done by using the vegan package with the metaMDS and envfit functions in R (55). A permanova implemented in mothur (56) was performed to determine the significant difference in the community structure between sites.
Canonical correspondence analysis (CCA) and analysis of similarities (ANOSIM) (57) were used to determine the fungal gene structures and test the correlation between fungal genetic diversity and taxonomic composition. The fungal genera identified in the pyrosequencing data were used as vectors in CCA. CCA and ANOSIM were done using the vegan package with the vegdist, envfit, and anosim functions in R. The data used in NMDS, CCA, ANOSIM, and HCA were based on either the relative abundance of each fungal group or OTU from pyrosequencing or the signal intensity from the GeoChip microarray. The scripts used in R to process the data are listed in Table S2 in the supplemental material.
Accession number.
The raw sequence data are available in the European Nucleotide Archive (ENA) at the European Bioinformatics Institute (EBI) under accession no. PRJEB6382 .
RESULTS
Information on pyrosequencing data.
Using 454 pyrosequencing, we obtained 431,595 raw sequences, with an average read length of 299 bp. A total of 316,206 high-quality sequences (73% of raw sequences) were retained after denoising and quality filtering. The number of sequences from each sample ranged from 2,476 to 15,747, with an average ± standard deviation of 6,324 ± 2,744 reads. A total of 2,400 sequences were randomly subsampled from each sample for comparable analysis between sites (see Table S4 in the supplemental material). The species accumulations (rarefaction) with sequencing efforts are shown in Fig. S1 in the supplemental material. The cumulative-sum scaling (CSS) analyses were consistent with the subsampling (see Table S4).
Fungal diversity and community composition after fire.
We observed a total of 2,007 OTUs (4,699 OTUs, including singletons) across all samples. The 2y site harbored the most diverse and even community, whereas the 152y site had the least diverse community (Fig. 1). Pairwise post hoc comparisons of time points since fire using Tukey's test revealed that the inverse diversity (1/D) differed significantly between the 2y and 152y sites (P < 0.02), 42y site (P < 0.001), and 60y-A site (P < 0.02) (Fig. 1b). The OTU richness (Chao1) at the 2y site significantly differed from that at the 60y-A site (P < 0.001) and 152y site (P < 0.001) (Fig. 1a). Sites with intermediate fire histories (42y and 60y) did not differ in richness or diversity. The observed OTU richness and diversity indices tended to decrease with increasing time since fire (Fig. 1).
FIG 1.
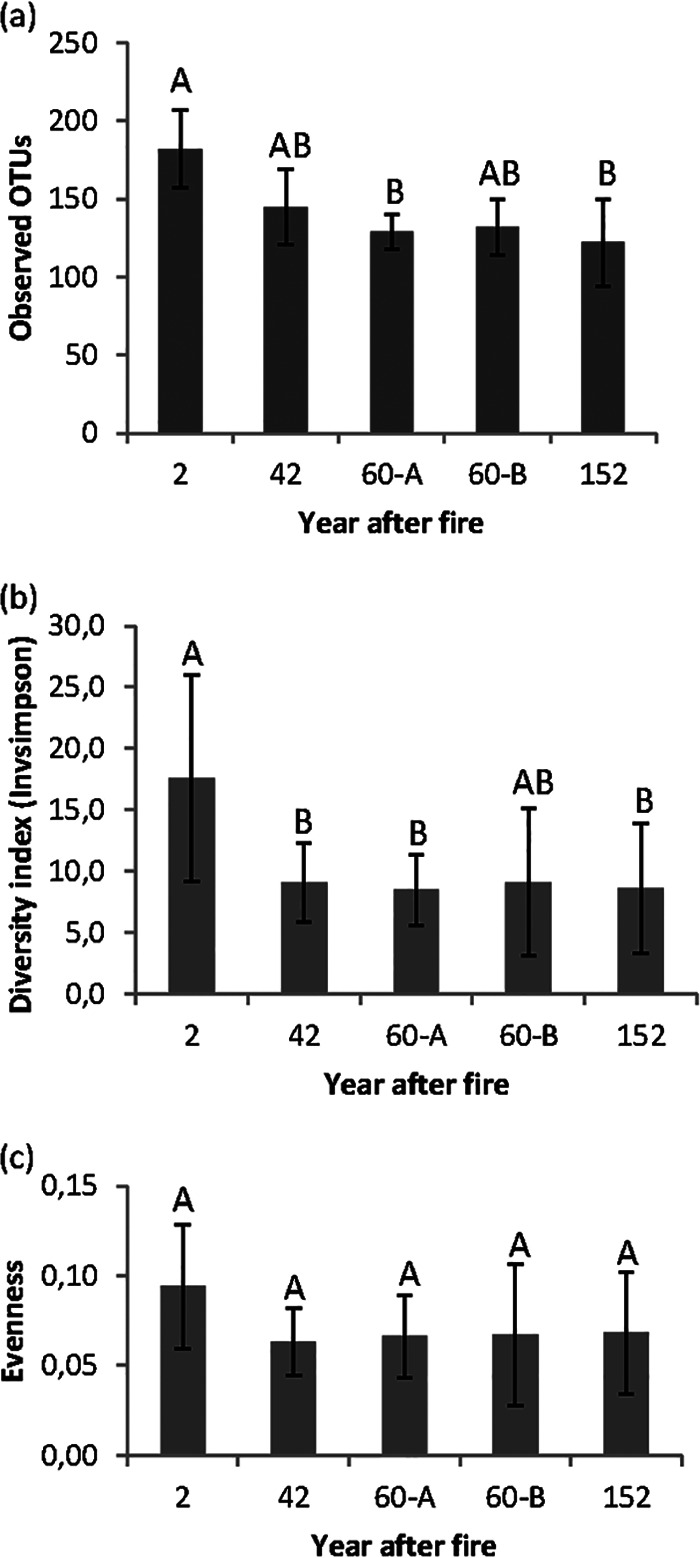
Community richness, diversity, and evenness estimators across the five sites differing in fire histories. Shown are the Observed OTU richness (a), inverse Simpson's diversity (Invsimpson) (b), and evenness based on Simpson's diversity index (c). The bars represent the standard deviations, and different letters in each panel represent Tukey's significance at a P value of 0.05.
The OTUs were classified into four fungal phyla. The phylum Ascomycota was the most species rich, whereas Basidiomycota dominated the sequence counts (Fig. 2a). The majority of the OTUs (42% of the total 2,007 OTUs) belonged to the Ascomycota, covering 90,919 sequence reads (29% of the total 313,514 reads). Basidiomycota represented 28% (568 OTUs) of the total OTUs but accounted for more than half (52% [162,169]) of the sequences. Zygomycota and Chytridiomycota made up 2% and 0.4% of the OTUs and accounted for 1.2% and 2.2% of the sequence count, respectively. Fifteen percent (48,408) of the fungal sequences were not classified into any fungal phylum (Fig. 2b). Ascomycota dominated at the site with the most recent fire (2y), whereas Basidiomycota were more abundant at sites with fires occurring longer ago (i.e., ≥42 years ago) (Fig. 3). Regression analyses of the relative abundances indicated that ascomycetes decreased with increasing time since fire (P = 0.031, R2 = 0.60), whereas basidiomycetes increased (P = 0.028, R2 = 0.15). More than 30% of the OTUs (627 OTUs) were assigned to the genus or species level and accounted for half of the sequences. Two hundred fifty OTUs belonged to the Ascomycota, which accounted for 9% of the total sequence reads, while 291 OTUs belonged to the Basidiomycota, which accounted for 44% of the reads.
FIG 2.

Bar charts showing the phylum-level assignment for operational taxonomic units (OTUs) from five sites differing in fire histories as the numbers of OTUs (n = 2,007) and sequence reads (n = 313,514) (a) and the relative proportions of OTUs and sequence reads (b).
FIG 3.
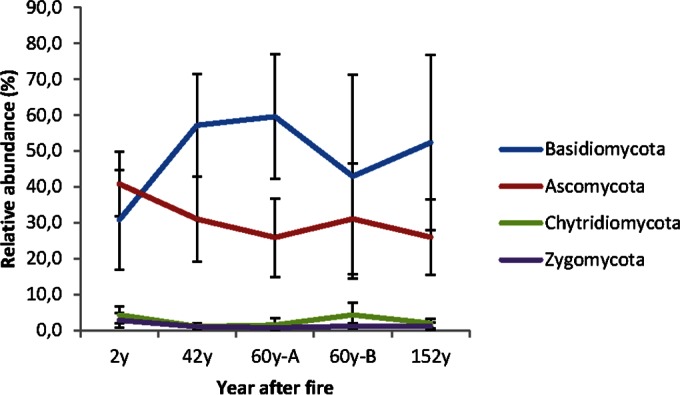
Distribution of phylogenetic groups in the five sites differing in fire histories. The bars represents the standard deviations.
The fungal community composition differed between the sites. Differences in community composition across the chronosequence were primarily driven by changes in the abundance of basidiomycetes and ascomycetes (Fig. 3). The ectomycorrhizal (ECM) genus Cortinarius was the most abundant (18.5% of sequences, 136 OTUs) across all sampling sites, followed by the ECM genus Piloderma (14.2%, 12 OTUs), the ericoid mycorrhizal (ERM) genus Rhizoscyphus (5.6%, 59 OTUs), the ECM genus Suillus (2.2%, 32 OTUs), the ECM genus Lactarius (2.2%, 20 OTUs), the ECM genus Russula (1.9%, 8 OTUs), the largely ERM genus Meliniomyces (1.0%, 18 OTUs), and the soil-inhabiting zygomycetous genus Mortierella (1.1%) (see Table S3 in the supplemental material). The abundance of Cortinarius increased with increasing time after fire and reached the maximum at the 152y site (P < 0.001, R2 = 0.524). The abundance of Piloderma was highest at 42 years after fire but decreased constantly afterwards (see Fig. S2 in the supplemental material). The abundance of the two genera differed significantly between the 2y and 152y sites (P < 0.001). Interestingly, Russula was found at the 60y-B site only. The abundance of the ERM genus Rhizoscyphus remained stable across the sites (∼5.3%). Among the top 50 most abundant fungal genera, with an abundance of >0.01%, ECM fungi dominated and ranged from 24.7% to 46.6% between the sites, followed by ERM (8.4% to 5.9%) and putative saprotrophs (6.4% to 1.9%) (Fig. 4; see also Fig. S2 and Table S3 in the supplemental material). Genera with unknown affinities comprised 37% to 61% of the sequences at each site. The abundance of ECM was the lowest 2y after fire, and putative saprotrophs were most common at that site. Thereafter, the abundance of ECM increased, and that of saprotrophs decreased (Fig. 4).
FIG 4.
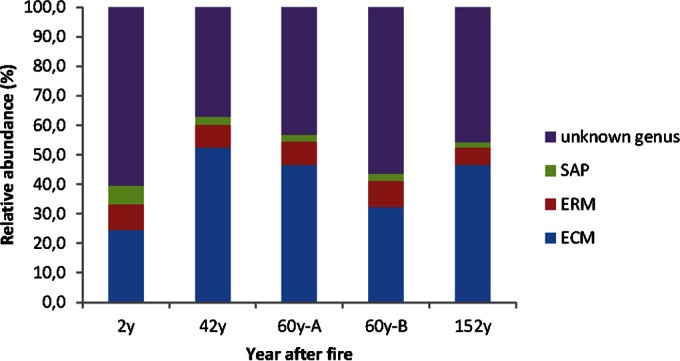
Relative abundances of ectomycorrhizae (ECM), ericoid mycorrhizae (ERM) and saprotrophs (SAP) among the top 50 most abundant genera in each site differing in fire histories.
Only 187 (9.3%) of the 2,007 OTUs were shared among all sites. The number and proportion of OTUs unique to each site ranged from 96 to 192 and 11.1% to 21.1% (see Fig. S3 in the supplemental material). The site with most recent fire had the highest number (192) of unique OTUs. The NMDS analyses based on relative OTU abundance indicated a clear separation in fungal communities between the sites with more recent fires and those occurring longer ago (Fig. 5a). A subsequent permanova test also indicated that the 2y and 152 y communities were different from each other (P < 0.02) and from those with other periods of time after fire (P < 0.02). Communities at sites with intermediate fire histories (i.e., the 42y and 60y sites) were similar and did not differ in community composition (P = 0.3).
FIG 5.

Nonmetric multidimensional scaling (NMDS) plots (environmental variables as vectors) showing differences in fungal community structure (a) and functional gene structure (b) with time since fire. OTUs from 454 pyrosequencing and signal intensity of genes from GeoChip were used to visualize the community structure and functional gene structure, respectively. σ: ■, 2y; |, 42y; ▲, 60y-A; △, 60y-B; ◆, 152y.
Fungal gene diversity and structure after fire.
In total, 3,042 fungal gene probes (69,476 probes in total) were detected using the GeoChip 4.0 microarray at the three sites (2y, 60y-B, and 152y) included in the functional analyses (see Table S5 in the supplemental material). Gene probes originating from bacteria, archaea, and viruses were excluded from this analysis. The number of functional genes and gene diversity (measured by 1/D) varied between the sites (Table 1; see also Fig. S5 in the supplemental material). The 2y site had the highest number of detected genes (mean ± standard deviation, 2,286 ± 42 genes) and the highest gene diversity. The 60y-B site had the lowest number of genes detected (mean ± standard deviation, 2,041 ± 129 genes) and the lowest estimated gene diversity. While the number of detected genes and gene diversity at the 2y site differed significantly from those of the 60y sites (Tukey's test, P = 0.03 for both) (Table 1; see also Fig. S5), neither of the 60y sites differed from the 152y site.
TABLE 1.
Number of genes involved in different processes detected at sites with different fire histories
| Gene category | No. of genes detected (mean ± SD)a |
No. of genes in each group | ||
|---|---|---|---|---|
| 2 yr after fire | 60 y after fire | 152 yr after fire | ||
| Transport | 313 ± 8 | 275 ± 20 | 274 ± 7 | 416 |
| Cellulose | 177 ± 4 | 166 ± 7 | 171 ± 7 | 238 |
| Pectin | 181 ± 4 | 162 ± 13 | 174 ± 7 | 235 |
| Lignin | 161 ± 3 | 148 ± 15 | 148 ± 5 | 217 |
| Transporter | 119 ± 2 | 106 ± 7 | 111 ± 4 | 160 |
| Aromatic carboxylic acid | 83 ± 3 | 76 ± 4 | 85 ± 4 | 125 |
| Chitin | 82 AB ± 3b | 76 A ± 4 | 86 B ± 2 | 122 |
| Hemicellulose | 97 A ± 4 | 87 AB ± 4 | 83 B ± 1 | 122 |
| Phospholipid | 83 ± 4 | 71 ± 3 | 82 ± 2 | 110 |
| Starch | 65 ± 2 | 58 ± 5 | 65 ± 3 | 91 |
| Chlorinated aromatics | 52 ± 3 | 45 ± 2 | 54 ± 2 | 70 |
| Other aromatics | 52 ± 1 | 47 ± 3 | 49 ± 2 | 65 |
| Cutin | 40 ± 1 | 38 ± 2 | 38 ± 2 | 49 |
| Glyoxylate cycle | 35 ± 2 | 32 ± 2 | 32 ± 2 | 48 |
| Polycyclic aromatics | 27 ± 1 | 25 ± 3 | 29 ± 3 | 37 |
| Protein | 29 ± 2 | 24 ± 2 | 27 ± 2 | 35 |
| Tannins | 25 ± 1 | 25 ± 4 | 25 ± 1 | 32 |
| Cyanide | 21 ± 1 | 17 ± 1 | 19 ± 2 | 28 |
| Lactose | 18 ± 1 | 16 ± 2 | 19 ± 1 | 27 |
| Ergot | 11 ± 1 | 10 ± 1 | 11 ± 1 | 18 |
| Detoxification | 9 ± 1 | 7 ± 1 | 7 ± 1 | 11 |
| Nitroaromatics | 9 ± 1 | 7 ± 1 | 7 ± 1 | 10 |
| Melanin | 7 ± 1 | 8 ± 1 | 8 ± 1 | 10 |
| Aromatic alpha hydroxy acid | 6 ± 1 | 5 ± 1 | 6 ± 1 | 8 |
| O-Methylsterigmatocystin oxidoreductase | 4 ± 1 | 4 ± 1 | 4 ± 2 | 8 |
| Toxin | 3 ± 1 | 3 ± 1 | 3 ± 1 | 4 |
| Enniatin | 3 ± 1 | 1 ± 1 | 2 ± 1 | 4 |
| Vanillin/lignin | 2 ± 1 | 1 ± 0 | 2 ± 1 | 2 |
| NAc | 548 ± 12 | 478 ± 33 | 503 ± 10 | 740 |
| Total no. of genes | 2,286 A ± 42 | 2,041 B ± 129 | 2,146 AB ± 52 | 3,042 |
The values represent the mean and standard deviation of three replicates from each age after fire.
Values within a row followed by different letters are significantly different at 0.05 in numbers of genes between each period of time after fire.
NA, not available.
We visualized the functional gene structure between sites generated using the GeoChip 4.0 microarray using NMDS (Fig. 5b). A subsequent analysis of the similarities (ANOSIM) confirmed that sites with different fire histories indeed possessed distinct compositions of functional genes (R2 = 0.999, P = 0.003). The hierarchical cluster analysis (HCA) based on the abundance of all functional genes revealed a difference in functional genes between the sites and clustered genes into 15 subclusters (Fig. 6). The 2y and 152y sites clustered together, indicating similar gene profiles, whereas both differed from the 60y-B site (P = 0.003 for both; Fig. 6).
FIG 6.

Hierarchical cluster analysis of functional genes in the three sites differing in fire histories. The numbers in the left column represent subclusters 1 to 15, and the numbers after the site name represent replicates 1 to 3 at each site.
Functional genes involved in different processes.
Overall, the subclusters (Fig. 6) included genes involved in distinct biogeochemical processes (Table 1). Nearly half of the detected genes were present in all samples. Genes encoding enzymes for hemicellulose degradation were more abundant at the 2y site than at the 152y site (P = 0.04), and genes encoding enzymes for chitin breakdown were more abundant at the 152y site than at the 60y site (P = 0.03) (Table 1). Some of the genes in subclusters 2, 4, 6, 8, and 9, which were derived from HCA (Fig. 6), were site specific (see Fig. S4 in the supplemental material). For example, genes in subcluster 2 (135 genes) were observed at the 60y and 152y sites only. Genes in this subcluster mainly encode proteins in the following functional categories: iron transport (26 genes) and degradation of lignin (20), cellulose (18), pectin (14), and chitin (10). Genes in subcluster 4 (107 genes) that represent functions similar to those in subcluster 2 were absent at the 152y site only. Genes in subclusters 6 (202 genes) and 9 (207 genes) were present only at the 2y and 152y sites, respectively. The two subclusters comprised similar gene categories, with high frequencies of genes encoding proteins involved in the breakdown of hemicellulose (12) and transport (42) in subcluster 6 and aromatic carboxylic acid (21) and chitin (17) in subcluster 9. One hundred seventeen genes in cluster 8 were not present at the 60y site.
Relationship between fungal community composition and functional gene structure.
The numbers of fungal OTUs and detected functional genes were positively correlated (Spearman correlation R2 = 0.356, P = 0.01); i.e., the more OTUs in a sample, the more functional genes were detected. The CCA using the 50 most common fungal genera as vectors highlighted that 12 genera were correlated (P < 0.01) with the functional gene content at the three sites (Fig. 7). Saprobic and common soil-inhabiting genera (Mortierella, Mycena, Lecythophora, and Coccomyces) were correlated with gene structure at the 2y site (Fig. 7). Of these, Mortierella and Mycena were among the most common genera at the 2y site (see Table S3 in the supplemental material). The ECM genus Cortinarius was associated with functional genes at the 152y site (Fig. 7) and strongly dominated at this site (see Fig. S2 in the supplemental material). The ECM genus Hygrophorus was correlated with the functional genes at the 60y-B site, even though it was not among the top 20 most common genera at this site (Fig. 7; see also Fig. S1 in the supplemental material). ECM fungi were clearly associated with 152y site and the saprobes (SAP) with the 2y site when the groups of ECM, ERM, and SAP were used as vectors (Fig. 7).
FIG 7.

CCA showing the relationship between fungal gene structures based on the signal intensity of genes from GeoChip and fungal community composition at the genus level. The top 50 genera and the fungal lifestyles from pyrosequencing data in the three sites differing in fire histories were used as vectors. σ: ■, 2y; △, 60y-B; ◆, 152y.
Environmental factors contributing to fungal community composition and function.
Many environmental parameters were strongly correlated with the taxonomic and gene structures of the fire sites (Fig. 5a and b). Soil pH and temperature were positively correlated with the community composition at the 2y site (Fig. 5a and b). The community at the 60y-A site correlated with low pH (Fig. 5a). Tree root biomass (TRootBiom) was linked to the fungal community at the 2y and 152y sites (Fig. 5a and b), and soil N and C contents, water content, and fungal biomass (as estimated from ergosterol content) were associated with the communities at the 60y sites.
DISCUSSION
Our pyrosequencing and gene microarray data from a fire chronosequence demonstrated that fungal abundance and community structure were clearly site related. Fungal richness and diversity were higher at the recently burned forest sites than at sites with fire events that occurred longer ago. The fungal communities at the sites with the most recent (2y) and distant (152y) fires differed. Genera and species from different functional groups dominated at these stages of postfire forest development (Fig. 4; see also Fig. S2 in the supplemental material). Interestingly, functional gene diversity (as measured by 1/D) did not differ between the 2y and 152y sites, suggesting that the fungal communities shared similar gene profiles and that there are functional similarities among fungal communities with differing compositions.
Mycorrhizal fungi are crucial in SOM transformations (58–60). The ECM fungus Paxillus involutus can convert SOM using a mechanism similar to that employed by brown-rot fungi using Fenton chemistry (61). Unique sets of genes that have been retained during fungal evolution may allow ECM fungi to decompose lignocellulose (62). In our study, the numbers of lignin-degrading genes detected at different sites did not differ among sites with different fire histories (Table 1). However, a limited number of genes encoding chitin- and hemicellulose-degrading enzymes differed between the oldest and other sites (Table 1). Chitinase-encoding genes were more abundant at the site with the oldest fire occurring, whereas hemicellulose genes were more abundant at sites with more recent fires. Chitin is decomposed primarily by N-limited microbiota for a source of N, and differences in the chitin utilization dynamics can indicate a discrepancy in microbial biomass or a difference in microbial communities (63). Chitin in boreal forest soils is mostly derived from dead fungal hyphae and is relatively easily available for fungi (64). However, when drawing conclusions regarding community functions, it is important to bear in mind that the GeoChip microarray data reflect the microbial communities carrying those genes and can be used only to estimate the potential functional capacity at the sites.
Soil properties and plant diversity are key factors shaping microbial communities (65). Increased quantities of decomposable and burned material increase soil pH due to the production of K and Na oxides, hydroxides, and carbonates through ash deposition (66). In our study, soil pH was correlated with the fungal community structure at the 2y site (Fig. 5). The increased soil pH may favor bacterial growth and result in a relative decline in fungal biomass (14). Fire is also believed to cause significant losses of fungal biomass in the organic horizons (18). We hypothesize that this reduction in fungal biomass, associated with a litter pulse from the dead vegetation in a ground fire, creates empty niches (67) that can be rapidly colonized by fungi specialized in the early stages of substrate decomposition (68–70). In our study, saprotrophic and ascomycete fungi were most common 2 years after fire, suggesting a response to the greater availability of decomposable material. The remaining organic fractions of C and N become more recalcitrant, and the mycorrhizal abundance increases with increasing time from the fire, after the easily decomposable compounds are consumed (3, 9). High proportions of fungal sequences (31 to 61%) at any of our study sites were not assigned to a functional group, and the largest proportion of unknown sequences was observed at the 2y site. It is unclear to which functional groups (ERM, ECM, and SAP) these unidentified sequences belong; further research into the function and diversity of soil fungal communities is clearly needed.
The postfire changes in vegetation may more strongly shape the soil microbial community that represents the direct effects of the fire disturbance (71). Previous studies have reported that the loss of host plants after fire clearly reduces mycorrhizal fungal diversity (17, 72). Our sites were dominated by P. sylvestris and included an abundant ericaceous understory, which can host both ECM and ERM fungi (73). The increase in fungal biomass during succession and the symbiotic associations between trees and mycorrhizal fungi favor the replacement of saprotrophic fungi (74). The loss of vegetation cover after fire can contribute to factors (i.e., loss of water, organic matter, etc.) that lead to prolonged elevated soil temperatures (13, 14). Changes in both temperature and water content after fire may control microbial growth and metabolism, which in turn alter the community function. Soil C and N contents and tree root biomass are also important controls of fungal communities after fire (14). In our study, sites with different fire histories housed distinct fungal communities in terms of both composition and function, although the importance of the changes in environmental conditions remains to be elucidated.
Cortinarius strongly dominated the fungal community at the 152y site, and almost 45% of all OTUs belonged to this genus. This dominance strongly contributes to the low diversity and evenness at the 152y site. The genus Cortinarius can be important in the decomposition of complex organic matter and may mobilize organically bound N in boreal forest soils (75). This genus can also possess genes needed to degrade lignin-like recalcitrant organic substrates (75). Species of the ECM genus Piloderma are abundant in coniferous forests (76) and favor low-N conditions (77). However, in our analyses, Piloderma did not correlate with soil N content, as its abundance varied among the sites with similar N concentrations (see Table S1 in the supplemental material). Interestingly, the ECM genus Russula was detected at the 60y-B site only (see Fig. S2 in the supplemental material), and the occurrence of Russula seems to contribute to the decrease in the abundance of other genera at this site. Koide et al. (78) provided evidence for antagonism among ECM fungi, in which the abundance of some species of Russula was negatively correlated with the abundance of other ECM. It is possible that ECM fungi compete for resources, such as water, N, and P, resulting in competitive exclusion of the weaker competitors.
Our fungal GeoChip microarray data clearly distinguished the communities at the 2y, 60y, and 152y sites. The three replicate samples from each site clustered closely together, and the sites were separated from each other by gene structure. This indicates differences in gene profiles present at the different sites. Approximately half of the genes in the array were present in all samples. The remaining genes suggested site-specific differences and clustered samples to site-specific clusters (Fig. 6). Genes encoding chitin-degrading enzymes (chitinases) were most abundant at the 152y site, and genes encoding hemicellulose-degrading enzymes were most abundant at the 2y and 60y sites. Analyses of composition and function revealed positive correlations, suggesting that greater biological diversity leads to greater functional potential. However, the redundancies in the genes represented in the array (Table 1) compromised our ability to detect functional differences. Based on our DNA-based microarrays, the gene profiles at all sites are diverse, and only a few differences are due to the presence/absence of certain genes or their abundance. Evidently, other metagenomic approaches to assay the functional attributes of the fungal communities are necessary to better link the abundance of functional genes to their expression.
In conclusion, our study highlights several important aspects of soil microbial ecology. First, despite the relatively high redundancy in functional gene profiles, we showed that the successional stage with higher biological diversity also represents the most diverse gene profile. This observation is important when conservation of ecosystem services and functions of soil biota are considered. Second, we demonstrated that soil pH and temperature were clearly correlated with fungal communities, but the community composition shifts from an ascomycete- to a basidiomycete-dominated one with a greater abundance of ECM. This shift is slow and seems to require decades to occur. Third, the genes involved in the organic matter degradation in the mature forest, where ECM fungi were the most abundant, were as common at the youngest site, where saprotrophic fungi had a relatively higher abundance. We postulate a new hypothesis that nutrient acquisition by ECM fungi in mature forest soil may use genetic mechanisms similar to those of saprobes in fire-disturbed ecosystems.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by grants from the Academy of Finland (project no. 263858, 130984, 218094, and 255576), ICOS project (grants ICOS 271878, ICOS-Finland 281255, and ICOS-ERIC 281250), the Estonian Research Council (grant PUT715), and the Research Funds of the University of Helsinki (grant 490127). A.J. was partly supported by the USDA Forest Service Cooperative Agreement 11-CA-11330136. In addition, this study was part of the Academy of Finland Finnish Centre of Excellence program (project 1118615), FiDiPro (project no. 138116), the European Social Fund and Estonia Research Council grant Mobilitas (MJD94), and an Estonia Research Council grant (PUT715).
The use of trade names in this publication does not constitute an official endorsement of these products by the USDA or any of its subsidiary agencies.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02063-15.
REFERENCES
- 1.Bonan GB, Shugart HH. 1989. Environmental factors and ecological processes in boreal forests. Annu Rev Ecol Syst 20:1–28. doi: 10.1146/annurev.es.20.110189.000245. [DOI] [Google Scholar]
- 2.Karhu K, Fritze H, Hämäläinen K, Vanhala P, Jungner H, Oinonen M, Sonninen E, Tuomi M, Spetz P, Kitunen V, Liski J. 2010. Temperature sensitivity of soil carbon fractions in boreal forest soil. Ecology 91:370–376. doi: 10.1890/09-0478.1. [DOI] [PubMed] [Google Scholar]
- 3.Clemmensen KE, Bahr A, Ovaskainen O, Dahlberg A, Ekblad A, Wallander H, Stenlid J, Finlay RD, Wardle DA, Lindahl BD. 2013. Roots and associated fungi drive long-term carbon sequestration in boreal forest. Science 339:1615–1618. doi: 10.1126/science.1231923. [DOI] [PubMed] [Google Scholar]
- 4.Schmidt MW, Noack AG. 2000. Black carbon in soils and sediments: analysis, distribution, implications, and current challenges. Global Biogeochem Cycles 14:777–793. doi: 10.1029/1999GB001208. [DOI] [Google Scholar]
- 5.Turetsky MR, Kane ES, Harden JW, Ottmar RD, Manies KL, Hoy E, Kasischke ES. 2011. Recent acceleration of biomass burning and carbon losses in Alaskan forests and peatlands. Nat Geosci 4:27–31. doi: 10.1038/ngeo1027. [DOI] [Google Scholar]
- 6.Ali AA, Blarquez O, Girardin MP, Hely C, Tinquaut F, El Guellab A, Valsecchi V, Terrier A, Bremond L, Genries A, Gauthier S, Bergeron Y. 2012. Control of the multimillennial wildfire size in boreal North America by spring climatic conditions. Proc Natl Acad Sci U S A 109:20966–20970. doi: 10.1073/pnas.1203467109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winsborough C, Basiliko N. 2010. Fungal and bacterial activity in northern peatlands. Geomicrobiol J 27:315–320. doi: 10.1080/01490450903424432. [DOI] [Google Scholar]
- 8.Taylor DL, Herriott IC, Stone KE, McFarland JW, Booth MG, Leigh MB. 2010. Structure and resilience of fungal communities in Alaskan boreal forest soils. Can J For Res 40:1288–1301. doi: 10.1139/X10-081. [DOI] [Google Scholar]
- 9.González-Pérez JA, González-Vila FJ, Almendros G, Knicker H. 2004. The effect of fire on soil organic matter–a review. Environ Int 30:855–870. doi: 10.1016/j.envint.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 10.Mataix-Solera J, Guerrero C, García-Orenes F, Bárcenas GM, Torres MP. 2009. Forest fire effects on soil microbiology, p 133–175. In Cerdà A, Robichaud PR (ed), Fire effects on soils and restoration strategies. CRC Press, Boca Raton, FL. [Google Scholar]
- 11.Taş N, Prestat E, McFarland JW, Wickland KP, Knight R, Berhe AA, Jorgenson T, Waldrop MP, Jansson JK. 2014. Impact of fire on active layer and permafrost microbial communities and metagenomes in an upland Alaskan boreal forest. ISME J 8:1904–1919. doi: 10.1038/ismej.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arocena J, Opio C. 2003. Prescribed fire-induced changes in properties of sub-boreal forest soils. Geoderma 113:1–16. doi: 10.1016/S0016-7061(02)00312-9. [DOI] [Google Scholar]
- 13.Switzer JM, Hope GD, Grayston SJ, Prescott CE. 2012. Changes in soil chemical and biological properties after thinning and prescribed fire for ecosystem restoration in a Rocky Mountain Douglas-fir forest. Forest Ecol Manag 275:1–13. doi: 10.1016/j.foreco.2012.02.025. [DOI] [Google Scholar]
- 14.Köster K, Berninger F, Lindén A, Köster E, Pumpanen J. 2014. Recovery in fungal biomass is related to decrease in soil organic matter turnover time in a boreal fire chronosequence. Geoderma 235:74–82. [Google Scholar]
- 15.Bååth E, Frostegård Å Pennanen T, Fritze H. 1995. Microbial community structure and pH response in relation to soil organic matter quality in wood-ash fertilized, clear-cut or burned coniferous forest soils. Soil Biol Biochem 27:229–240. doi: 10.1016/0038-0717(94)00140-V. [DOI] [Google Scholar]
- 16.Jonsson L, Dahlberg A, Nilsson M, Zackrisson O, Kåren O. 1999. Ectomycorrhizal fungal communities in late-successional Swedish boreal forests, and their composition following wildfire. Mol Ecol 8:205–215. doi: 10.1046/j.1365-294x.1999.00553.x. [DOI] [Google Scholar]
- 17.Smith JE, McKay D, Brenner G, McIver J, Spatafora JW. 2005. Early impacts of forest restoration treatments on the ectomycorrhizal fungal community and fine root biomass in a mixed conifer forest. J Appl Ecol 42:526–535. doi: 10.1111/j.1365-2664.2005.01047.x. [DOI] [Google Scholar]
- 18.Jonsson T, Kokalj S, Finlay R, Erland S. 1999. Ectomycorrhizal community structure in a limed spruce forest. Mycol Res 103:501–508. doi: 10.1017/S0953756298007461. [DOI] [Google Scholar]
- 19.Brown SP, Callaham MA Jr, Oliver AK, Jumpponen A. 2013. Deep Ion Torrent sequencing identifies soil fungal community shifts after frequent prescribed fires in a southeastern U.S. forest ecosystem. FEMS Microbiol Ecol 86:557–566. doi: 10.1111/1574-6941.12181. [DOI] [PubMed] [Google Scholar]
- 20.Oliver AK, Callaham MA Jr, Jummponen A. 2015. Soil fungal communities respond compositionally to recurring frequent prescribed burning in a managed southeastern U.S. forest ecosystem. Forest Ecol Manag 345:1–9. doi: 10.1016/j.foreco.2015.02.020. [DOI] [Google Scholar]
- 21.Cairney JW, Bastias BA. 2007. Influences of fire on forest soil fungal communities. Can J For Res 37:207–215. doi: 10.1139/x06-190. [DOI] [Google Scholar]
- 22.Buscardo E, Rodríguez-Echeverría S, Martín MP, De Angelis P, Pereira JS, Freitas H. 2010. Impact of wildfire return interval on the ectomycorrhizal resistant propagules communities of a Mediterranean open forest. Fungal Biol 114:628–636. doi: 10.1016/j.funbio.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 23.Richter S, Kipfer T, Wohlgemuth T, Calderon Guerrero C, Ghazoul J, Moser B. 2012. Phenotypic plasticity facilitates resistance to climate change in a highly variable environment. Oecologia 169:269–279. doi: 10.1007/s00442-011-2191-x. [DOI] [PubMed] [Google Scholar]
- 24.Bent E, Kiekel P, Brenton R, Taylor DL. 2011. Root-associated ectomycorrhizal fungi shared by various boreal forest seedlings naturally regenerating after a fire in interior Alaska and correlation of different fungi with host growth responses. Appl Environ Microbiol 77:3351–3359. doi: 10.1128/AEM.02575-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rincón A, Santamaría B, Ocaña L, Verdú M. 2014. Structure and phylogenetic diversity of post-fire ectomycorrhizal communities of maritime pine. Mycorrhiza 24:131–141. doi: 10.1007/s00572-013-0520-0. [DOI] [PubMed] [Google Scholar]
- 26.Clemmensen KE, Bahr A, Ovaskainen O, Dahlberg A, Ekblad A, Wallander H, Stenlid J, Finlay RD, Wardle DA, Lindahl BD. 2013. Roots and associated fungi drive long-term carbon sequestration in boreal forest. Science 339:1615–1618. doi: 10.1126/science.1231923. [DOI] [PubMed] [Google Scholar]
- 27.Holden SR, Gutierrez A, Treseder KK. 2013. Changes in soil fungal communities, extracellular enzyme activities, and litter decomposition across a fire chronosequence in Alaskan boreal forests. Ecosystems 16:34–46. doi: 10.1007/s10021-012-9594-3. [DOI] [Google Scholar]
- 28.Fierer N, Breitbart M, Nulton J, Salamon P, Lozupone C, Jones R, Robeson M, Edwards RA, Felts B, Rayhawk S, Knight R, Rohwer F, Jackson RB. 2007. Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl Environ Microbiol 73:7059–7066. doi: 10.1128/AEM.00358-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jumpponen A, Jones K. 2009. Massively parallel 454 sequencing indicates hyperdiverse fungal communities in temperate Quercus macrocarpa phyllosphere. New Phytol 184:438–448. doi: 10.1111/j.1469-8137.2009.02990.x. [DOI] [PubMed] [Google Scholar]
- 30.Weedon JT, Kowalchuk GA, Aerts R, van Hal J, van Logtestijn R, Taş N, Röling WFM, van Bodegom PM. 2012. Summer warming accelerates sub-arctic peatland nitrogen cycling without changing enzyme pools or microbial community structure. Global Change Biol 18:138–150. doi: 10.1111/j.1365-2486.2011.02548.x. [DOI] [Google Scholar]
- 31.He Z, Gentry TJ, Schadt CW, Wu L, Liebich J, Chong SC, Huang Z, Wu W, Gu B, Jardine P, Criddle C, Zhou J. 2007. GeoChip: a comprehensive microarray for investigating biogeochemical, ecological and environmental processes. ISME J 1:67–77. doi: 10.1038/ismej.2007.2. [DOI] [PubMed] [Google Scholar]
- 32.Xu M, Wu W, Wu L, He Z, Van Nostrand JD, Deng Y, Luo J, Carley J, Ginder-Vogel M, Gentry TJ, Gu B, Watson D, Jardine PM, Marsh TL, Tiedje JM, Hazen T, Criddle CS, Zhou J. 2010. Responses of microbial community functional structure to pilot-scale uranium in situ bioremediation. ISME J 4:1060–1070. doi: 10.1038/ismej.2010.31. [DOI] [PubMed] [Google Scholar]
- 33.Zhou J, Kang S, Schadt CW, Garten CT Jr. 2008. Spatial scaling of functional gene diversity across various microbial taxa. Proc Natl Acad Sci U S A 105:7768–7773. doi: 10.1073/pnas.0709016105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tu Q, Yu H, He Z, Deng Y, Wu L, Van Nostrand JD, Zhou A, Voordeckers J, Lee Y, Qin Y, Hemme CL, Shi Z, Xue K, Yuan T, Wang A, Zhou J. 2014. GeoChip 4: a functional gene-array-based high-throughput environmental technology for microbial community analysis. Mol Ecol Resour 14:914–928. [DOI] [PubMed] [Google Scholar]
- 35.Bergeron Y. 1991. The influence of island and mainland lakeshore landscapes on boreal forest fire regimes. Ecology 72:1980–1992. doi: 10.2307/1941553. [DOI] [Google Scholar]
- 36.Liski EP, Nummi T. 1995. Variation in soil organic carbon and thickness of soil horizons within a boreal forest stand–effects of trees and implications for sampling. Silva Fennica 29:255–266. doi: 10.14214/sf.a9212. [DOI] [Google Scholar]
- 37.Ihrmark K, Bödeker I, Cruz-Martinez K, Friberg H, Kubartova A, Schenck J, Strid Y, Stenlid J, Brandström-Durling M, Clemmensen KE, Lindahl BD. 2012. New primers to amplify the fungal ITS2 region–evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol Ecol 82:666–677. doi: 10.1111/j.1574-6941.2012.01437.x. [DOI] [PubMed] [Google Scholar]
- 38.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huse SM, Welch DM, Morrison HG, Sogin ML. 2010. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol 12:1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Needleman SB, Wunsch CD. 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 42.Tedersoo L, Nilsson RH, Abarenkov K, Jairus T, Sadam A, Saar I, Bahram M, Bechem E, Chuyong G, Kõljalg U. 2010. 454 pyrosequencing and Sanger sequencing of tropical mycorrhizal fungi provide similar results but reveal substantial methodological biases. New Phytol 188:291–301. doi: 10.1111/j.1469-8137.2010.03373.x. [DOI] [PubMed] [Google Scholar]
- 43.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chao A. 1984. Nonparametric estimation of the number of classes in a population. Scand J Stat 11:265–270. [Google Scholar]
- 45.Gihring TM, Green SJ, Schadt CW. 2012. Massively parallel rRNA gene sequencing exacerbates the potential for biased community diversity comparisons due to variable library sizes. Environ Microbiol 14:285–290. doi: 10.1111/j.1462-2920.2011.02550.x. [DOI] [PubMed] [Google Scholar]
- 46.Paulson JN, Stine OC, Bravo HC, Pop M. 2013. Differential abundance analysis for microbial marker-gene surveys. Nat Methods 10:1200–1202. doi: 10.1038/nmeth.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu L, Liu X, Schadt CW, Zhou J. 2006. Microarray-based analysis of subnanogram quantities of microbial community DNAs by using whole-community genome amplification. Appl Environ Microbiol 72:4931–4941. doi: 10.1128/AEM.02738-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang Y, He Z, Wu L, Deng Y, Li G, Zhou J. 2010. Development of a common oligonucleotide reference standard for microarray data normalization and comparison across different microbial communities. Appl Environ Microbiol 76:1088–1094. doi: 10.1128/AEM.02749-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He Z, Van Nostrand JD, Deng Y, Zhou J. 2011. Development and applications of functional gene microarrays in the analysis of the functional diversity, composition, and structure of microbial communities. Front Environ Sci Engin China 5:1–20. doi: 10.1007/s11783-011-0301-y. [DOI] [Google Scholar]
- 50.Team RC. 2013. R: a language and environment for statistical computing, version 3.0.2. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 51.Warnes GR, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, Lumley T, Maechler M, Magnusson A, Moeller S. 2013. gplots: various R programming tools for plotting data. R Package version 2.11.0. R Foundation for Statistical Computing, Vienna, Austria: https://cran.r-project.org/web/packages/gplots/index.html. [Google Scholar]
- 52.Millar RB, Anderson MJ. 2004. Remedies for pseudoreplication. Fish Res 70:397–407. doi: 10.1016/j.fishres.2004.08.016. [DOI] [Google Scholar]
- 53.Bates D, Maechler M, Bolker B, Walker S. 2014. lme4: linear mixed-effects models using Eigen and S4. R Foundation for Statistical Computing, Vienna, Austria: https://cran.r-project.org/web/packages/lme4/citation.html. [Google Scholar]
- 54.Hothorn T, Bretz F, Westfall P. 2008. Simultaneous inference in general parametric models. Biom J 50:346–363. doi: 10.1002/bimj.200810425. [DOI] [PubMed] [Google Scholar]
- 55.Oksanen J. 2011. Multivariate analysis of ecological communities in R: vegan tutorial. R package version: 2.0-2.1. University of Oulu, Oulu, Finland: http://cc.oulu.fi/∼jarioksa/opetus/metodi/vegantutor.pdf. [Google Scholar]
- 56.Schloss PD. 2008. Evaluating different approaches that test whether microbial communities have the same structure. ISME J 2:265–275. doi: 10.1038/ismej.2008.5. [DOI] [PubMed] [Google Scholar]
- 57.Clarke KR. 1993. Non-parametric multivariate analyses of changes in community structure. Aust J Ecol 18:117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x. [DOI] [Google Scholar]
- 58.Phillips LA, Ward V, Jones MD. 2013. Ectomycorrhizal fungi contribute to soil organic matter cycling in sub-boreal forests. ISME J 8:699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lindahl BD, Tunlid A. 2015. Ectomycorrhizal fungi–potential organic matter decomposers, yet not saprotrophs. New Phytol 205:1443–1447. doi: 10.1111/nph.13201. [DOI] [PubMed] [Google Scholar]
- 60.Talbot JM, Bruns TD, Smith DP, Branco S, Glassman SI, Erlandson S, Vilgalys R, Peay KG. 2013. Independent roles of ectomycorrhizal and saprotrophic communities in soil organic matter decomposition. Soil Biol Biochem 57:282–291. doi: 10.1016/j.soilbio.2012.10.004. [DOI] [Google Scholar]
- 61.Rineau F, Roth D, Shah F, Smits M, Johansson T, Canbäk B, Olsen PB, Persson P, Grell MN, Lindquist E, Grigoriev IV, Lange L, Tunlid A. 2012. The ectomycorrhizal fungus Paxillus involutus converts organic matter in plant litter using a trimmed brown-rot mechanism involving Fenton chemistry. Environ Microbiol 14:1477–1487. doi: 10.1111/j.1462-2920.2012.02736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kohler A, Kuo A, Nagy LG, Morin E, Barry KW, Buscot F, Canbäk B, Choi C, Cichocki N, Clum A, Colpaert J, Copeland A, Cost MD, Dore J, Floundas D, Gay G, Girlanda M, Henrissat B, Herrmann S, Hess J, Högberg N, Johansson T, Khouja HR, LaButti K, Levasseur A, Lindquist EA, Lipzen A, Marmeisse R, Martino E, Murat C, Ngan CY, Nehls U, Plett JM, Pringle A, Ohm RA, Perotto S, Peter M, Riley R, Rineau F, Ruytinx J, Salamov A, Shah F, Sun H, Tarkka M, Tritt A, Veneault-Fourrey C, Zuccaro A, Mycorrhizal Genomics Initiative Consortium, Tunlid A, Grigoriev IV, et al. . 2015. Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nat Genet 47:410–415. doi: 10.1038/ng.3223. [DOI] [PubMed] [Google Scholar]
- 63.Zeglin LH, Kluber LA, Myrold DD. 2013. The importance of amino sugar turnover to C and N cycling in organic horizons of old-growth Douglas-fir forest soils colonized by ectomycorrhizal mats. Biogeochemistry 112:679–693. doi: 10.1007/s10533-012-9746-8. [DOI] [Google Scholar]
- 64.Fernandez CW, Koide RT. 2012. The role of chitin in the decomposition of ectomycorrhizal fungal litter. Ecology 93:24–28. doi: 10.1890/11-1346.1. [DOI] [PubMed] [Google Scholar]
- 65.Drenovsky RE, Vo D, Graham KJ, Scow KM. 2004. Soil water content and organic carbon availability are major determinants of soil microbial community composition. Microb Ecol 48:424–430. doi: 10.1007/s00248-003-1063-2. [DOI] [PubMed] [Google Scholar]
- 66.Peay KG, Garbelotto M, Bruns TD. 2010. Evidence of dispersal limitation in soil microorganisms: isolation reduces species richness on mycorrhizal tree islands. Ecology 91:3631–3640. doi: 10.1890/09-2237.1. [DOI] [PubMed] [Google Scholar]
- 67.Hutchinson GE. 1957. Concluding remarks. Cold Spring Harb Symp Quant 22:415–427. [Google Scholar]
- 68.Odum EP. 1969. The strategy of ecosystem development. Science 164:262–270. [DOI] [PubMed] [Google Scholar]
- 69.Pianka ER. 1970. On r-and K-selection. Am Nat 104:592–597. doi: 10.1086/282697. [DOI] [Google Scholar]
- 70.Fritze H, Pennanen T, Pietikäinen J. 1993. Recovery of soil microbial biomass and activity from prescribed burning. Can J For Res 23:1286–1290. doi: 10.1139/x93-164. [DOI] [Google Scholar]
- 71.Hart SC, DeLuca TH, Newman GS, MacKenzie MD, Boyle SI. 2005. Post-fire vegetative dynamics as drivers of microbial community structure and function in forest soils. Forest Ecol Manag 220:166–184. doi: 10.1016/j.foreco.2005.08.012. [DOI] [Google Scholar]
- 72.Rashid A, Ahmed T, Ayub N, Khan A. 1997. Effect of forest fire on number, viability and post-fire re-establishment of arbuscular mycorrhizae. Mycorrhiza 7:217–220. doi: 10.1007/s005720050183. [DOI] [Google Scholar]
- 73.Vrålstad T, Schumacher T, Taylor AF. 2002. Mycorrhizal synthesis between fungal strains of the Hymenoscyphus ericae aggregate and potential ectomycorrhizal and ericoid hosts. New Phytol 153:143–152. doi: 10.1046/j.0028-646X.2001.00290.x. [DOI] [Google Scholar]
- 74.Cutler NA, Chaput DL, van der Gast CJ. 2014. Long-term changes in soil microbial communities during primary succession. Soil Biol Biochem 69:359–370. doi: 10.1016/j.soilbio.2013.11.022. [DOI] [Google Scholar]
- 75.Bödeker I, Clemmensen KE, Boer W, Martin F, Olson Å, Lindahl BD. 2014. Ectomycorrhizal Cortinarius species participate in enzymatic oxidation of humus in northern forest ecosystems. New Phytol 203:245–256. doi: 10.1111/nph.12791. [DOI] [PubMed] [Google Scholar]
- 76.Dunham SM, Larsson K, Spatafora JW. 2007. Species richness and community composition of mat-forming ectomycorrhizal fungi in old-and second-growth Douglas-fir forests of the HJ Andrews Experimental Forest, Oregon, USA. Mycorrhiza 17:633–645. [DOI] [PubMed] [Google Scholar]
- 77.Högberg MN, Yarwood SA, Myrold DD. 2014. Fungal but not bacterial soil communities recover after termination of decadal nitrogen additions to boreal forest. Soil Biol Biochem 72:35–43. doi: 10.1016/j.soilbio.2014.01.014. [DOI] [Google Scholar]
- 78.Koide RT, Xu B, Sharda J, Lekberg Y, Ostiguy N. 2005. Evidence of species interactions within an ectomycorrhizal fungal community. New Phytol 165:305–316. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.