

Abstract
The diversity-oriented chemoenzymatic synthesis of α-dystroglycan (α-DG) core M1 O-mannose glycans has been achieved via a three-step sequential one-pot multienzyme (OPME) glycosylation of a chemically prepared disaccharyl serine intermediate. The high flexibility and efficiency of this chemoenzymatic strategy was demonstrated for the synthesis of three more complex core M1 O-mannose glycans for the first time along with three previously reported core M1 structures.
α-Dystroglycan (α-DG), a heavily glycosylated protein found in muscle and brain tissue, is a key component of the dystroglycan-glycoprotein complex that links the extracellular matrix to the intracellular cytoskeleton.1–4 In addition to conventional N-glycans and O-GalNAc glycans, the “uncommon” O-mannose glycans abundant on the mucin domain of α-DG play essential roles in muscle structure and function. Defects in O-mannosylation can cause several congenital muscular dystrophies (CMDs) and promote metastasis of many types of cancers.1,2,5–7 Moreover, α-DG is a host cell surface receptor for a number of arenaviruses, including lymphocytic choriomeningitis virus (LCMV) and Lassa fever virus (LFV).1,2,6
So far, more than 50 mammalian O-mannosylated proteins have been identified, over 20 O-mannose glycan structures have been well characterized, and it is estimated that the O-mannose glycans account for up to 30% of all O-glycans in mammalian brain tissue.1 However, α-DG is the only O-mannosylated protein which has been intensively studied. The core M1 structures are account for at least 50% of the total O-mannose glycans in brain, and the sialyl tetrasaccharide structure (III in Figure 1) has been identified as the most abundant form of core M1 glycans.1
Figure 1.

Structures of core M1 O-mannose glycans. Abbreviation: LN, LacNAc; sLNAc, sialyl LacNAc (Neu5Ac); sLNGc, sialyl LacNAc (Neu5Gc); HNK-1, human natural killer-1; Lex, Lewis x; sLex, sialyl Lewis x. Please see reference 1 for core structure nomenclature.
Although the biological significance of O-mannose glycans has been well documented, the biosynthetic pathway and the precise function of this diverse glycan library have not yet been fully understood.1–6. Therefore, the synthesis of structurally well-defined O-mannose glycans in sufficient amount will not only provide carbohydrate standards for the identification of these glycans from natural sources, but also provide the probes for the elucidation of their biosynthetic pathway and structure-activity relationship (SAR) studies.
Despite the tremendous advances made in the chemical glycosylation processes in the past few decades, the stereoselective formation of the sialic acid linkage is still challenging.7–11 The overall synthetic strategy of core M1 O-mannosyl amino acids was further complicated by the lability of sialic acid and fucose residues12,13, and the racemization-prone amino acid moiety14, 15. Therefore, despite the O-mannose glycan structure III (Figure 1) was first identified by Endo and co-workers in 199716, and over 20 structurally distinct O-mannose glycans have been reported thereafter, only four of them have been synthesized by 5 research groups around the world.17–22 The previous chemical or chemoenzymatic synthesis mainly focused on the synthesis of core M1 tetrasaccharide structure III17–21 or the phosphorylated core M3 trisaccharide22. However, the Neu5Gc-, Lex- and sLex-containing more complex core M1 structures IV, VI, and VII (Figure 1) have not been synthesized in any forms. Therefore, there is an urgent need to develop a diversity-oriented synthesis to access these structurally distinct complex O-mannose glycans. We have successfully achieved this using a sequential three-step one-pot multienzyme (OPME) glycosylation process.
The retrosynthetic plan for the diversity-oriented chemoenzymatic synthesis of core M1 O-mannose glycans 2–7 is depicted in Scheme 1. The chemical synthesized disaccharide O-mannosyl serine 2 was chosen as the key intermediate and starting point for our sequential OPME synthesis of core M1 O-mannose glycans 3–7 (Scheme 1). The disaccharide O-mannosyl serine 2, which in turn could be prepared from disaccharide thioglycoside 1 by using strategy of late stage introduction of amino acid. Subsequently, the disaccharyl serine 2 can serve as the substrate for a sequential OPME glycosylation process to afford the core M1 O-mannose glycans 3–7 using OPME β1–4 galactosylation (OPME 1)23,24, α2–3-sialylation(OPME 2)25 and α1–3-fucosylation (OPME 3)24,26 reactions (Scheme 1).
Scheme 1.
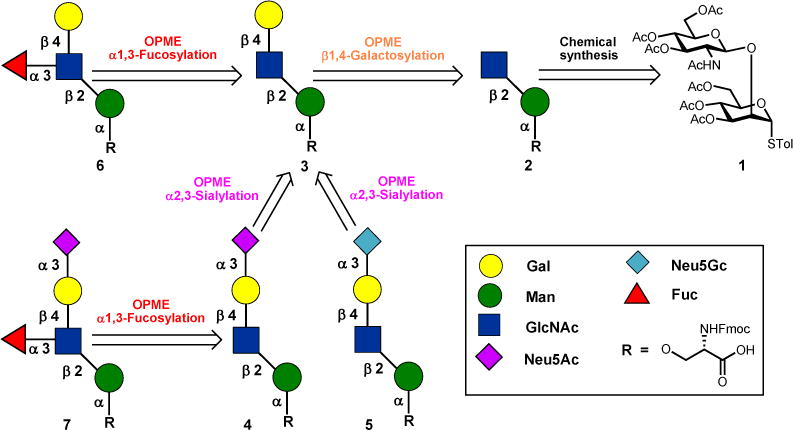
Retrosynthetic plan for core M1 α-DG glycans 2–7
The chemical synthesis of disaccharide O-mannosyl serine 2 was commenced with preparation of bifunctional thiomannoside 10 (Scheme 2). The readily available thiomannoside 827 was transformed into the corresponding 2,6-diol 9 in one step in 89% yield using Ley’s diketone protection protocol28,29. The C6–OH of 9 was selectively protected as a tert-butyldimethylsilyl (TBDMS) ether to give mannoside 10 in 93% yield in the presence of TBDMSCl and imidazole at room temperature. The resulting thiomannoside 10 was then glycosylated with a known glucosamine trichloroacetimidate donor 1130 in the presence of trimethylsilyl trifluoromethanesulfonate (TMSOTf) to give disaccharide 12 in 95% yield.
Scheme 2.
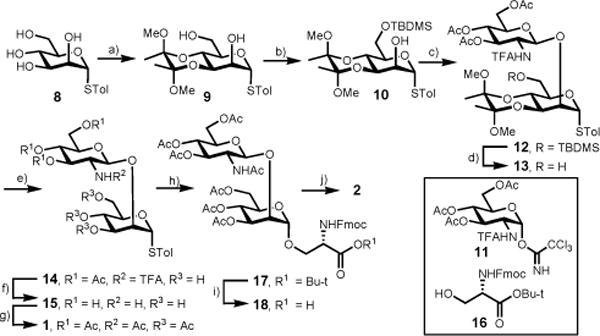
Chemical synthesis of disaccharyl serine 2. Reagents and conditions: a) 2,3-butanedione(1.1 eqiv), trimethyl orthoformate (3.3 equiv), (±) camphorsulfonic acid (CSA, 0.11 equiv), 110 °C, 12 h, 89%; b) TBDMSCl (1.5 equiv), imidazole (3.0 equiv), DMF, rt, overnight, 93%; c) 11 (1.5 equiv) TMSOTf (0.2 equiv), 4 Å MS, CH2Cl2, −30 °C, 30 min, 95%; d) pyridine-HF, rt, overnight, 94%; e) TFA/H2O (9:1, v/v), 30 min, 91%; f) MeOH/NaOMe pH 11, 20 min, then 1 M LiOH, 1 h; g) Ac2O, pyridine, rt, overnight, 68 % for two steps; h) 16 (1.5 equiv) NIS (1.5 equiv), AgOTf (0.2 equiv), CH2Cl2, 4 Å MS, −25 °C, 1 h, 94%; i) TFA/H2O (10:1, v/v), rt, 30 min; j) MeOH/NaOMe pH 8.5, rt, 40 min, 72% for two steps.
The protecting group manipulation of 12 to give disaccharide donor 1 was executed prior to the introduction of the amino acid moiety in a four-step procedure. Briefly, the TBDMS silyl in 12 group was removed using HF-pyridine to furnish disaccharide 13 in 94% yield. The C3,C4-diketone deprotection was achieved by treating 13 with trifluoracetic acid/water (9:1, v/v) for 30 minutes to provide the disaccharide 14 in 91% yield. Finally, the deacetylation including the removal of the N-trifluoroacetyl group was performed under basic condition, and the resulting disaccharide 15 was peracetylated to afford thioglycoside 1 in 68% yield for two steps.
Coupling of disaccharide donor 1 with serine derivative 1631 in the presence of N-iodocussinimide (NIS) and catalytic amount of silver trifluoromethanesulfonate (AgOTf) produced the α-linked disaccharyl serine 17 as the only product in 94% yield. The nascent glycosidic linkage was determined as α since the coupling constant (1JCH = 173.4 Hz) between the anomeric carbon and hydrogen atom was in the range of α-manno configurate O-glycoside.32,33 The fully protected disaccharyl serine 17 was converted to the Fmoc-protected disaccharyl serine 2 in 72% yield for two steps including trifluoroacetic acid mediated deprotection of tert-butyl ester and deacetylation under Zemplén condition (Scheme 2).
With the chemically synthesized disaccharyl serine 2 in hand, two OPME glycosylation (OPME 1, OPME β1–4-galactosylation and OPME 2, OPME α2–3-sialylation) were investigated for the biomimetic stepwise chain elongation to afford trisaccharide O-mannose glycan 3, tetrasaccharide O-mannose glycans 4 and 5, respectively (Scheme 3).
Scheme 3.
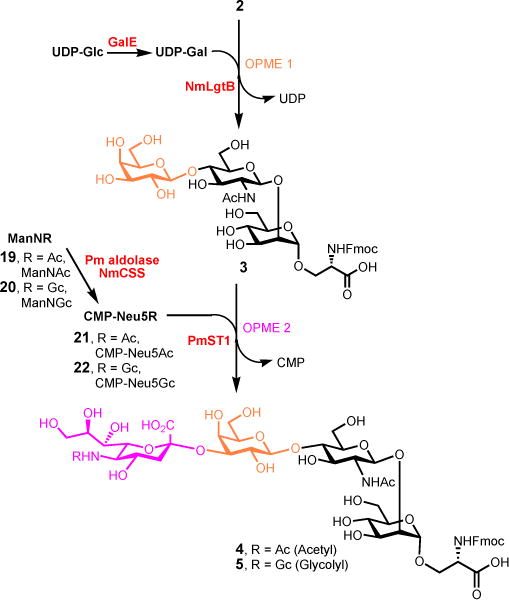
Sequential OPME synthesis of trisaccharyl serine 3, tetrasaccharyl serine 4 and 5. Reagents and conditions: OPME 1, UDP-Glc (1.3 equiv), MgCl2 (20 mmol), Tris-HCl buffer (100 mmol, pH 7.5), EcGalE, NmLgtB, 37 °C, 16 h, 89% for 3 in one-pot; OPME 2, ManNAc (2.0 equiv, for 4), ManNGc (2.0 equiv, for 5), pyruvate (4.0 equiv), CTP (2.0 equiv), MgCl2 (20 mmol), Tris-HCl buffer (100 mmol, pH 8.5), Pm aldolase, NmCSS, PmST1, 37 °C, 30 min, 92% for 4, 95% for 5.
The OPME β1–4-galactosylation of disaccharyl serine acceptor 2 was performed on preparative scales (100 mg) in 100 mM Tris-HCl buffer (pH 7.5) in the presence of UDP-Glc, MgCl2 (20 mmol), an Escherichia coli UDP-galactose-4-epimerase (EcGalE)34 and a Neisseria meningitidis β1–4GalT (NmLgtB)23 at 37 °C for 16 hours. In this one-pot two-enzyme system, the commercially available UDP-Glc was epimerized by EcGalE to afford UDP-Gal which was then used by NmLgtB to form the required β1–4-linkage. To our delight, the disaccharyl serine 2 can be well tolerated by NmLgtB to afford the desired trisaccharyl serine 3 in 89% yield. Previously, Paulson and co-workers found that the UDP-Glc also can be used by NmLgtB to give the glucosylated disaccharide product in 13% yield in the absence of GalE, while no glucosylated by-product was detected when the NmLgtB-catalyzed reaction was coupled with GalE.35 The structure of trisaccharide 3 was confirmed by extensive 1D and 2D NMR studies (SI), as the chemical shifts of H2 (3.53 ppm) and H4 (3.89 ppm) of the galactose residue were consistent with previous report [H2 (3.55 ppm) and H4 (3.92 ppm)]18, in contrast, the chemical shifts of H2 and H4 of glucose residue were usually in the range of 3.31 ppm and 3.41 ppm, respectively36.
Having successfully demonstrated the OPME β1–4-galactosylation using disaccharyl serine 2 as acceptor, we next anticipated that the OPME α2–3-sialylation25 might also be capable of using this galactosyl trisaccharide 3 as substrate. As shown in Scheme 3, the one-pot three-enzyme sialylation of trisaccharide 3 (150 mg) was performed in 100 mM Tris-HCl buffer (pH 8.5) in the presence of N-acetyl-mannosamine (ManNAc, 19), sodium pyruvate, cytidine 5′-triphosphate (CTP), MgCl2 (20 mmol), a Pasteurella multocida sialic acid aldolase (Pm aldolase)37, a Neisseria meningitidis CMP-sialic acid synthetase (NmCSS)38 and a Pasteurella multocida multifunctional α2–3-sialyltransferase (PmST1)25 at 37 °C for 30 min. The Pm aldolase is responsible for converting ManNAc to N-acetylneuraminic acid (Neu5Ac) in the presence of sodium pyruvate. The Neu5Ac was then converted to CMP-Neu5Ac (21) catalyzed by NmCSS in the presence of cytidine 5′-triphosphate (CTP), and the in situ generated CMP-Neu5Ac (21) was utilized by PmST1 to form the desired α2–3-linked sialoside 4 in 92% yield (Scheme 3). Following the same approach, the N-glycolylneuraminic acid (Neu5Gc)-containing tetrasaccharyl serine 5 was also prepared in one-pot in 95% yield using N-glycolyl-mannosamine (ManNGc, 20) as a sialic acid precursor (Scheme 3). It is noteworthy that the Neu5Gc-containing O-mannose tetrasaccharide 5 was synthesized for the first time.
For the synthesis of fucosyl O-mannose glycans 6 and 7, a one-pot two-enzyme system (OPME 3)24,26 was employed to stereo- and regioselective introducing α1–3-linked fucose to the trisaccharide acceptor 3 and tetrasaccharide acceptor 4, respectively. As shown in Scheme 4, in this system, L-fucose was activated by the bifunctional L-fucokinase/GDP-fucose pyrophosphorylase (FKP) from Bacteroides fragilis39 in the presence of adenosine 5′-triphosphate (ATP) and guanosine 5’- triphosphate (GTP) to form GDP-Fuc via a fucose-1-phosphate (Fuc-1-P) intermediate. The in situ generated GDP-Fuc was then taken by a recombinant N-His6-tagged C-terminal truncated Helicobacter pylori α1–3-fucosyltransferase (Hp α1–3FTΔ66)26,40 to form the desired Lex-containing O-mannose glycan 6 (200 mg) in 85% yield after purification. The synthesis of sialyl Lex-containing O-mannose glaycan 7 from tetrasaccharide 4 was performed following the same procedure as described above for 6 (Scheme 4). The pentasaccharide 7 (35 mg) was obtained in 74% yield. It is noteworthy that both Lex- and sialyl Lex-containing O-mannose glycans 6 and 7 were synthesized for the first time.
Scheme 4.
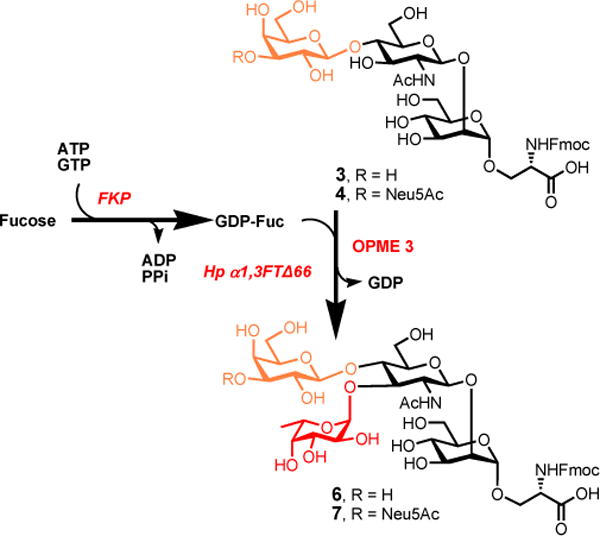
Enzymatic synthesis of fucosyl O-mannose glycans 6 and 7 using OPME fucosylation. Reagents and conditions: L-fucose (1.3 equiv), ATP (1.3 equiv), GTP (1.3 equiv), MnCl2 (20 mM), Tris-HCl buffer (100 mM, pH 7.5), FKP, Hp α1–3FTΔ66, 37 °C, 12 h, 85% for 6, 74% for 7.
In summary, we have developed a highly efficient diversity-oriented chemoenzymatic strategy for the rapid assembly of core M1 O-mannose glycans 2–7 as their Fmoc-Ser-linked derivatives. The success of this strategy relies on the stepwise elongation of a chemically produced disaccharide intermediate 2 using three highly efficient OPME glycosylation reactions. The concise chemical synthesis of disaccharide 2 by means of Ley’s diketone protection group and introducing the amino acid moiety at late stage provide a novel entry for gram-scale production of core M1 disaccharides without the aid of mammalian enzymes. It is worth to notice that the Neu5Gc-, Lex- and sLex-containing O-mannose glycans 5–7 were synthesized for the first time.
Supplementary Material
Acknowledgments
This work was financially supported by the Major State Basic Research Development Program of China (973 Program, No. 2012CB822102 to H. C. and F. W.), the National High Technology Research and Development Program of China (2012AA021504 to H.C.), National Natural Science Foundation of China (Grant Nos. 21172135, 21372145 to H. C.), Development Fund for Collaborative Innovation Center of Glycoscience of Shandong University and NIH grant R01HD065122 (to X. C.).
Footnotes
Electronic Supplementary Information (ESI) available: Experimental details, characterization data, and copies of NMR (1H and 13C) spectra of all key intermediates and final products. See DOI: 10.1039/b000000x/
Contributor Information
Xi Chen, Email: xiichen@ucdavis.edu.
Fengshan Wang, Email: fswang@sdu.edu.cn.
Hongzhi Cao, Email: hzcao@sdu.edu.cn.
Notes and references
- 1.Praissman JL, Wells L. Biochemistry. 2014;53:3066. doi: 10.1021/bi500153y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Live D, Wells L, Boons GJ. ChemBioChem. 2013;14:2392. doi: 10.1002/cbic.201300417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stalnaker SH, Stuart R, Wells L. Curr Opin Struct Biol. 2011;21:603. doi: 10.1016/j.sbi.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manya H. Trends Glycosci Glycotechnol. 2011;23:272. doi: 10.4052/tigg.23.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thiel C, Korner C. Glycoconjugate J. 2013;30:77. doi: 10.1007/s10719-012-9447-5. [DOI] [PubMed] [Google Scholar]
- 6.Dobson CM, Hempel SJ, Stalnaker SH, Stuart R, Wells L. Cell Mol Life Sci. 2013;70:2849. doi: 10.1007/s00018-012-1193-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boltje TJ, Buskas T, Boons G-J. Nat Chem. 2009;1:611. doi: 10.1038/nchem.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muthana S, Cao H, Chen X. Curr Opin Chem Biol. 2009;13:573. doi: 10.1016/j.cbpa.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu X, Schmidt RR. Angew Chem Int Ed. 2009;48:1900. doi: 10.1002/anie.200802036. [DOI] [PubMed] [Google Scholar]
- 10.Mydock LK, Demchenko AV. Org Biomol Chem. 2010;8:497. doi: 10.1039/b916088d. [DOI] [PubMed] [Google Scholar]
- 11.Hsu C-H, Hung S-C, Wu C-Y, Wong C-H. Angew Chem Int Ed. 2011;50:11872. doi: 10.1002/anie.201100125. [DOI] [PubMed] [Google Scholar]
- 12.Kunz H, Unverzagt C. Angew Chem Int Ed. 1988;27:1697. [Google Scholar]
- 13.Cao H, Huang S, Cheng J, Li Y, Muthana S, Son B, Chen X. Carbohydr Res. 2008;343:2863. doi: 10.1016/j.carres.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Fenza A, Tancredi M, Galoppini C, Rovero P. Tetrahedron Lett. 1998;39:8529. [Google Scholar]
- 15.Zhang Y, Muthana SM, Farnsworth D, Ludek O, Adams K, Barchi JJ, Jr, Gildersleeve JC. J Am Chem Soc. 2012;134:6316. doi: 10.1021/ja212188r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiba A, Matsumura K, Yamada H, Inazu T, Shimizu T, Kusunoki S, Kanazawa I, Kobata A, Endo T. J Biol Chem. 1997;272:2156. doi: 10.1074/jbc.272.4.2156. [DOI] [PubMed] [Google Scholar]
- 17.Yu J, Westerlind U. ChemBioChem. 2014;15:939. doi: 10.1002/cbic.201300537. [DOI] [PubMed] [Google Scholar]
- 18.Sardzik R, Green AP, Laurent N, Both P, Fontana C, Voglmeir J, Weissenborn MJ, Haddoub R, Grassi P, Haslam SM, Widmalm G, Flitsch SL. J Am Chem Soc. 2012;134:4521. doi: 10.1021/ja211861m. [DOI] [PubMed] [Google Scholar]
- 19.Matsuo I, Isomura M, Ajisaka K. Tetrahedron Lett. 1999;40:5047. [Google Scholar]
- 20.Seifert J, Ogawa T, Kurono S, Ito Y. Glycoconjugate J. 2000;17:407. doi: 10.1023/a:1007112232131. [DOI] [PubMed] [Google Scholar]
- 21.Seifert J, Ogawa T, Ito Y. Tetrahedron Lett. 1999;40:6803. [Google Scholar]
- 22.Mo KF, Fang T, Stalnaker SH, Kirby PS, Liu M, Wells L, Pierce M, Live DH, Boons G-J. J Am Chem Soc. 2011;133:14418. doi: 10.1021/ja205473q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lau K, Thon V, Yu H, Ding L, Chen Y, Muthana MM, Wong D, Huang R, Chen X. Chem Comm. 2010;46:6066. doi: 10.1039/c0cc01381a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen C, Zhang Y, Xue M, Liu X, Li Y, Chen X, Wang PG, Wang F, Cao H. Chem Commun. 2015 doi: 10.1039/c5cc01330e. 51:7689. [DOI] [PubMed] [Google Scholar]
- 25.Yu H, Chokhawala H, Karpel R, Yu H, Wu BY, Zhang JB, Zhang YX, Jia Q, Chen X. J Am Chem Soc. 2005;127:17618. doi: 10.1021/ja0561690. [DOI] [PubMed] [Google Scholar]
- 26.Sugiarto G, Lau K, Yu H, Vuong S, Thon V, Li Y, Huang S, Chen X. Glycobiology. 2011;21:387. doi: 10.1093/glycob/cwq172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang ZY, Ollmann IR, Ye XS, Wischnat R, Baasov T, Wong C-H. J Am Chem Soc. 1999;121:734. [Google Scholar]
- 28.Ley SV, Polara A. J Org Chem. 2007;72:5943. doi: 10.1021/jo0703451. [DOI] [PubMed] [Google Scholar]
- 29.Grice P, Ley SV, Pietruszka J, Osborn HMI, Priepke HWM, Warriner SL. Chem -Eur J. 1997;3:431. [Google Scholar]
- 30.Makino A, Kurosaki K, Ohmae M, Kobayashi S. Biomacromolecules. 2006;7:950. doi: 10.1021/bm050895d. [DOI] [PubMed] [Google Scholar]
- 31.Knerr PJ, van der Donk WA. J Am Chem Soc. 2012;134:7648. doi: 10.1021/ja302435y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teumelsan N, Huang X. J Org Chem. 2007;72:8976. doi: 10.1021/jo7013824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Chen C, Jin L, Tan H, Wang F, Cao H. Carbohydr Res. 2015;401:109. doi: 10.1016/j.carres.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 34.Chen X, Zhang W, Wang JQ, Fang JW, Wang PG. Biotechnol Prog. 2000;16:595. doi: 10.1021/bp000052s. [DOI] [PubMed] [Google Scholar]
- 35.Blixt O, Brown J, Schur MJ, Wakarchuk W, Paulson JC. J Org Chem. 2001;66:2442. doi: 10.1021/jo0057809. [DOI] [PubMed] [Google Scholar]
- 36.Hounsell EF. Prog Nucl Mag Res Sp. 1995;27:445. [Google Scholar]
- 37.Li Y, Yu H, Cao H, Lau K, Muthana S, Tiwari VK, Son B, Chen X. Appl Microbiol Biotechnol. 2008;79:963. doi: 10.1007/s00253-008-1506-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu H, Yu H, Karpel R, Chen X. Bioorg Med Chem. 2004;12:6427. doi: 10.1016/j.bmc.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 39.Zhao G, Guan W, Cai L, Wang PG. Nat Protoc. 2010;5:636. doi: 10.1038/nprot.2010.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun HY, Lin SW, Ko TP, Pan JF, Liu CL, Lin CN, Wang AH, Lin CH. J Biol Chem. 2007;282:9973. doi: 10.1074/jbc.M610285200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.