

Abstract
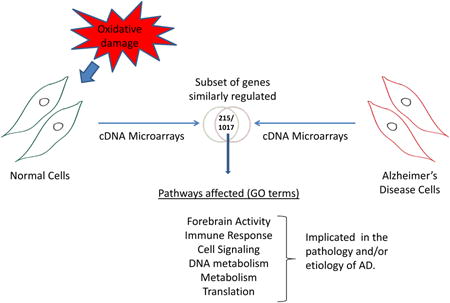
Alzheimer's disease (AD) is a major health problem in the US, affecting one in eight Americans over the age of 65. The number of elderly suffering from AD is expected to continue to increase over the next decade, as the average age of the US population increases. The risk factors for and etiology of AD are not well understood; however, recent studies suggest that exposure to oxidative stress may be a contributing factor. Here, microarray gene expression signatures were compared in AD patient-derived fibroblasts and normal fibroblasts exposed to hydrogen peroxide or menadione (to simulate conditions of oxidative stress). Using the 23K Illumina cDNA microarray to screen expression of > 14,000 human genes, we identified a total of 1017 genes that are chronically up- or down-regulated in AD fibroblasts, 215 of which were also differentially expressed in normal human fibroblasts within 12 h after exposure to hydrogen peroxide or menadione. Pathway analysis of these 215 genes and their associated pathways reveals cellular functions that may be critically dysregulated by oxidative stress and play a critical role in the etiology and/or pathology of AD. We then examined the AD fibroblasts for the presence of oxidative DNA damage and found increased accumulation of 8-oxo-guanine. These results indicate the possible role of oxidative stress in the gene expression profile seen in AD.
Graphical abstract
Introduction
Alzheimer's disease (AD) is a complex, heterogeneous disorder primarily affecting the elderly, characterized by progressive loss of neuronal cells and a variable degree of cognitive loss. AD is a major public health issue in the US, with an estimated 5.4 million Americans affected by and/or being treated for AD during 2011, at a cost of nearly $183 billion dollars [1]. The exact causes of AD are poorly understood, but a complex etiology is likely, possibly involving multiple genetic, epigenetic, viral and environmental factors [2]. Genetic studies suggest that mutations in the genes encoding presenilin-1, presenilin-2 and the beta-amyloid precursor protein are strong risk factors for AD in the relatively small fraction of AD patients who present with early onset familial disease. However, the vast majority of AD cases are non-familial, sporadic late onset AD, and it is generally thought that genetic predisposition plays little or no role in late onset AD [3-6].
Several studies suggest that DNA damage accumulates in older neurons, and that persistent DNA damage may play a role in late onset AD. For example, DNA strand breaks are 2-fold more abundant in nuclear DNA from cortical neurons in AD brains than in normal control brains [7] and higher levels of the oxidative DNA base modifications 8-oxo-guanine (8-OdG), 8-hydroxyadenine, 5-hydroxycytosine, 5-hydroxyuracil and 4,6-diamino-5-formamidopyrimidine are found in nuclear and mitochondrial DNA in AD brains [8-11]. Oxidative DNA damage is considered a possible early biomarker for AD [12-17] as well as its precursor syndrome, Mild Cognitive Impairment (MCI) [18]. Recent studies also demonstrate impaired oxidative metabolism in fibroblasts from AD patients [19, 20] and protein oxidation, lipid peroxidation, and other markers of oxidative damage are detected in AD brains [21].
Base excision repair (BER), the primary mechanism for repairing oxidative damage to DNA bases [22], is deficient in brains of AD and MCI patients [23-26], which may explain the higher load of oxidative DNA damage in AD tissues. However, other factors may also play a role, including altered patterns of transcription, translation, oxidant defence systems or protein processing, leading to dysfunctional regulatory networks and additional transcriptional defects [2, 27-32].
As discussed above, much evidence supports the hypothesis that oxidative macromolecular damage plays a role in AD. Here, we investigated this idea by comparing the gene expression profile of normal human fibroblasts exposed to oxidative stress with the gene expression profile of sporadic AD-patient derived fibroblast cell lines. For this study, primary human fibroblasts were derived from normal donors age and sex-matched to donors with AD, and the normal fibroblasts were exposed briefly to hydrogen peroxide (H2O2) or menadione and analysed 0, 1, 6 or 12 h after the end of treatment. cDNA microarray technology was used to analyze expression of >14,000 genes in the oxidatively-stressed and untreated normal fibroblasts and in untreated AD patient-derived fibroblasts. AD cells were also analyzed by flow cytometry after exposure to a fluorescein-labelled antibody to 8-OdG. The results of these studies support the hypothesis that AD cells experience chronic oxidative stress, which alters gene expression and likely contributes to AD etiology and/or pathology.
Materials and Methods
Cell culture
Primary cell lines were obtained from Coriell Cell Repositories and maintained at 37°C and 5% CO2. Normal fibroblast cell lines used were AG05448 and AG09857. AD cell lines were: AG05770 and AG07374. All lines were grown in AmnioMAX II Complete media (Invitrogen, CA, USA). U2OS was grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin.
Experimental Treatment
Cells were treated with either 25 μM menadione or 100 μM H2O2, prepared in media without serum. Treatment with H2O2 was carried out for 15 minutes whereas menadione treatment was for 1 h. Post-treatment, the cells were washed once with 10 mL PBS. Conditioned media was returned to the plates then the cells were cultured at 37°C until they were harvested. Cells were harvested after 0, 1, 6, or 12 h. The control cells were manipulated as the experimental except no drug was added (i.e., mock incubation).
Flow Cytometry
Treatment of the cells was carried out as above. Cells were trypsinized and collected in Phosphate buffered saline (PBS) with 10% serum. They were then spun at 400 × g, 4°C for 5 min and washed twice with PBS. Cells were passed through a needle and fixed overnight using 70% ethanol. Fixed cells were washed the next day with PBS. They were incubated in 1 mL PBS with horse serum at 4°C for 30 min. They were further washed in PBS and incubated in 2 N HCl for 20 min. The cells were passed through a needle again and collected by centrifugation as above. Buffer containing 0.1M sodium borate (pH 8.5) was then added for 2 min, followed by centrifugation. The supernatant was collected and incubated with the primary antibody to 8-OdG (Millipore; 1:250) for 2 h at 4°C. The primary antibody was removed by washing 3 times with PBS. The cells were then incubated with a FITC-coupled secondary antibody at 1:1000 dilution for 1 h at 4°C, followed by 3 washes using PBS. The cells were suspended in 1 mL PBS and analysed on an Accuri C6 flow cytometer. The data were analysed using FLOJO software.
Microarray analysis
Total RNA was extracted from normal and AD fibroblasts using QIAGEN RNeasy Mini Kit according to the manufacturer's protocol. The quantity of recovered RNA was assessed using a NanoDrop ND-1000 spectrophotometer. RNA from the 2 cell lines was pooled prior to analysis [33].
A 0.5 μg aliquot of total RNA from the pooled normal or AD cell lines was labeled using the Illumina Total Prep RNA Amplification kit (Ambion). A total of 0.85 μg of biotin-labeled cRNA was hybridized for 16 h to the 23 k gene Illumina's Sentrix HumanRef-8 v2 Expression BeadChips (Illumina). The arrays were washed, blocked and then hybridized. Biotinylated cRNA was detected with streptavidin-Cy3 and quantitated using Illumina's BeadStation 500GX Genetic Analysis Systems scanner. Image processing and data extraction were performed using BeadStudio ver. 15 (Illumina).
Microarray data were analyzed using DIANE 6.0, a spread sheet-based microarray analysis program based on the JMP6 package. Raw microarray data were subjected to Z normalization and tested for significant changes. Genes were determined to be differentially expressed after calculating the Z-ratio and false discovery rate and filtered by ANOVA test and detection values. Individual genes with P-value <0.02, Z-ratio >1.5 and false discovery rate <0.03 were considered significantly changed with one way ANOVA by sample p values less or equal to 0.05. Gene set enrichment analysis was used to identify biological and phenotypic changes using the gene expression change of all genes on the array to identify the most significantly changed terms with a Z-test score not more than 0.05 and each selected phenotype term having at least three genes on the array [34, 35]. The functional gene sets used were from the mSIG and Gene Ontology (GO) databases while the disease gene sets were from the GAD database.
Quantitative PCR
Nine genes of interest were chosen based on microarray data, their relevance to AD onset or pathology or their expression level. Total RNA was extracted and used to prepare cDNA with an iScript cDNA synthesis kit (Biorad). RT-PCR using TAQMAN probes (see Table 1) was conducted as per manufacturer's instructions using the Applied Biosystems 7900 HT Real-time PCR system. Each assay was replicated four times and CT values were averaged. Control reactions revealed no significant contamination or cross-contamination of samples.
Results
Experimental Design
Experimental systems for studying AD often use tissue from autopsied brains or peripheral lymphoblasts. In this study, cell lines derived from sporadic AD patient fibroblasts and normal human fibroblasts (controls) from donors age- and sex-matched to the AD patient donors, were used as sources of mRNA for gene expression analysis [36]. The normal cell lines were AG04558, AG09857 and the AD cell lines were AG07374, AG05770. The popoluation doubling and the passage number were the same for the different cells used. Control cells were exposed to 100 μM H2O2 for 15 min or 25 μM menadione for 1 h, and harvested 0, 1, 6, or 12 h after treatment (Figure. S1). Gene expression was also analysed in untreated control cells and untreated AD cells after mock incubations. mRNA was prepared and pooled from two biologically-independent samples for each experimental condition, because this minimized data variability [33]. mRNA was hybridized in triplicate to the 23 k gene Sentrix HumanRef-8 v2 Expression BeadChips (Illumina). Of 14,422 genes analyzed, a total of 1,017 genes (approximately 7%) were up- or down-regulated in AD and oxidatively-stressed normal cells that had a Z-score >1.5 across all the time points measured (see Methods for experimental details and statistical analysis).
Identification of normal fibroblast genes modulated by oxidative stress
Normal human fibroblasts AG04558 and AG09857 were exposed to H2O2 (15 min) or menadione (1 h) after which the cells were incubated and harvested for mRNA as described above. When AD cells, relative to untreated normal control cells, were analysed after mock incubations, more genes were down-regulated than up-regulated at both time points (Figures 1A and 1B). At the 0 h time point for the stressed control cells (stressed control versus untreated control, Figure 1A), fewer genes were down-regulated than up-regulated while the reverse was observed at the 12 h time point (Figure 1B). Using a Z-score cut-off of 1.5, 275 or 752 genes were differentially expressed at 0 and 12 h time points, respectively (Figure 1A and 1B). These data suggest that oxidative stress has a greater tendency to down-regulate gene expression than to up-regulate gene expression in normal human fibroblasts.
Figure 1. The response of normal human fibroblasts to oxidative stress.
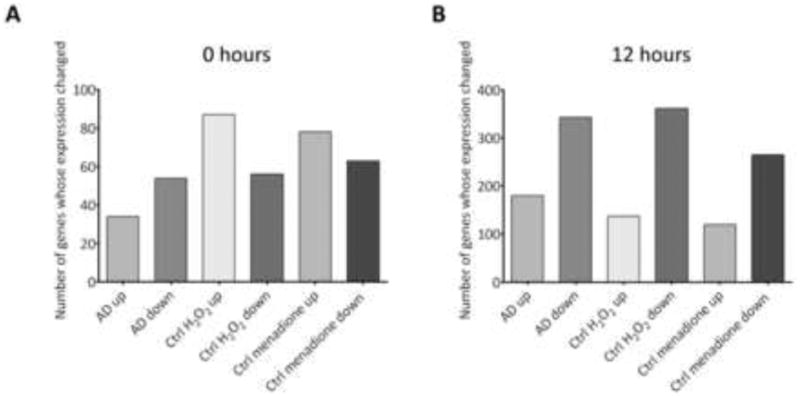
(A) Bar graphs plotting number of genes up- (up) or down-regulated (down) 0 h after treatment with 100 μM H2O2, or 25 μM menadione (normal fibroblasts) or after mock incubation (AD cells and control cells) (B) Same as (A), except 12 hours after treatment or mock treatment.
Common patterns of gene expression associated with oxidative stress and AD
To investigate whether oxidative stress plays a role in AD etiology or pathology, the expression profile of normal cells exposed to hydrogen peroxide or menadione was compared with the expression profile of untreated AD cells; in each case, relative expression values in untreated normal cells served as the reference point. For all four samples/conditions, data were examined at the 0, 1, 6, and 12 h time points after treatment (or mock treatment, for AD cells and untreated normal cells). Data for the 0 and 12 h time points are summarized in Figure 2 and data for the 1 and 6 h time points are summarized in Figure S2. Data for the 0, 1 and 6 h time points were relatively uninformative, because few genes met the cut-off criteria (Z-score > 1.5) and of those, few were up- or down-regulated in both normal cells and AD cells within 6 h after oxidative stress (Figure 2A, S2A and S2B).
Figure 2. Gene expression profiles associated with AD or oxidative stress.
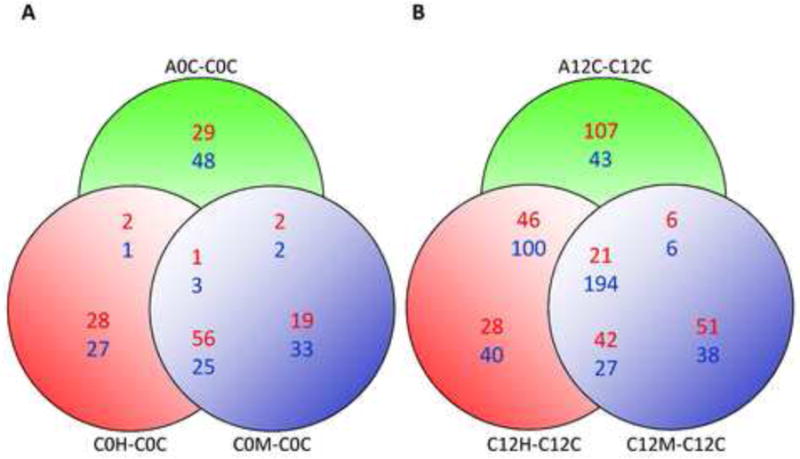
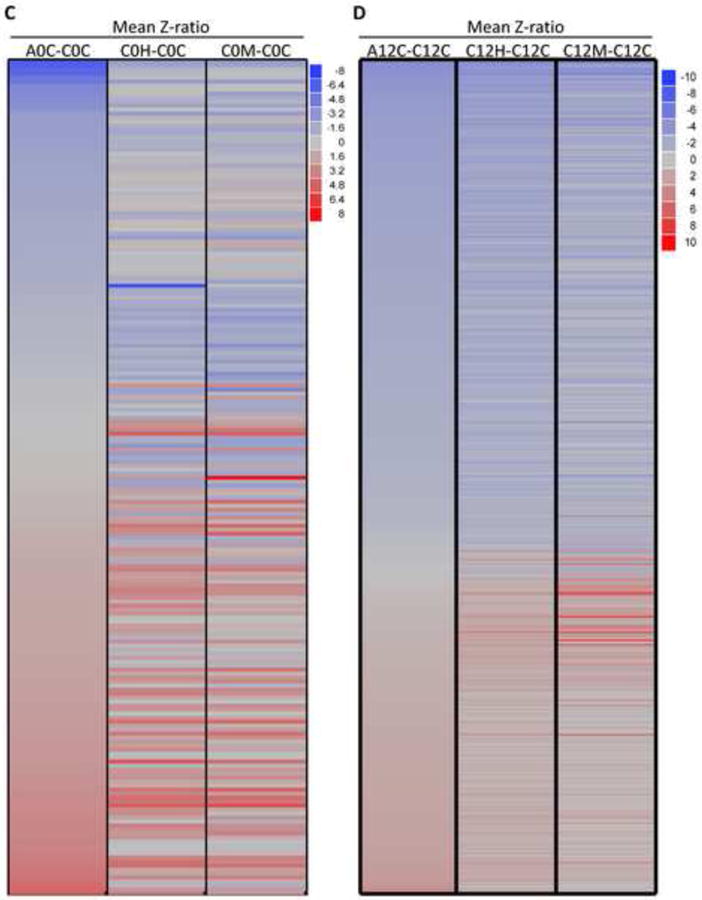
(A) Venn diagram showing the number of genes co-regulated in unstressed AD cells (A0C-C0C); H2O2 treated normal fibroblasts (C0H-C0C) or menadione-treated normal fibroblasts (C0M-COC) using cells harvested immediately after the end of treatment or mock treatment. Up-regulated genes are shown in red and down-regulated genes are shown in blue. (B) Same as (A), except using cells harvested 12 h after the end of treatment or mock treatment. (C) Heat map/Cell plot summarizing gene expression ratios immediately after the end of treatment or mock treatment. The color key on the right maps color intensity to the range of positive and negative expression ratios. (D) Same as (C), except using cells harvested 12 h after the end of treatment or mock treatment.
In contrast, at the 12 h time point, 752 genes met the criteria for differential expression (i.e., Z-score > 1.5). Of these, 523 genes were differentially-expressed in AD cells, 498 were differentially-expressed in hydrogen peroxide-treated normal cells, and 385 were differentially-expressed in menadione-treated normal cells. These data, and the degree of overlap between the three gene sets, are presented in the Venn diagrams in Figures 2A and 2B. The Venn diagrams illustrate the finding that 215 genes (41%) appear to be similarly co-regulated in H2O2 –treated normal cells, menadione -treated normal cells and untreated AD cells (Figure 2B). Cell plots showing gene expression ratios vs gene identity were also used to summarize and analyse these data into gene clusters (Figures 2C, 2D, S2C and S2D), revealing an overall trend similar to the trends described above. The results of this analysis support the idea that AD cells experience chronic oxidative stress, and that this state could influence patterns of gene expression associated with AD.
Gene ontology analysis reveals co-regulation of similar pathways in oxidatively stress normal cells and AD cells
To better understand the global impact of oxidative stress and AD on cellular gene expression networks and cell functions, Gene Set Enrichment Analysis was performed to map the experimentally-determined gene expression profile associated with AD onto specific metabolic pathways and cell functions [37, 38]. In this analysis, the 215 co-differentially-expressed genes (Figure 2B) were clustered according to gene ontology (GO) terms [34, 35], and pathways in which three or more genes were up- or down-regulated were identified. Initial analysis of GO terms corresponding to the 12 h data set is shown in Figures 3A and 3B, with more detailed listing of selected affected pathways shown in Figure 3C. A total of 571 pathways were predicted to be differentially-impacted under all three experimental conditions (Figures 3A and 3B), 337 pathways were predicted to be differentially-impacted in AD cells, 356 in hydrogen peroxide-treated cells and 327 in menadione-treated cells.
Figure 3. Oxidative stress triggers overlap of gene ontology changes that resemble Alzheimer's disease expression profile.
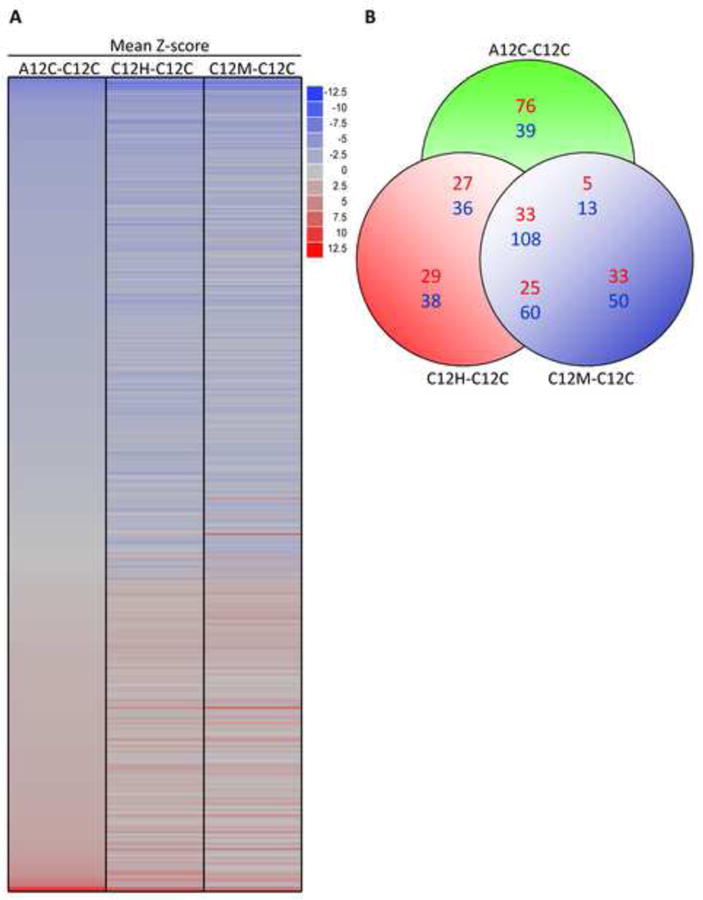
(A) Heat map summarizing GO terms linked to significant gene expression ratios shown in Figure 2A (see text for details). (B) Venn diagram number of GO-terms/pathways co-regulated in unstressed AD cells (A12C-C12C); H2O2 treated normal fibroblasts (C12H-C2C) or menadione-treated normal fibroblasts (C12M-C2C) using cells harvested 12 h after the end of treatment or mock treatment. The color key on the right maps color intensity to the range of positive and negative expression ratios. (C) Selected prominent GO terms/pathways from (A), with corresponding Z-scores, thought to have potential relevance to AD.
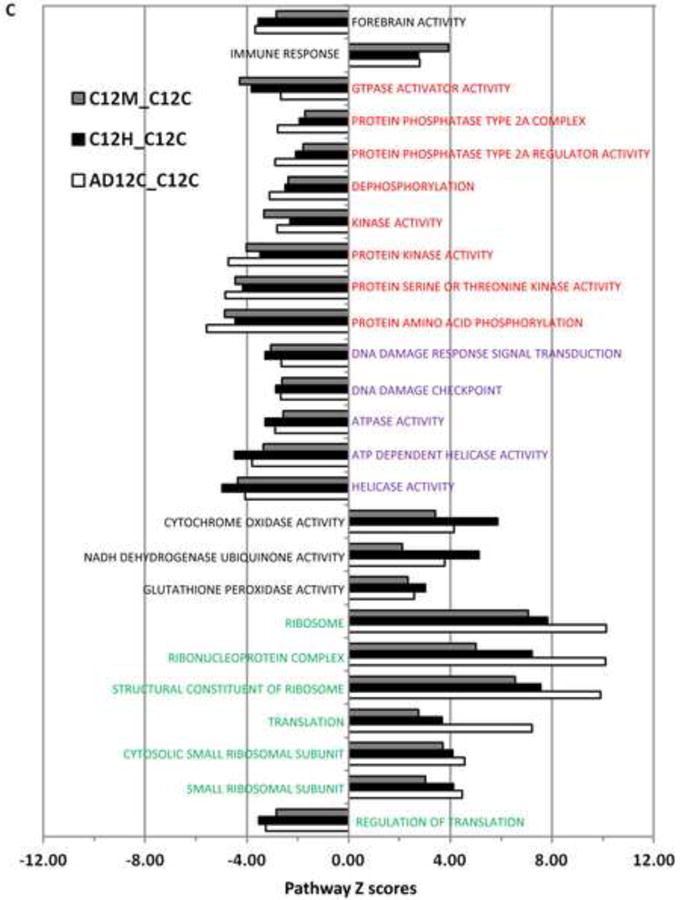
The overlap between the three sets of pathways shown in Figure 3A included 141 GO terms/pathways (Figures 3B and S4), many of which have been implicated previously as critical to AD pathology (Figure S4). A subset of these pathways and their Z-scores are shown in Figure 3C. We identified four groups of GO terms/cellular functions that were consistently dysregulated across all three datasets (Figure 3C and S4). The first group of GO terms included up-regulation of translation, ribosomal genes and down-regulation of translation regulation. The second group included metabolism and detoxification of reactive oxygen species (ROS) and up-regulation of cytochrome oxidase, NADH dehydrogenase and glutathione peroxidase. The third group included helicases, ATPase activity and pathways that response to DNA damage. The fourth group of down-regulated GO terms included kinase activity, protein phosphorylation and protein dephosphorylation. Other GO terms associated with altered pathways patterns included forebrain activity (down-regulated) and immune response (up-regulated).
Estimating oxidative DNA damage in AD fibroblasts
To test whether AD cells experience chronic oxidative stress, 8-OdG was quantified in fixed cultured AD and control cells using a commercially available monoclonal antibody against 8-OdG, a fluorescein isothiocyanate (FITC)-labeled secondary antibody and flow cytometry. As control experiments, AD cells were pre-treated with RNase and/or DNase and normal cells were treated with H2O2 or menadione (Figures 4A, 4B, S5 and data not shown). The results showed that endogenous 8-OdG was higher in AD cells than in untreated normal fibroblasts (Figures 4C and 4D). Consistent with this, an elevated level of 8-OdG was previously reported in brain and peripheral cells from AD patients [13, 18-20].
Figure 4. Flow cytometric quantitative analysis of 8-OdG in AD fibroblasts grown in culture.
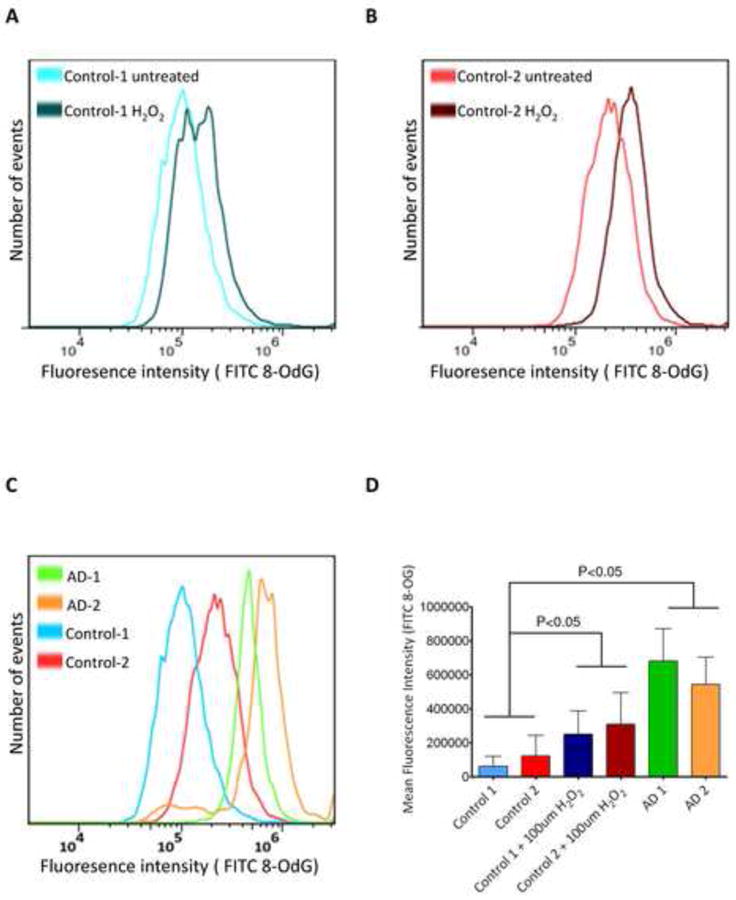
(A) Quantified FITC intensity as indicator of 8-OdG in fixed normal fibroblasts (control 1) after exposure to 100 μM H2O2 for 15 min. (B) As in (A), except using untreated normal fibroblasts (control 1). (C) As in (A), except using untreated normal fibroblasts (control 1), untreated normal fibroblasts (control 2), untreated AD fibroblasts (AG07374) and untreated AD fibroblasts (AG05770). (D) Quantification of the data shown in (C) using the mean value across different cell lines. Error bars show ±SEM, n=2 per cell line.
Validation of microarray
qPCR experiments were performed to validate the gene expression ratios measured by microarray in this study. For this purpose, the following genes were selected, based on their relevance to AD onset or pathology, their expression level or a literature search: IL-6, CDK6, PTPN12 (Protein phosphatase type 2A complex), TROVE 2 and EIF4G3 (translation factors), DDEF2/ASAP2 (a GTPASE activator), DLC1 (involved in forebrain development) and ATM. In all cases, the PCR data confirmed the results of the microarray study (Figure 5).
Figure 5. Validation of microarray data.
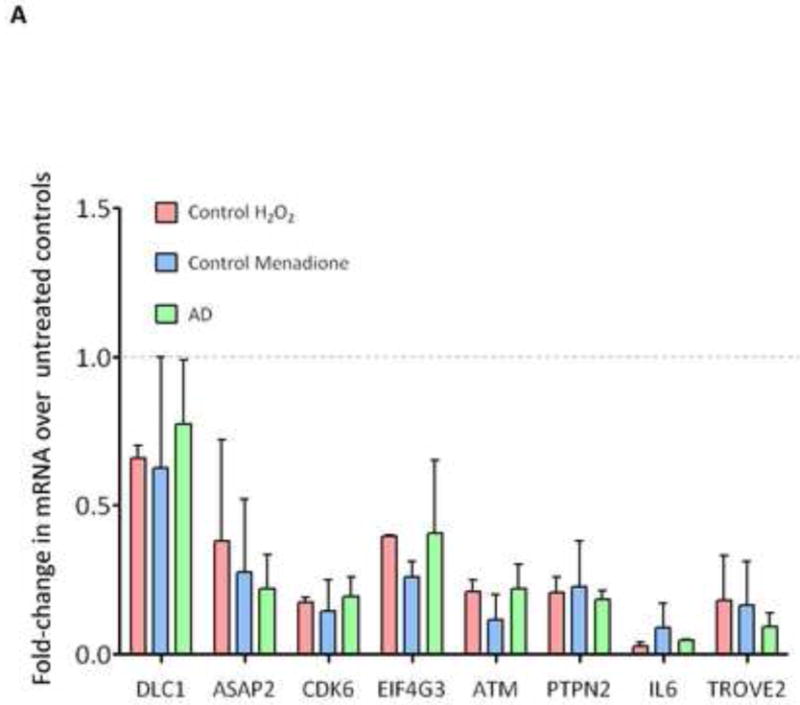
Fold-change in expression of the indicated genes was determined using quantitative PCR. Error bars show ±SD, n=3
Discussion
The goal of this study was to investigate the possible role of oxidative stress in the pathology of AD. We report that a similar group of genes and pathways are differentially expressed in AD patient-derived fibroblasts and in oxidatively-stressed fibroblasts from age- and sex- matched normal donors relative to AD patient donors. We also provide direct evidence that AD cells experience chronic oxidative stress, as measured by the elevated number of unrepaired 8-OdG lesions in AD fibroblasts grown in culture. The findings directly link oxidative stress to molecular characteristics of AD cells, and therefore generate a putative link to AD etiology and/or pathology.
Several other research groups have used microarray technology to study gene expression in AD cells [2, 39-43]. The present study differs from most previous studies, in that gene expression was analyzed in AD patient-derived fibroblasts. It was previously reported that the gene expression profile of tangle-bearing neurons in AD brains have approximately three-times more down-regulated genes (18%) than up-regulated genes (6%) [40], a result confirmed here when gene expression ratios in AD fibroblasts or oxidatively-stressed normal fibroblasts were compared to gene expression ratios in untreated normal fibroblasts (Figure 1C). Furthermore, the present study confirms earlier studies that suggested possible involvement of specific genes and pathways in AD etiology and/or pathology; thus, the credibility of our study and of previous studies is strengthened. Together, the findings support the idea that oxidative stress influences patterns of gene expression in AD cells, and may play a significant role in AD etiology and/or pathology. Nunomura and co-workers demonstrated that RNA oxidation reflected a snapshot of the oxidative state of the cell since RNA is rapidly turned over [44]. The expression pattern we report in AD cells could be a result of oxidative damage to RNA resulting in the corruption of gene expression. It is interesting that despite extensive literature that exists to support the role of mitochondrial dysfunction in the AD pathogenesis [45-47], we did not observe a change in the expression of genes that regulate mitochondrial function. It is important to note that neither of the two treatments gave a complete overlap in the expression pattern with the AD cells. We speculate that the treatment length, concentration and other factors could have contributed to this and thus to the lack of mitochondrial gene expression changes in our study.
It is also interesting to note the lack of evidence that BER is up-regulated in response to oxidative stress in the context of AD. This calls into question whether expression of BER genes is a valid surrogate marker for oxidative stress [48] or DNA damage, an idea that has been questioned previously, because it is generally recognized that BER genes are constitutively-expressed, and are not induced by DNA damage [49]. However, the basal level at which BER genes are expressed varies with tissue type and with age; so, inadequate BER capacity might still play a role in AD or other aging-related diseases. We believe this question warrants further study.
Finally, we would like to note that in our measurement of 8-OdG lesions in the cultured fibroblasts, we failed to increase this level in the control cells to the endogenous 8-OdG levels found in the AD cells even after treatment with oxidative stress (Figure 4D). This could be one reason for why we do not observe a complete overlap in the expression patterns between control cells treated with oxidative stress and the AD cells The increased endogenous levels of oxidative DNA damage in the AD cells likely is due to the chronic stress exposure that they are under.
Below is a brief discussion of some of the more interesting gene expression findings from our analysis. For some of the genes, there is prior literature on them with respect to AD, whereas others are novel genes that we propose may have a role in the response to oxidative stress in AD cells. In Figure 6, we summarize some of the more interesting genes that we find commonly altered after oxidative stress treatment to normal cells or in untreated AD cells relative to untreated control cells. We used Pubmatrix [50] to identify which of these genes had been previously reported in the literature with AD and those gene products are depicted in red.
Figure 6. Summary of Pubmatrix search.
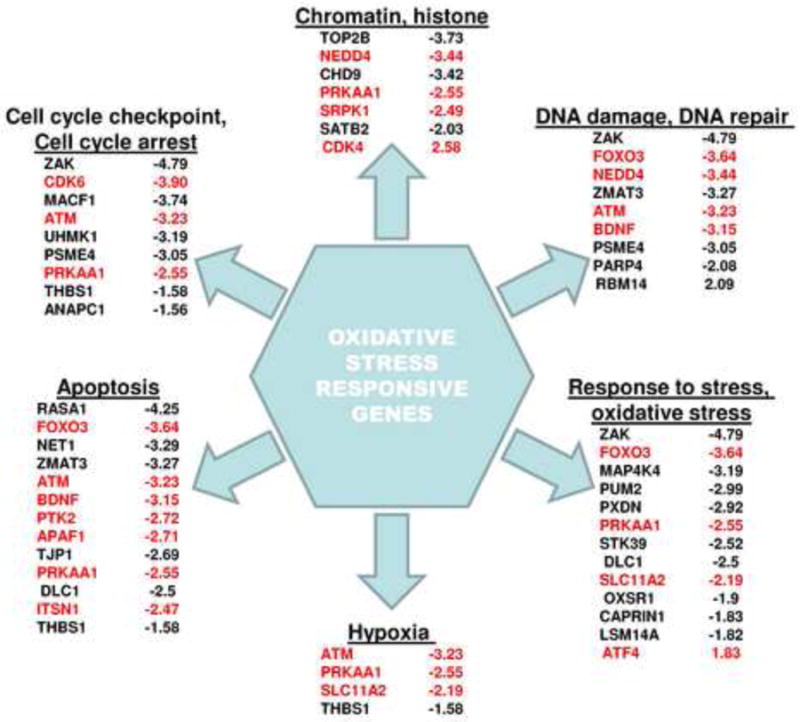
Summary of some of the more interesting genes that we find commonly altered after oxidative stress treatment to normal cells or in untreated AD cells relative to untreated control cells. Pubmatrix [50] was used to identify which of these genes had been previously reported in the literature with AD and those gene products are depicted in red.
IL-6
IL-6 is an inflammatory cytokine secreted by lymphocyte cells during acute infection [51]. Previous reports indicate that IL-6 is involved in the pathology of AD [52-54]. In particular, a polymorphism in the 3′ flanking region of the IL-6 gene is specifically associated with late onset AD and with low risk of AD, results that have been interpreted as evidence that IL-6 expression correlates negatively with AD risk [55], at least in some populations. Interestingly, the same polymorphism has been linked to increased risk of sporadic AD in a different subpopulation of white Americans [56]. In the present study, expression of IL-6 was lower in AD cells and oxidatively-stressed normal fibroblasts (Figure 3B, S3, and Figure 5). Another recent study proposed that IL-6 promotes removal of amyloid-β deposits [57]. Thus, it is possible that IL-6 stimulates clearing of deleterious AD-associated plaques by microglia, explaining the antagonism between IL-6 and AD risk. Future studies of the effect of oxidative stress on expression of IL-6 might be interesting, and it may be premature to rule out the possibility that IL-6 could be a useful therapeutic target for AD.
ATM
Ataxia-Telangiectasia (A-T) is a progressive cerebellar ataxia characterized by neurological manifestations, conjunctival and cutaneous telangiectasia and recurrent sino-pulmonary infections [58]. The gene mutated in A-T encodes a kinase, ATM, which plays an important role in the DNA damage signaling cascade (DDR) [59]. Furthermore, lack of ATM is associated with high levels of ROS, which in turn activates the ATM-dependent DDR in oxidatively-stressed ATM-proficient cells [60, 61]. Interestingly, ATM-/- mice have an abnormally high number of lysosomes, fueling speculation that A-T and AD are in some way related or similar [62]. The present study shows that ATM mRNA is less abundant in AD fibroblasts and oxidatively-stressed fibroblasts than in untreated normal fibroblasts. However, additional studies are needed to confirm these results and further to determine the significance of this observation.
PRKAA1
Mammalian AMP-activated protein kinase, AMPK, is a heterotrimeric complex containing a catalytic subunit, αl or α2, and two regulatory subunits, β and γ. PRKAA1 and PRKAA1 code for the αl and α2 subunits of AMPK. AMPKα1 is expressed in multiple tissues like brain, heart, liver, kidney etc, whereas AMPKα2 is more abundant in skeletal muscle and heart and less so in liver and kidney [63]. AMPK plays a major role in cellular energy homeostasis by regulating lipid and glucose metabolism in response to AMP levels and other stressors [64]. AMPK is activated after DNA damage by ionizing radiation [65], regulates ROS, autophagy, cell proliferation, and mitochondrial functions [64]. In AD, AMPK has been reported to be over activated and it directly regulates tau phosphorylation [66]. Here we report loss of PRKAA1 mRNA after oxidative stress and in AD fibroblasts. As a result, AD cells may not have a normal DNA damage response and additionally would need to preferentially rely upon PRKAA2, which may have altered tissue expression patterns as well as other catalytic differences.
BDNF
Brain-derived neurotrophic factor, BDNF, is a widely expressed brain growth factor that plays important roles in neuronal viability, synaptic plasticity, neurogenesis and aging [67, 68]. Mixed findings have been reported for the association of AD and BDNF levels. Some reports state that BDNF levels are elevated in AD [69, 70] whereas others have reported lower BDNF levels in AD patients [69-73]. Here, we report that BDNF mRNA is down-regulated after exogenous oxidative stress. Therefore, our results suggest that BDNF expression goes down after oxidative stress and support the hypothesis that loss of BDNF, in response to oxidative stress, may make neurons more sensitive to oxidant-induced cell death.
NEDD4
NEDD4 (neural-precursor cell-expressed developmentally down-regulated gene 4) is a HECT(homology to E6-AP carboxy terminus) E3 ubiquitin ligase, which shows high expression in the embryonic mouse CNS [74]. E3 ligases are involved in the ubiquitination of several neuronal proteins, which controls their endocytosis and endocytic sorting [75-79]. It is thought that the endocytic-lysosomal pathway plays an important role in neurodegeneration and protein aggregation is a hallmark of AD [80]. After DNA damage that blocks transcription, NEDD4 is known to bind to and ubiquitinate RNA polymerase II, which is necessary for DNA repair [81]. In our study, we find a dramatic down-regulation of the NEDD4 gene transcription in the AD samples and in the control cells when treated with oxidative stress. This in turn could lead lysosomal dysfunction, leading to protein aggregation in AD, and could also lead to aberrant DNA repair of transcription blocking lesions.
ZAK
The zipper sterile-a-motif kinase (ZAK) is a serine/threonine kinase that activates the c-Jun amino-terminal kinase (JNK) and the mitogen-activated-protein kinase (MAPK p38) pathways [82]. These kinases respond to a variety of stressors and some cellular endpoints include apoptosis, cell cycle, and metabolism. It has been reported that transfection of a kinase-dead ZAK leads to disruption of actin stress fibers [83]. Another function of ZAK is to arrest cell proliferation by causing a G2 cell cycle arrest [83]. Specifically, it has been shown that ZAK participates in the G2 arrest of cells after DNA damage by gamma radiation [84]. In our study, we observed significant down-regulation of ZAK in AD and normal human fibroblasts after oxidative stress. There is no literature on ZAK and AD but loss of ZAK may compromise the ability of AD neurons to induce an appropriate cell cycle checkpoint after damage.
ZMAT3
ZMAT3 or Wig1 was discovered as a p53 transcriptional target [85, 86]. Later studies demonstrated the ability of ZMAT3 to bind to double stranded RNA, besides DNA, and is thought to play a role similar to that of miRNA [87-89]. Interestingly, its expression was found to be highest in the brain [89]. Furthermore, ZMAT3's expression was induced in the nervous system by a number of stress agents, including ischemia, treatment with methamphetamine, onset of disease in a model of ALS, and by L-DOPA in a Parkinson model [90-94]. These data indicate a role for ZMAT3 in cellular stress responses and pathological conditions in the nervous system. ZMAT expression was down-regulated in our study, suggesting a role for this protein in the oxidative stress response in both normal and AD cells.
Top2B
Topoisomerase IIβ is a type II topoisomerase, an enzyme involved in cutting and resealing double stranded DNA, and important for DNA replication and transcription [95]. Top2B is expressed in post-mitotic cells, implying its importance in the maintenance of tissue homeostasis [96-98]. A previous report suggested a role for this enzyme in neural cell differentiation and another reported aberrant lamination in the cerebral cortex of mouse embryos lacking DNA Top2B [99, 100]. Our microarray analysis revealed a down-regulation of Top2B transcripts; whether it plays a role in the response to oxidative stress in AD cells will need additional research.
Lastly, AD is a complex disease with many cellular pathways and proteins deregulated. How all these events contribute to the pathology observed in AD warrants intensive research efforts. AD is the scourge of our generation and its toll on society will only increase with time. Here we have identified oxidative-stress responsive mRNAs of previously accepted AD-associated genes and novel genes that may play a role in the complex pathology of AD. In the future, we hope to further characterize if some of the altered genes identified here show alterations in primary AD tissues.
Supplementary Material
Figure S1 Overview of experimental design.
Figure S2: Oxidative stress triggers gene expression profile changes that resemble Alzheimer's disease expression profile
(A) Venn diagram demonstrating the overlapping gene expressions in the various sets: A1C-C1C- gene expression changes in AD cells compared to control cells without treatment at 1 h; C1H-C1C - gene expression changes in control cells treated with H2O2 compared to control cells without treatment at 1 h; C1M-C1C -gene expression changes in control cells treated with menadione as compared to control cells without treatment at 1 h. Numbers in red represent number of genes up-regulated and in blue represent the number of genes down-regulated (B) Venn diagram demonstrating the overlapping gene expressions in the various sets. A6C-C6C - gene expression changes in AD cells compared to control cells without treatment at 6 h, C6H-C6C - gene expression changes in control cells treated with H2O2 compared to control cells without treatment at 6 h; C6M-C6C -gene expression changes in control cells treated with menadione as compared to control cells without treatment at 6 h. (C) Cell plot of the gene expression profiles at 1 h. The color key on the right indicates the extent of change in the gene expression. Blue bars represents down-regulation of gene expression whereas red bars represents up-regulation of gene expression (D) Cell plot of the gene expression profiles at 6 h. The color key on the right indicates the extent of change in the gene expression. Blue bars represent down-regulation of gene expression whereas red bars represent up-regulation of gene expression.
Figure S3: Gene expression profile of overlapping genes in the 12 h arrays
Cell plot of the 215 significant genes that changed expression in the three sets; 1 - gene expression changes in AD cells compared to control cells without treatment at 12 h; 2 - gene expression changes in control cells treated with H2O2 compared to control cells without treatment at 12 h; 3 -gene expression changes in control cells treated with menadione as compared to control cells without treatment at 12 h. The color key on the right indicates the extent of change in the gene expression. Blue bars represent down- regulation of gene expression whereas red bars represents up-regulation of gene expression.
Figure S4: Gene Ontology profile of overlapping pathways in the 12 h arrays
Cell plot of the 141 pathways that changed expression in the three sets; 1 - GO changes in AD cells compared to control cells without treatment at 12 h 2 - GO changes in control cells treated with H2O2 compared to control cells without treatment at 12 h; 3 -GO changes in control cells treated with menadione as compared to control cells without treatment at 12 h. The color key on the right indicates the extent of change in the GO. Blue bars represent down-regulation of a pathway as measured by GO and red bars represent up-regulation of a pathway as measured by GO
Figure S5: Sporadic AD cultured fibroblasts display increased oxidative damage.
Flow cytometric analysis measuring the FITC intensity (8-OdG) of U2OS cell line with the indicated treatments.
Table S1 List of probe sets used for qPCR validation.
Highlights.
Alzheimers fibroblasts (AD) have an oxidative stress phenotype
AD fibroblasts have increased 8-oxoG in their DNA
Many genes and pathways common to AD fibroblasts and stressed normal fibroblasts
Acknowledgments
We would like to thank Dr. Magda Masiak and Martin Borch Jensen for critically reading this manuscript. This work was supported by the National Institutes of Health Intramural Program of the National Institute on Aging (Z01-AG000733-14).
Footnotes
Author Contributions: MR and VB conceived and designed the study. MR, PS, MSK, CD and CK performed the experiments. MR, YZ, KB and DLC analysed the data. MR, DLC and VB interpreted the data. MR and DC wrote the manuscript with comments from VB.
Conflict of Interest: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alzheimer's Disease Facts and Figures 2011 [Google Scholar]
- 2.Colangelo V, Schurr J, Ball MJ, Pelaez RP, Bazan NG, Lukiw WJ. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J Neurosci Res. 2002;70:462–73. doi: 10.1002/jnr.10351. [DOI] [PubMed] [Google Scholar]
- 3.Czech C, Tremp G, Pradier L. Presenilins and Alzheimer's disease: biological functions and pathogenic mechanisms. Prog Neurobiol. 2000;60:363–84. doi: 10.1016/s0301-0082(99)00033-7. [DOI] [PubMed] [Google Scholar]
- 4.Price DL, Tanzi RE, Borchelt DR, Sisodia SS. Alzheimer's disease: genetic studies and transgenic models. Annu Rev Genet. 1998;32:461–93. doi: 10.1146/annurev.genet.32.1.461. [DOI] [PubMed] [Google Scholar]
- 5.Shastry BS. Molecular and cell biological aspects of Alzheimer disease. J Hum Genet. 2001;46:609–18. doi: 10.1007/s100380170011. [DOI] [PubMed] [Google Scholar]
- 6.St George-Hyslop PH. Molecular genetics of Alzheimer's disease. Biol Psychiatry. 2000;47:183–99. doi: 10.1016/s0006-3223(99)00301-7. [DOI] [PubMed] [Google Scholar]
- 7.Mullaart E, Boerrigter ME, Ravid R, Swaab DF, Vijg J. Increased levels of DNA breaks in cerebral cortex of Alzheimer's disease patients. Neurobiol Aging. 1990;11:169–73. doi: 10.1016/0197-4580(90)90542-8. [DOI] [PubMed] [Google Scholar]
- 8.Gabbita SP, Lovell MA, Markesbery WR. Increased nuclear DNA oxidation in the brain in Alzheimer's disease. J Neurochem. 1998;71:2034–40. doi: 10.1046/j.1471-4159.1998.71052034.x. [DOI] [PubMed] [Google Scholar]
- 9.Lyras L, Cairns NJ, Jenner A, Jenner P, Halliwell B. An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer's disease. J Neurochem. 1997;68:2061–9. doi: 10.1046/j.1471-4159.1997.68052061.x. [DOI] [PubMed] [Google Scholar]
- 10.Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer's disease. Ann Neurol. 1994;36:747–51. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer's disease. J Neurochem. 2005;93:953–62. doi: 10.1111/j.1471-4159.2005.03053.x. [DOI] [PubMed] [Google Scholar]
- 12.Cohen G. Oxidative stress, mitochondrial respiration, and Parkinson's disease. Ann N Y Acad Sci. 2000;899:112–20. doi: 10.1111/j.1749-6632.2000.tb06180.x. [DOI] [PubMed] [Google Scholar]
- 13.Naderi J, Lopez C, Pandey S. Chronically increased oxidative stress in fibroblasts from Alzheimer's disease patients causes early senescence and renders resistance to apoptosis by oxidative stress. Mech Ageing Dev. 2006;127:25–35. doi: 10.1016/j.mad.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 14.Su B, Wang X, Nunomura A, Moreira PI, Lee HG, Perry G, et al. Oxidative stress signaling in Alzheimer's disease. Curr Alzheimer Res. 2008;5:525–32. doi: 10.2174/156720508786898451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weissman L, de Souza-Pinto NC, Stevnsner T, Bohr VA. DNA repair, mitochondria, and neurodegeneration. Neuroscience. 2007;145:1318–29. doi: 10.1016/j.neuroscience.2006.08.061. [DOI] [PubMed] [Google Scholar]
- 16.Ozkul A, Akyol A, Yenisey C, Arpaci E, Kiylioglu N, Tataroglu C. Oxidative stress in acute ischemic stroke. J Clin Neurosci. 2007;14:1062–6. doi: 10.1016/j.jocn.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 17.Ansari MA, Scheff SW. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol. 2010;69:155–67. doi: 10.1097/NEN.0b013e3181cb5af4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coppede F, Migliore L. DNA damage and repair in Alzheimer's disease. Curr Alzheimer Res. 2009;6:36–47. doi: 10.2174/156720509787313970. [DOI] [PubMed] [Google Scholar]
- 19.Gibson G, Martins R, Blass J, Gandy S. Altered oxidation and signal transduction systems in fibroblasts from Alzheimer patients. Life Sci. 1996;59:477–89. doi: 10.1016/0024-3205(96)00327-x. [DOI] [PubMed] [Google Scholar]
- 20.Curti D, Rognoni F, Gasparini L, Cattaneo A, Paolillo M, Racchi M, et al. Oxidative metabolism in cultured fibroblasts derived from sporadic Alzheimer's disease (AD) patients. Neurosci Lett. 1997;236:13–6. doi: 10.1016/s0304-3940(97)00741-6. [DOI] [PubMed] [Google Scholar]
- 21.Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002;32:1050–60. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- 22.Fishel ML, Vasko MR, Kelley MR. DNA repair in neurons: so if they don't divide what's to repair? Mutat Res. 2007;614:24–36. doi: 10.1016/j.mrfmmm.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 23.Weissman L, Jo DG, Sorensen MM, de Souza-Pinto NC, Markesbery WR, Mattson MP, et al. Defective DNA base excision repair in brain from individuals with Alzheimer's disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007;35:5545–55. doi: 10.1093/nar/gkm605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lovell MA, Gabbita SP, Markesbery WR. Increased DNA oxidation and decreased levels of repair products in Alzheimer's disease ventricular CSF. J Neurochem. 1999;72:771–6. doi: 10.1046/j.1471-4159.1999.0720771.x. [DOI] [PubMed] [Google Scholar]
- 25.Lovell MA, Xie C, Markesbery WR. Decreased base excision repair and increased helicase activity in Alzheimer's disease brain. Brain Res. 2000;855:116–23. doi: 10.1016/s0006-8993(99)02335-5. [DOI] [PubMed] [Google Scholar]
- 26.Shao C, Xiong S, Li GM, Gu L, Mao G, Markesbery WR, et al. Altered 8-oxoguanine glycosylase in mild cognitive impairment and late-stage Alzheimer's disease brain. Free Radic Biol Med. 2008;45:813–9. doi: 10.1016/j.freeradbiomed.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rogaev EI, Sherrington R, Wu C, Levesque G, Liang Y, Rogaeva EA, et al. Analysis of the 5′ sequence, genomic structure, and alternative splicing of the presenilin-1 gene (PSEN1) associated with early onset Alzheimer disease. Genomics. 1997;40:415–24. doi: 10.1006/geno.1996.4523. [DOI] [PubMed] [Google Scholar]
- 28.Lukiw WJ, Bazan NG. Cyclooxygenase 2 RNA message abundance, stability, and hypervariability in sporadic Alzheimer neocortex. J Neurosci Res. 1997;50:937–45. doi: 10.1002/(SICI)1097-4547(19971215)50:6<937::AID-JNR4>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 29.Chow N, Cox C, Callahan LM, Weimer JM, Guo L, Coleman PD. Expression profiles of multiple genes in single neurons of Alzheimer's disease. Proc Natl Acad Sci U S A. 1998;95:9620–5. doi: 10.1073/pnas.95.16.9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Leeuwen FW, de Kleijn DP, van den Hurk HH, Neubauer A, Sonnemans MA, Sluijs JA, et al. Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer's and Down patients. Science. 1998;279:242–7. doi: 10.1126/science.279.5348.242. [DOI] [PubMed] [Google Scholar]
- 31.Yao PJ, Morsch R, Callahan LM, Coleman PD. Changes in synaptic expression of clathrin assembly protein AP180 in Alzheimer's disease analysed by immunohistochemistry. Neuroscience. 1999;94:389–94. doi: 10.1016/s0306-4522(99)00360-7. [DOI] [PubMed] [Google Scholar]
- 32.Martin LJ. Neuronal cell death in nervous system development, disease, and injury (Review) Int J Mol Med. 2001;7:455–78. [PubMed] [Google Scholar]
- 33.Kyng KJ, May A, Kolvraa S, Bohr VA. Gene expression profiling in Werner syndrome closely resembles that of normal aging. Proc Natl Acad Sci U S A. 2003;100:12259–64. doi: 10.1073/pnas.2130723100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 35.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang HM, Martins R, Gandy S, Etcheberrigaray R, Ito E, Alkon DL, et al. Use of cultured fibroblasts in elucidating the pathophysiology and diagnosis of Alzheimer's disease. Ann N Y Acad Sci. 1994;747:225–44. doi: 10.1111/j.1749-6632.1994.tb44412.x. [DOI] [PubMed] [Google Scholar]
- 37.Kim SY, Volsky DJ. PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics. 2005;6:144. doi: 10.1186/1471-2105-6-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schurman SH, Hedayati M, Wang Z, Singh DK, Speina E, Zhang Y, et al. Direct and indirect roles of RECQL4 in modulating base excision repair capacity. Hum Mol Genet. 2009;18:3470–83. doi: 10.1093/hmg/ddp291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer's disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci U S A. 2004;101:2173–8. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ginsberg SD, Hemby SE, Lee VM, Eberwine JH, Trojanowski JQ. Expression profile of transcripts in Alzheimer's disease tangle-bearing CA1 neurons. Ann Neurol. 2000;48:77–87. [PubMed] [Google Scholar]
- 41.Ricciarelli R, d'Abramo C, Massone S, Marinari U, Pronzato M, Tabaton M. Microarray analysis in Alzheimer's disease and normal aging. IUBMB Life. 2004;56:349–54. doi: 10.1080/15216540412331286002. [DOI] [PubMed] [Google Scholar]
- 42.Pasinetti GM. Use of cDNA microarray in the search for molecular markers involved in the onset of Alzheimer's disease dementia. J Neurosci Res. 2001;65:471–6. doi: 10.1002/jnr.1176. [DOI] [PubMed] [Google Scholar]
- 43.Yao PJ, Zhu M, Pyun EI, Brooks AI, Therianos S, Meyers VE, et al. Defects in expression of genes related to synaptic vesicle trafficking in frontal cortex of Alzheimer's disease. Neurobiol Dis. 2003;12:97–109. doi: 10.1016/s0969-9961(02)00009-8. [DOI] [PubMed] [Google Scholar]
- 44.Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S, et al. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer's disease. J Neurosci. 1999;19:1959–64. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moreira PI, Cardoso SM, Santos MS, Oliveira CR. The key role of mitochondria in Alzheimer's disease. J Alzheimers Dis. 2006;9:101–10. doi: 10.3233/jad-2006-9202. [DOI] [PubMed] [Google Scholar]
- 46.Moreira PI, Carvalho C, Zhu X, Smith MA, Perry G. Mitochondrial dysfunction is a trigger of Alzheimer's disease pathophysiology. Biochim Biophys Acta. 2010;1802:2–10. doi: 10.1016/j.bbadis.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 47.Coskun P, Wyrembak J, Schriner SE, Chen HW, Marciniack C, Laferla F, et al. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim Biophys Acta. 2012;1820:553–64. doi: 10.1016/j.bbagen.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rusyn I, Asakura S, Pachkowski B, Bradford BU, Denissenko MF, Peters JM, et al. Expression of base excision DNA repair genes is a sensitive biomarker for in vivo detection of chemical-induced chronic oxidative stress: identification of the molecular source of radicals responsible for DNA damage by peroxisome proliferators. Cancer Res. 2004;64:1050–7. doi: 10.1158/0008-5472.can-03-3027. [DOI] [PubMed] [Google Scholar]
- 49.Inoue M, Shen GP, Chaudhry MA, Galick H, Blaisdell JO, Wallace SS. Expression of the oxidative base excision repair enzymes is not induced in TK6 human lymphoblastoid cells after low doses of ionizing radiation. Radiat Res. 2004;161:409–17. doi: 10.1667/3163. [DOI] [PubMed] [Google Scholar]
- 50.Becker KG, Hosack DA, Dennis G, Jr, Lempicki RA, Bright TJ, Cheadle C, et al. PubMatrix: a tool for multiplex literature mining. BMC Bioinformatics. 2003;4:61. doi: 10.1186/1471-2105-4-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gadient RA, Otten UH. Interleukin-6 (IL-6)--a molecule with both beneficial and destructive potentials. Prog Neurobiol. 1997;52:379–90. doi: 10.1016/s0301-0082(97)00021-x. [DOI] [PubMed] [Google Scholar]
- 52.Bauer J, Strauss S, Schreiter-Gasser U, Ganter U, Schlegel P, Witt I, et al. Interleukin-6 and alpha-2-macroglobulin indicate an acute-phase state in Alzheimer's disease cortices. FEBS Lett. 1991;285:111–4. doi: 10.1016/0014-5793(91)80737-n. [DOI] [PubMed] [Google Scholar]
- 53.Bauer J, Ganter U, Strauss S, Stadtmuller G, Frommberger U, Bauer H, et al. The participation of interleukin-6 in the pathogenesis of Alzheimer's disease. Res Immunol. 1992;143:650–7. doi: 10.1016/0923-2494(92)80051-l. [DOI] [PubMed] [Google Scholar]
- 54.Ganter U, Strauss S, Jonas U, Weidemann A, Beyreuther K, Volk B, et al. Alpha 2-macroglobulin synthesis in interleukin-6-stimulated human neuronal (SH-SY5Y neuroblastoma) cells. Potential significance for the processing of Alzheimer beta-amyloid precursor protein. FEBS Lett. 1991;282:127–31. doi: 10.1016/0014-5793(91)80460-k. [DOI] [PubMed] [Google Scholar]
- 55.Papassotiropoulos A, Bagli M, Jessen F, Bayer TA, Maier W, Rao ML, et al. A genetic variation of the inflammatory cytokine interleukin-6 delays the initial onset and reduces the risk for sporadic Alzheimer's disease. Ann Neurol. 1999;45:666–8. doi: 10.1002/1531-8249(199905)45:5<666::aid-ana18>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 56.Bhojak TJ, DeKosky ST, Ganguli M, Kamboh MI. Genetic polymorphisms in the cathespin D and interleukin-6 genes and the risk of Alzheimer's disease. Neurosci Lett. 2000;288:21–4. doi: 10.1016/s0304-3940(00)01185-x. [DOI] [PubMed] [Google Scholar]
- 57.Chakrabarty P, Jansen-West K, Beccard A, Ceballos-Diaz C, Levites Y, Verbeeck C, et al. Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010;24:548–59. doi: 10.1096/fj.09-141754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McKinnon PJ. ATM and the molecular pathogenesis of ataxia telangiectasia. Annu Rev Pathol. 2012;7:303–21. doi: 10.1146/annurev-pathol-011811-132509. [DOI] [PubMed] [Google Scholar]
- 59.Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–53. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 60.Barzilai A, Rotman G, Shiloh Y. ATM deficiency and oxidative stress: a new dimension of defective response to DNA damage. DNA Repair (Amst) 2002;1:3–25. doi: 10.1016/s1568-7864(01)00007-6. [DOI] [PubMed] [Google Scholar]
- 61.Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010;330:517–21. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 62.Barlow C, Ribaut-Barassin C, Zwingman TA, Pope AJ, Brown KD, Owens JW, et al. ATM is a cytoplasmic protein in mouse brain required to prevent lysosomal accumulation. Proc Natl Acad Sci U S A. 2000;97:871–6. doi: 10.1073/pnas.97.2.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–85. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 64.Wang S, Song P, Zou MH. AMP-activated protein kinase, stress responses and cardiovascular diseases. Clin Sci (Lond) 2012;122:555–73. doi: 10.1042/CS20110625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sanli T, Rashid A, Liu C, Harding S, Bristow RG, Cutz JC, et al. Ionizing radiation activates AMP-activated kinase (AMPK): a target for radiosensitization of human cancer cells. Int J Radiat Oncol Biol Phys. 2010;78:221–9. doi: 10.1016/j.ijrobp.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 66.Vingtdeux V, Davies P, Dickson DW, Marambaud P. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer's disease and other tauopathies. Acta Neuropathol. 2011;121:337–49. doi: 10.1007/s00401-010-0759-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Diniz BS, Teixeira AL. Brain-derived neurotrophic factor and Alzheimer's disease: physiopathology and beyond. Neuromolecular Med. 2011;13:217–22. doi: 10.1007/s12017-011-8154-x. [DOI] [PubMed] [Google Scholar]
- 68.Hu Y, Russek SJ. BDNF and the diseased nervous system: a delicate balance between adaptive and pathological processes of gene regulation. J Neurochem. 2008;105:1–17. doi: 10.1111/j.1471-4159.2008.05237.x. [DOI] [PubMed] [Google Scholar]
- 69.Angelucci F, Spalletta G, di IF, Ciaramella A, Salani F, Colantoni L, et al. Alzheimer's disease (AD) and Mild Cognitive Impairment (MCI) patients are characterized by increased BDNF serum levels. Curr Alzheimer Res. 2010;7:15–20. doi: 10.2174/156720510790274473. [DOI] [PubMed] [Google Scholar]
- 70.Laske C, Stransky E, Leyhe T, Eschweiler GW, Wittorf A, Richartz E, et al. Stage-dependent BDNF serum concentrations in Alzheimer's disease. J Neural Transm. 2006;113:1217–24. doi: 10.1007/s00702-005-0397-y. [DOI] [PubMed] [Google Scholar]
- 71.Lee JG, Shin BS, You YS, Kim JE, Yoon SW, Jeon DW, et al. Decreased serum brain-derived neurotrophic factor levels in elderly korean with dementia. Psychiatry Investig. 2009;6:299–305. doi: 10.4306/pi.2009.6.4.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Laske C, Stransky E, Leyhe T, Eschweiler GW, Maetzler W, Wittorf A, et al. BDNF serum and CSF concentrations in Alzheimer's disease, normal pressure hydrocephalus and healthy controls. J Psychiatr Res. 2007;41:387–94. doi: 10.1016/j.jpsychires.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 73.Forlenza OV, Diniz BS, Gattaz WF. Diagnosis and biomarkers of predementia in Alzheimer's disease. BMC Med. 2010;8:89. doi: 10.1186/1741-7015-8-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kumar S, Harvey KF, Kinoshita M, Copeland NG, Noda M, Jenkins NA. cDNA cloning, expression analysis, and mapping of the mouse Nedd4 gene. Genomics. 1997;40:435–43. doi: 10.1006/geno.1996.4582. [DOI] [PubMed] [Google Scholar]
- 75.Fotia AB, Ekberg J, Adams DJ, Cook DI, Poronnik P, Kumar S. Regulation of neuronal voltage-gated sodium channels by the ubiquitin-protein ligases Nedd4 and Nedd4-2. J Biol Chem. 2004;279:28930–5. doi: 10.1074/jbc.M402820200. [DOI] [PubMed] [Google Scholar]
- 76.Jespersen T, Membrez M, Nicolas CS, Pitard B, Staub O, Olesen SP, et al. The KCNQ1 potassium channel is down-regulated by ubiquitylating enzymes of the Nedd4/Nedd4-like family. Cardiovasc Res. 2007;74:64–74. doi: 10.1016/j.cardiores.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 77.Kamynina E, Debonneville C, Bens M, Vandewalle A, Staub O. A novel mouse Nedd4 protein suppresses the activity of the epithelial Na+ channel. FASEB J. 2001;15:204–14. doi: 10.1096/fj.00-0191com. [DOI] [PubMed] [Google Scholar]
- 78.Sorkina T, Miranda M, Dionne KR, Hoover BR, Zahniser NR, Sorkin A. RNA interference screen reveals an essential role of Nedd4-2 in dopamine transporter ubiquitination and endocytosis. J Neurosci. 2006;26:8195–205. doi: 10.1523/JNEUROSCI.1301-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schwarz LA, Hall BJ, Patrick GN. Activity-dependent ubiquitination of GluA1 mediates a distinct AMPA receptor endocytosis and sorting pathway. J Neurosci. 2010;30:16718–29. doi: 10.1523/JNEUROSCI.3686-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Olzscha H, Schermann SM, Woerner AC, Pinkert S, Hecht MH, Tartaglia GG, et al. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell. 2011;144:67–78. doi: 10.1016/j.cell.2010.11.050. [DOI] [PubMed] [Google Scholar]
- 81.Anindya R, Aygun O, Svejstrup JQ. Damage-induced ubiquitylation of human RNA polymerase II by the ubiquitin ligase Nedd4, but not Cockayne syndrome proteins or BRCA1. Mol Cell. 2007;28:386–97. doi: 10.1016/j.molcel.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 82.Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol. 2002;3:663–72. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- 83.Yang JJ. Mixed lineage kinase ZAK utilizing MKK7 and not MKK4 to activate the c-Jun N-terminal kinase and playing a role in the cell arrest. Biochem Biophys Res Commun. 2002;297:105–10. doi: 10.1016/s0006-291x(02)02123-x. [DOI] [PubMed] [Google Scholar]
- 84.Tosti E, Waldbaum L, Warshaw G, Gross EA, Ruggieri R. The stress kinase MRK contributes to regulation of DNA damage checkpoints through a p38gamma-independent pathway. J Biol Chem. 2004;279:47652–60. doi: 10.1074/jbc.M409961200. [DOI] [PubMed] [Google Scholar]
- 85.Hellborg F, Qian W, Mendez-Vidal C, Asker C, Kost-Alimova M, Wilhelm M, et al. Human wig-1, a p53 target gene that encodes a growth inhibitory zinc finger protein. Oncogene. 2001;20:5466–74. doi: 10.1038/sj.onc.1204722. [DOI] [PubMed] [Google Scholar]
- 86.Varmeh-Ziaie S, Okan I, Wang Y, Magnusson KP, Warthoe P, Strauss M, et al. Wig-1, a new p53-induced gene encoding a zinc finger protein. Oncogene. 1997;15:2699–704. doi: 10.1038/sj.onc.1201454. [DOI] [PubMed] [Google Scholar]
- 87.Huang J, Wang HB, Yao L, Zheng XM, Radmark O. Dicer interacts with Wig-1 protein. Fen Zi Xi Bao Sheng Wu Xue Bao. 2008;41:376–82. [PubMed] [Google Scholar]
- 88.Vilborg A, Glahder JA, Wilhelm MT, Bersani C, Corcoran M, Mahmoudi S, et al. The p53 target Wig-1 regulates p53 mRNA stability through an AU-rich element. Proc Natl Acad Sci U S A. 2009;106:15756–61. doi: 10.1073/pnas.0900862106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vilborg A, Bersani C, Wilhelm MT, Wiman KG. The p53 target Wig-1: a regulator of mRNA stability and stem cell fate? Cell Death Differ. 2011;18:1434–40. doi: 10.1038/cdd.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Asanuma M, Miyazaki I, Higashi Y, Diaz-Corrales FJ, Shimizu M, Miyoshi K, et al. Suppression of p53-activated gene, PAG608, attenuates methamphetamine-induced neurotoxicity. Neurosci Lett. 2007;414:263–7. doi: 10.1016/j.neulet.2006.12.036. [DOI] [PubMed] [Google Scholar]
- 91.Gillardon F, Spranger M, Tiesler C, Hossmann KA. Expression of cell death-associated phospho-c-Jun and p53-activated gene 608 in hippocampal CA1 neurons following global ischemia. Brain Res Mol Brain Res. 1999;73:138–43. doi: 10.1016/s0169-328x(99)00251-x. [DOI] [PubMed] [Google Scholar]
- 92.Morimoto N, Nagai M, Miyazaki K, Ohta Y, Kurata T, Takehisa Y, et al. Induction of parkinsonism-related proteins in the spinal motor neurons of transgenic mouse carrying a mutant SOD1 gene. J Neurosci Res. 2010;88:1804–11. doi: 10.1002/jnr.22341. [DOI] [PubMed] [Google Scholar]
- 93.Shimizu M, Miyazaki I, Higashi Y, Eslava-Alva MJ, Diaz-Corrales FJ, Asanuma M, et al. Specific induction of PAG608 in cranial and spinal motor neurons of L-DOPA-treated parkinsonian rats. Neurosci Res. 2008;60:355–63. doi: 10.1016/j.neures.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 94.Tomasevic G, Shamloo M, Israeli D, Wieloch T. Activation of p53 and its target genes p21(WAF1/Cip1) and PAG608/Wig-1 in ischemic preconditioning. Brain Res Mol Brain Res. 1999;70:304–13. doi: 10.1016/s0169-328x(99)00146-1. [DOI] [PubMed] [Google Scholar]
- 95.Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3:430–40. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 96.Capranico G, Tinelli S, Austin CA, Fisher ML, Zunino F. Different patterns of gene expression of topoisomerase II isoforms in differentiated tissues during murine development. Biochim Biophys Acta. 1992;1132:43–8. doi: 10.1016/0167-4781(92)90050-a. [DOI] [PubMed] [Google Scholar]
- 97.Tsutsui K, Tsutsui K, Okada S, Watanabe M, Shohmori T, Seki S, et al. Molecular cloning of partial cDNAs for rat DNA topoisomerase II isoforms and their differential expression in brain development. J Biol Chem. 1993;268:19076–83. [PubMed] [Google Scholar]
- 98.Watanabe M, Tsutsui K, Tsutsui K, Inoue Y. Differential expressions of the topoisomerase II alpha and II beta mRNAs in developing rat brain. Neurosci Res. 1994;19:51–7. doi: 10.1016/0168-0102(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 99.Lyu YL, Wang JC. Aberrant lamination in the cerebral cortex of mouse embryos lacking DNA topoisomerase IIbeta. Proc Natl Acad Sci U S A. 2003;100:7123–8. doi: 10.1073/pnas.1232376100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tsutsui K, Tsutsui K, Sano K, Kikuchi A, Tokunaga A. Involvement of DNA topoisomerase IIbeta in neuronal differentiation. J Biol Chem. 2001;276:5769–78. doi: 10.1074/jbc.M008517200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Overview of experimental design.
Figure S2: Oxidative stress triggers gene expression profile changes that resemble Alzheimer's disease expression profile
(A) Venn diagram demonstrating the overlapping gene expressions in the various sets: A1C-C1C- gene expression changes in AD cells compared to control cells without treatment at 1 h; C1H-C1C - gene expression changes in control cells treated with H2O2 compared to control cells without treatment at 1 h; C1M-C1C -gene expression changes in control cells treated with menadione as compared to control cells without treatment at 1 h. Numbers in red represent number of genes up-regulated and in blue represent the number of genes down-regulated (B) Venn diagram demonstrating the overlapping gene expressions in the various sets. A6C-C6C - gene expression changes in AD cells compared to control cells without treatment at 6 h, C6H-C6C - gene expression changes in control cells treated with H2O2 compared to control cells without treatment at 6 h; C6M-C6C -gene expression changes in control cells treated with menadione as compared to control cells without treatment at 6 h. (C) Cell plot of the gene expression profiles at 1 h. The color key on the right indicates the extent of change in the gene expression. Blue bars represents down-regulation of gene expression whereas red bars represents up-regulation of gene expression (D) Cell plot of the gene expression profiles at 6 h. The color key on the right indicates the extent of change in the gene expression. Blue bars represent down-regulation of gene expression whereas red bars represent up-regulation of gene expression.
Figure S3: Gene expression profile of overlapping genes in the 12 h arrays
Cell plot of the 215 significant genes that changed expression in the three sets; 1 - gene expression changes in AD cells compared to control cells without treatment at 12 h; 2 - gene expression changes in control cells treated with H2O2 compared to control cells without treatment at 12 h; 3 -gene expression changes in control cells treated with menadione as compared to control cells without treatment at 12 h. The color key on the right indicates the extent of change in the gene expression. Blue bars represent down- regulation of gene expression whereas red bars represents up-regulation of gene expression.
Figure S4: Gene Ontology profile of overlapping pathways in the 12 h arrays
Cell plot of the 141 pathways that changed expression in the three sets; 1 - GO changes in AD cells compared to control cells without treatment at 12 h 2 - GO changes in control cells treated with H2O2 compared to control cells without treatment at 12 h; 3 -GO changes in control cells treated with menadione as compared to control cells without treatment at 12 h. The color key on the right indicates the extent of change in the GO. Blue bars represent down-regulation of a pathway as measured by GO and red bars represent up-regulation of a pathway as measured by GO
Figure S5: Sporadic AD cultured fibroblasts display increased oxidative damage.
Flow cytometric analysis measuring the FITC intensity (8-OdG) of U2OS cell line with the indicated treatments.
Table S1 List of probe sets used for qPCR validation.