

Abstract
The aim of the present study was to analyze the clinical characteristics of primary Sjogren's syndrome (pSS) with neuromyelitis optica spectrum disorder (NMOSD). We retrospectively reviewed the medical records of 616 patients who were admitted to the Peking Union Medical College Hospital from 1985 to 2013. Of these patients, 43 developed NMOSD. The median duration of symptoms was 60 months and 72% of the patients experienced neurological complications onset in the pSS with NMOSD group. Twenty-one out of 43 patients had neuromyelitis optica (NMO), and 22 exhibited a limited form of NMO. Serum anti-aquaporin-4 (AQP4) antibody positivity was detected in 89.3% of the patients. A total of 60.5% of the patients (26 patients) complained of dry mouth, 72.1% were positive for objective xerostomia, 53.5% complained of dry eyes, and 74.4% had a positive ocular test. Biopsy of the minor salivary glands was performed in 33 patients, 28 of whom (84.8%) had a lymphocytic focus score of ≥1. Anti-Ro/SSA or anti-La/SSB antibodies were detected in 41 patients (95.3%). Compared with the pSS patients without NMOSD, the incidences of xerophthalmia, xerostomia, arthritis, interstitial lung disease, and renal tubular acidosis were significantly lower in the patients with NMOSD.
NMOSD is a neurologic complication of pSS. The presence of anti-AQP4 antibody may be a predictor for pSS patients with NMOSD. Neurological manifestations are prominent in these patients. In clinical scenarios involving pSS or NMOSD, rheumatologists and neurologists should be aware of this association and perform the appropriate tests.
INTRODUCTION
Primary Sjogren's syndrome (pSS) is a chronic systemic autoimmune disease that is characterized by exocrine involvement.1 Approximately 20% to 25%2,3 of patients also have neurological manifestations, but the exact prevalence of central nervous system (CNS) involvement remains controversial. Neuromyelitis optica (NMO), also known as Devic's syndrome, is a severely disabling CNS disorder that is thought to have an autoimmune etiology and predominantly affects the optic nerves and spinal cord.4 However, NMO is now recognized as a spectrum disease that affects other regions of the CNS and includes more diverse clinical presentations because of the identification of a disease-specific autoantibody against aquaporin-4 (AQP4).5 With increased numbers of emerging pSS patients with neuromyelitis optica spectrum disorder (NMOSD) cases reports,6–8 studies of large Chinese populations have been rare. The aim of the present study was to assess the clinical characteristics, seroimmunological correlations, and risk factors for pSS with NMOSD in a Chinese cohort at a single center. To our knowledge, this is the largest pSS patients with NMOSD cohort in the literature.
MATERIALS AND METHODS
Patients
We retrospectively reviewed the clinical charts of 616 Chinese patients who were diagnosed with pSS and admitted to Peking Union Medical College Hospital (PUMCH) in Beijing, China, between January 1985 and December 2013, as shown in Figure 1. The diagnosis of pSS was based on the revised version of the diagnostic criteria of the American-European Consensus Group.9 Clinical symptoms of sicca complex, including dry mouth, recurrent parotid enlargement, and rampant caries, were evaluated. Ocular involvement was documented by the Schirmer test or the Rose Bengal score.10 Objective xerostomia was confirmed by an abnormal salivary scintigraphy11 or unstimulated salivary flow. Biopsy samples of the minor salivary glands with lymphocytic focus scores of at least 1 were considered suggestive of Sjogren's syndrome.12 Screening for autoantibodies to Ro/SSA and La/SSB was systematically performed by Ouchterlony double-gel immunodiffusion and in some cases by Western blotting. All tests were performed at the clinical rheumatology immunology laboratory at PUMCH.
FIGURE 1.

Inclusion and exclusion criteria.
Of the 616 pSS patients, 43 were identified as having NMOSD during the study period. Patients were considered to have NMO/NMOSD simultaneously if they fulfilled the Wingerchuk criteria5,13 (Figure 2). Longitudinal extensive transverse myelitis (LETM) was defined as T2 enhancement on spinal magnetic resonance imaging (MRI) in 3 or more contiguous vertebral segments. Optic neuritis (ON) was diagnosed by a board-certified neurologist or neuro-ophthalmologist. Depending on the clinical findings, patients with NMOSD underwent brain or spinal MRI and cerebrospinal fluid (CSF) assessment. Indirect immunofluorescence analysis was performed to detect anti-AQP4 antibody at the PUMCH clinical neuroimmunological laboratory. Transfection of HEK-293 cells with AQP4 was performed as originally reported by Lennon et al.14,15 We selected the remaining pSS patients without NMOSD as controls. Extraglandular manifestations other than neurologic involvement were also recorded for all patients. A comparison of the clinical features of the pSS patients with and without NMOSD was performed. The institutional review board of PUMCH approved this study. The requirement for written informed consent was waived because this study was retrospective and only involved the review of records.
FIGURE 2.

Definitions of NMO and NMOSD. ∗To be diagnosed, a patient needs to fulfill the first 2 criteria and 2 out of 3 of the supporting criteria. ∗∗Any of the 5 clinical scenarios listed are indicative of NMOSD. LETM = longitudinal extensive transverse myelitis; MS = multiple sclerosis; NMO = neuromyelitis optica; NMOSD = neuromyelitis optica spectrum disorder; ON = optic neuritis.
Statistical Analysis
Statistical analysis was performed using SPSS statistical package version 18.0 (IBM, Armonk, NY). Categorical data are presented as numbers (percentages), and continuous variables are presented as the mean and standard deviation (SD) or the median and interquartile range, depending on the type of distribution. The chi-square and Fisher's exact tests were used to analyze categorical variables, and the independent samples t test was used to compare quantitative data between the groups. P values of <0.05 were considered to be statistically significant.
RESULTS
Demographic Characteristics
A total of 43 pSS patients were identified with NMOSD between January 1985 and December 2013. There were 38 females (88.4%) and 5 males (11.6%), corresponding to a ratio of 7.6 to 1. The mean age at onset of disease was 35.3 ± 12.2 years (range 14–69 years), and the median duration of symptoms before diagnosis was 60.2 months (range 1–252 months). Neurologic complications were the first symptoms in 31 patients (72.1%) in the pSS with NMOSD group. Only 12 patients (27.9%) exhibited sicca symptoms before the onset of neurologic manifestations.
Glandular Features
Of the 43 patients, 26 (60.5%) developed dry mouth, 10 had rampant caries, and 3 presented with recurrent parotid enlargement. A total of 31 patients (72.1%) were positive for objective xerostomia. Twenty-three patients (53.5%) complained of dry eyes, and 32 (74.4%) had a positive ocular test result. Biopsy of the minor salivary glands was performed in 33 patients, and 28 (84.8%) had a lymphocytic focus score of 1 or more. Anti-Ro/SSA or anti-La/SSB antibodies were detected in 41 patients (95.3%).
Neurological Findings
Of the 43 pSS patients with NMOSD, 32 (74.4%) had optic nerve involvement including visual loss (29 patients, 67.4%), visual field defects (13 patients, 30.2%), or diplopia (3 patients, 7.0%). Spinal cord involvement was identified in 36 patients (83.7%), 32 (74.4%) of whom had LETM. Peripheral nervous system involvement was noted in 5 patients (11.6%). The median time between visual and spinal cord involvement was 5.5 months, and 14 patients (32.6%) had both visual and spinal cord manifestations within 1 year. Upon referral to our hospital, there were 21 patients (48.8%) with NMO and 22 (51.2%) with limited forms of NMO (idiopathic single or recurrent events of LETM, 11 patients, 25.6%; recurrent or simultaneous bilateral ON, 11 patients, 25.6%). A comparison of the clinical characteristics among the 3 groups is shown in Table 1. Eleven patients were experiencing their first NMOSD attack, whereas 32 (74.4%) cases involved a recurrent course (range of 1–6 recurrences).
TABLE 1.
Demographic and Clinical Features of pSS Patients With NMOSD

Brain and spinal MRI were performed for all patients. Brain lesions were found in 19 patients, with the areas surrounding the third and fourth ventricles being more frequently involved. A brain stem lesion was noted in 11 patients (25.6%). The spinal cord lesions were predominantly located in the cervical (29 patients, 67.4%) and thoracic cords (27 patients, 62.8%) (Figure 3). Thirty-four patients underwent lumbar puncture. The CSF pressure was 135.9 ± 50.8 mm H2O (range 75–210 mm H2O), and it was elevated in 6 patients and depressed in 4. The CSF protein level in these patients was 0.61 g/L (range 0.25–2.13 g/L), and an elevated protein level was noted in 21 patients (61.8%). Activated lymphocytes were found in 10/34 patient samples (29.4%). CSF oligoclonal bands were identified in 12/34 (35.3%) patient sample.
FIGURE 3.

Representative cerebral and spinal cord MRI images of 4 pSS patients with NMOSD. (A) Abnormal T2-weighted signal at the midbrain. (B) Sagittal T2-weighted spinal cord MRI showing a high signal intensity long cord lesion; (B1) cervical 1–4, (B2) thoracic 1–8, and (B3) thoracic 10-lumbar1. MRI = magnetic resonance imaging; NMOSD = neuromyelitis optica spectrum disorder.
A total of 25/28 patients (89.3%) were positive for serum anti-AQP4 antibody. In addition, this antibody was detected in the CSF of 8/9 seropositive patients (88.9%), and it was not detected in the sera or CSF of 3 patients.
Extraglandular Manifestations Other Than Neurologic Involvement
Among the 43 pSS patients with NMOSD, 9 (20.9%) had arthritis, 5 patients (11.6%) had interstitial lung disease, and 3 (7.0%) had purpura. Extraglandular manifestations also included Raynaud's phenomenon (1 patient, 2.3%), myositis (1 patient, 2.3%), and renal tubular acidosis (1 patient, 2.3%).
Comparison of Clinical Features Between pSS Patients With and Without NMOSD
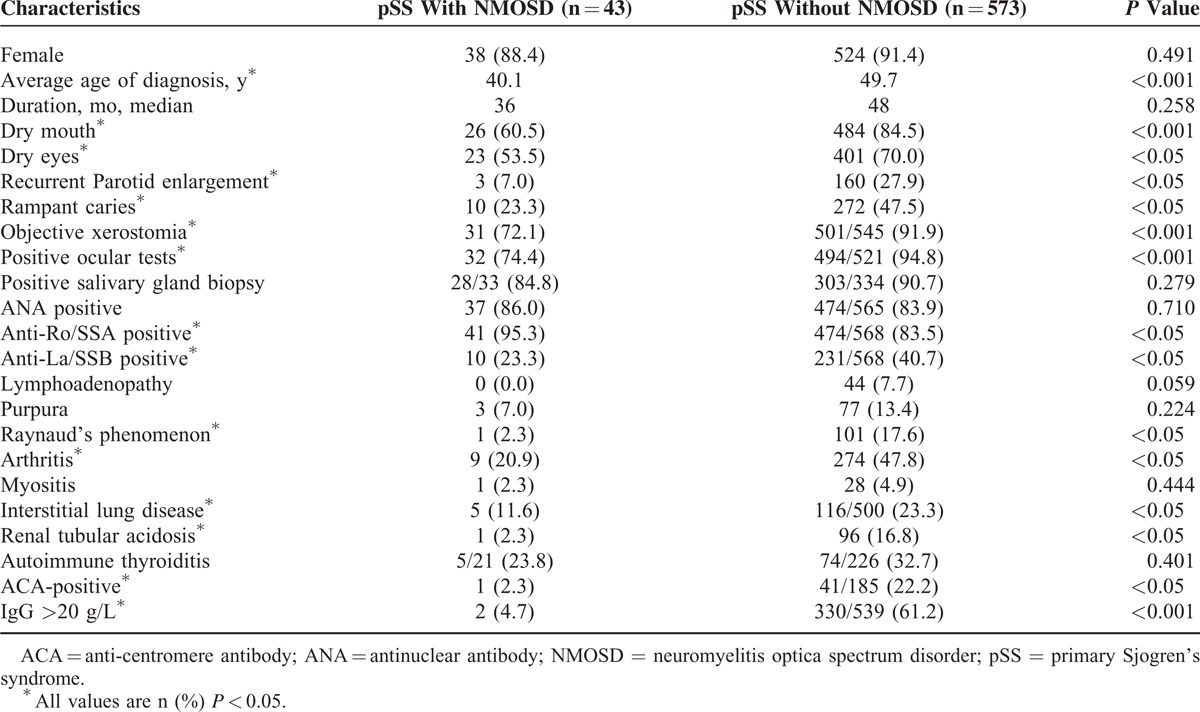
A total of 573 pSS patients without NMOSD were selected as controls. The clinical features were compared between the pSS patients with and without NMOSD (Table 2). Significant differences were found for dry mouth (P < 0.001), dry eyes (P = 0.024), recurrent parotid enlargement (P = 0.003), rampant caries (P = 0.002), objective xerostomia (P < 0.001), positive ocular test (P < 0.001), Raynaud's phenomenon (P = 0.009), arthritis (P = 0.001), interstitial lung disease (P < 0.05), and renal tubular acidosis (P < 0.05). There were no significant differences in gender or the duration of symptoms between the 2 groups.
TABLE 2.
Clinical Manifestations Between pSS Patients With and Without NMOSD
DISCUSSION
pSS is a systemic autoimmune disease that typically presents with xerophthalmia and xerostomia. Generalized exocrine dysfunction is also typical. Neurological involvement associated with pSS is classified as peripheral nervous system or CNS involvement, but the particular CNS manifestations are still under debate. NMOSD is an inflammatory demyelinating spectrum disorder of the CNS that is characterized by severe attacks of ON and myelitis. In 10% to 40% of patients, this condition is complicated by an autoimmune disease, such as thyroiditis, systemic lupus erythematosus, or pSS5. Few studies have attempted to address the relationship between pSS and NMOSD.
Discriminating between pSS and NMOSD is difficult because sicca symptoms can occur after the onset of neurological symptoms. Alhomoud et al16 have reported that up to 33% of patients who present with pSS with CNS involvement lack sicca symptoms at the time of presentation but that they eventually develop these symptoms over a 5-year follow-up period. In our cohort, neurological manifestations preceded the sicca symptoms in 72.1% of the patients, which is consistent with the findings of other studies.17–19 This delay in symptoms may lead to the underestimation of the prevalence of pSS in patients with NMOSD. Neurological manifestations were prominent in our study, whereas the frequency of other organ lesions was low. Accurate diagnosis and management requires a thorough history, physical examination, and laboratory tests, and careful clinical follow-up is recommended.
NMO-IgG is a disease-specific autoantibody for NMO14. The NMO antigen is AQP4, which is the predominant water channel protein that is expressed on astrocytes in the CNS15. Carvalho DC19 reviewed 37 cases of pSS patients with NMOSD whose anti-AQP4 antibody serostatus was informed, and showed that it was positive in 32 cases (86.5%). Similarly, 89.3% of the pSS patients with NMOSD were positive for serum anti-AQP4 antibody in our series. CSF positivity for anti-AQP4 antibody was found in 88.9% of the seropositive patients. We also tested 20 patients who had been diagnosed with pSS without NMOSD during the same period, none of whom were positive for this antibody. These findings suggest that anti-AQP4 antibody is also an important marker for pSS patients with NMOSD with a relatively high specificity and sensitivity of detection. Moreover, this marker can be detected in patients with NMOSD in the early stages of the disease who are undetectable by MRI.20 Thus, the anti-AQP4 antibody may be a predictor of the occurrence of pSS with NMOSD. Park et al21 have reported that the frequency of anti-AQP4 antibody positivity is significantly higher in anti-Ro/SSA antibody-positive patients with NMOSD than in anti-Ro/SSA antibody-negative patients (90% vs 32.6%, P < 0.001). In our experience, anti-Ro/SSA antibody positivity is also closely related to systemic autoimmune diseases, especially pSS. However, we found anti-AQP4 antibody positivity rates of 88% and 100% for the anti-Ro/SSA-positive and anti-Ro/SSA-negative patients, respectively (P = 0.145). This discrepancy is likely because of differences in subjects and the limited number of anti-Ro/SSA negative pSS-NMOSD patients in this study (2 patients).
pSS is characterized by mononuclear infiltration and the destruction of salivary and lachrymal glands. Similarly, invasion of visceral organs or vasculitic lesions by mononuclear infiltrates can lead to extraglandular manifestations. However, the precise pathogenesis pSS patients with NMOSD remain unclear. The co-association could be due to common genetic and/or environmental factors that cause a predisposition to autoimmunity. Javed22 confirmed that 20 NMOSD patients who underwent minor salivary gland biopsy all had evidence of minor salivary gland inflammation. Moreover, a positive labial biopsy is common in pSS patients. In addition, inflammatory lesions in NMO occur in areas with high levels of AQP4 expression, such as the spinal cord and optic nerves.23 AQP4 is expressed at low levels in the salivary glands, whereas anti-aquaporin-5 (AQP5) is expressed at high levels and plays a major role in salivary gland secretion.24 The protein sequence identity between AQP4 and AQP5 is approximately 50%.25 A subset of autoreactive immune cells recognize homologous portions of AQP4 and AQP5, thereby causing inflammation in both the CNS and salivary glands, suggesting a common pathophysiological mechanism between pSS and NMOSD.
To our knowledge, no studies have directly evaluated the optimal therapy for pSS patients with NMOSD. Previous data on the risk of relapse of NMO patients with AQP4 autoantibody positivity have led us to recommend immunosuppressive therapy because of the association with visual impairment and the high risk (>60%) of neurological relapse within 1 year.26,27 Responders to corticosteroid pulse therapy are more commonly positive for anti-AQP4 antibody than nonresponders.28 Currently, corticosteroid pulse therapy for early induction and the use of an immunosuppressant (such as cyclophosphamide, azathioprine) for maintenance are recommended. Rituximab and tocilizumab have also demonstrated sustained clinical efficacy in treatment-resistant patients.29,30 In patients with relapsing disease, lifelong therapy should be considered.31 However, the standard for the management of pSS patients with NMOSD awaits long-term observation.
Because of the retrospective nature of this study, our results may have been affected by selection bias. The proportion of NMOSD patients in the pSS group is higher compared with previous reports. PUMCH is a university-based hospital and referral center for complicated patients nationwide, and our neurologic department specializes in inflammatory disorders (eg, multiple sclerosis, ON, myelopathy). In addition, the ratio of renal and pulmonary involvement was relatively high in the control group, which may have been because the control group patients were all inpatients, suggesting the presence of relatively severe disease.
In conclusion, NMOSD is a neurologic complication of pSS, and these 2 conditions may share a common pathophysiological mechanism. pSS patients with NMOSD have a distinctive clinical profile that is characterized by relatively severe neurological involvement and mild xerophthalmia, xerostomia, and other extraglandular manifestations. The presence of anti-AQP4 antibody may be a predictor for these patients. Rheumatologists and neurologists should be aware of this association and perform appropriate tests. The early diagnosis of pSS with NMOSD is important for choosing suitable therapies and improving patient prognosis.
Footnotes
Abbreviations: ACA = anti-centromere antibody, ANA = antinuclear antibody, AQP4 = aquaporin-4, AQP5 = aquaporin-5, CNS = central nervous system, CSF = cerebrospinal fluid, LETM = longitudinal extensive transverse myelitis, MRI = magnetic resonance imaging, NMO = neuromyelitis optica, NMOSD = neuromyelitis optica spectrum disorder, ON = optic neuritis, pSS = primary Sjogren's syndrome.
LQ and QW contributed equally to this article.
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Moutsopoulos HM. Sjogren's syndrome: autoimmune epithelitis. Clin Immunol Immunopathol 1994; 72:162–165. [DOI] [PubMed] [Google Scholar]
- 2.Delalande S, de Seze J, Fauchais AL, et al. Neurologic manifestations in primary Sjogren syndrome: a study of 82 patients. Medicine (Baltimore) 2004; 83:280–291. [DOI] [PubMed] [Google Scholar]
- 3.Pizova NV. The Sjogren's syndrome and multiple sclerosis: similarity and differences. Zh Nevrol Psikhiatr Im S S Korsakova 2012; 112 (Pt 2):69–74. [PubMed] [Google Scholar]
- 4.Wingerchuk DM, Hogancamp WF, O’Brien PC, et al. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology 1999; 53:1107–1114. [DOI] [PubMed] [Google Scholar]
- 5.Wingerchuk DM, Lennon VA, Lucchinetti CF, et al. The spectrum of neuromyelitis optica. Lancet Neurol 2007; 6:805–815. [DOI] [PubMed] [Google Scholar]
- 6.Kahlenberg JM. Neuromyelitis optica spectrum disorder as an initial presentation of primary Sjogren's syndrome. Semin Arthritis Rheum 2011; 40:343–348. [DOI] [PubMed] [Google Scholar]
- 7.Rabadi MH, Kundi S, Brett D, et al. Neurological pictures. Primary Sjogren syndrome presenting as neuromyelitis optica. J Neurol Neurosurg Psychiatry 2010; 81:213–214. [DOI] [PubMed] [Google Scholar]
- 8.Colaci M, Cassone G, Manfredi A, et al. Neurologic complications associated with Sjogren's disease: case reports and modern pathogenic dilemma. Case Rep Neurol Med 2014; 2014:590292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vitali C, Bombardieri S, Jonsson R, et al. Classification criteria for Sjogren's syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis 2002; 61:554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Bijsterveld OP. Diagnosis and differential diagnosis of keratoconjunctivitis sicca associated with tear gland degeneration. Clin Exp Rheumatol 1990; 8 (Suppl 5):3–6. [PubMed] [Google Scholar]
- 11.Schall GL, Anderson LG, Wolf RO, et al. Xerostomia in Sjogren's syndrome. Evaluation by sequential salivary scintigraphy. JAMA 1971; 216:2109–2116. [PubMed] [Google Scholar]
- 12.Daniels TE, Whitcher JP. Association of patterns of labial salivary gland inflammation with keratoconjunctivitis sicca. Analysis of 618 patients with suspected Sjogren's syndrome. Arthritis Rheum 1994; 37:869–877. [DOI] [PubMed] [Google Scholar]
- 13.Wingerchuk DM, Lennon VA, Pittock SJ, et al. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006; 66:1485–1489. [DOI] [PubMed] [Google Scholar]
- 14.Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004; 364:2106–2112. [DOI] [PubMed] [Google Scholar]
- 15.Lennon VA, Kryzer TJ, Pittock SJ, et al. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 2005; 202:473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alhomoud IA, Bohlega SA, Alkawi MZ, et al. Primary Sjogren's syndrome with central nervous system involvement. Saudi Med J 2009; 30:1067–1072. [PubMed] [Google Scholar]
- 17.Tristano AG. A case of Sjogren's syndrome with acute transverse myelitis and polyneuropathy in a patient free of sicca symptoms. Clin Rheumatol 2006; 25:113–114. [DOI] [PubMed] [Google Scholar]
- 18.Manabe Y, Sasaki C, Warita H, et al. Sjogren's syndrome with acute transverse myelopathy as the initial manifestation. J Neurol Sci 2000; 176:158–161. [DOI] [PubMed] [Google Scholar]
- 19.Carvalho DC, Tironi TS, Freitas DS, et al. Sjogren syndrome and neuromyelitis optica spectrum disorder co-exist in a common autoimmune milieu. Arq Neuropsiquiatr 2014; 72:619–624. [DOI] [PubMed] [Google Scholar]
- 20.Min JH, Kim HJ, Kim BJ, et al. Brain abnormalities in Sjogren syndrome with recurrent CNS manifestations: association with neuromyelitis optica. Mult Scler 2009; 15:1069–1076. [DOI] [PubMed] [Google Scholar]
- 21.Park JH, Hwang J, Min JH, et al. Presence of anti-Ro/SSA antibody may be associated with anti-aquaporin-4 antibody positivity in neuromyelitis optica spectrum disorder. J Neurol Sci 2015; 348:132–135. [DOI] [PubMed] [Google Scholar]
- 22.Javed A, Balabanov R, Arnason BG, et al. Minor salivary gland inflammation in Devic's disease and longitudinally extensive myelitis. Mult Scler 2008; 14:809–814. [DOI] [PubMed] [Google Scholar]
- 23.Pittock SJ, Weinshenker BG, Lucchinetti CF, et al. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol 2006; 63:964–968. [DOI] [PubMed] [Google Scholar]
- 24.Delporte C, Steinfeld S. Distribution and roles of aquaporins in salivary glands. Biochim Biophys Acta 2006; 1758:1061–1070. [DOI] [PubMed] [Google Scholar]
- 25.Altschul SF, Madden TL, Schaffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakashima I, Fujihara K, Miyazawa I, et al. Clinical and MRI features of Japanese patients with multiple sclerosis positive for NMO-IgG. J Neurol Neurosurg Psychiatry 2006; 77:1073–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weinshenker BG, Wingerchuk DM, Vukusic S, et al. Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol 2006; 59:566–569. [DOI] [PubMed] [Google Scholar]
- 28.Estiasari R, Matsushita T, Masaki K, et al. Comparison of clinical, immunological and neuroimaging features between anti-aquaporin-4 antibody-positive and antibody-negative Sjogren's syndrome patients with central nervous system manifestations. Mult Scler 2012; 18:807–816. [DOI] [PubMed] [Google Scholar]
- 29.Kim SH, Huh SY, Lee SJ, et al. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol 2013; 70:1110–1117. [DOI] [PubMed] [Google Scholar]
- 30.Kieseier BC, Stuve O, Dehmel T, et al. Disease amelioration with tocilizumab in a treatment-resistant patient with neuromyelitis optica: implication for cellular immune responses. JAMA Neurol 2013; 70:390–393. [DOI] [PubMed] [Google Scholar]
- 31.Wingerchuk DM, Weinshenker BG. Neuromyelitis optica. Curr Treat Options Neurol 2008; 10:55–66. [DOI] [PubMed] [Google Scholar]