

Uniparental disomy (UPD) can be the cause of atypical disease manifestations in patients with Mendelian disorders. Consideration of this disease mechanism proved essential in our evaluation of a 3-year-old boy, referred to the NIH Clinical Center for further evaluation of Gaucher disease (GD).
Case report
The patient was born at term to non-consanguineous parents with a paternal family history of autosomal dominant Charcot-Marie-Tooth disease type I (CMT1). His medical history included moderate sensorineural hearing loss, Wolff-Parkinson-White arrhythmia, bicuspid aortic valve, and motor delay, ambulating at 26 months. At 17 months, hepatosplenomegaly was noted and a bone marrow biopsy led to the diagnosis of type 3 GD. Molecular analysis revealed homozygosity for the NM_000157.2:c.1448T>C (L444P) mutation in the glucocerebrosidase gene (GBA). He began enzyme replacement therapy (Imiglucerase, 120 U/kg biweekly), with significant resolution of his organomegaly.
On examination, he had mild hepatosplenomegaly, bilateral pes cavus, externally rotated hips, genu valgus, hammertoes, decreased muscle tone, and mild foot drop. Reflexes were depressed in the upper extremities and absent in knees and ankles. Ophthalmologic evaluation revealed nystagmus, dysconjugate gaze, slowed, hypometric horizontal saccades, and decreased pupillary constriction (Adie-like). Nerve conduction studies were 13 to 18 m/second in all extremities, consistent with a childhood demyelinating pattern; the father’s velocities were 17 to 22 m/second. Cranial MRI was normal.
GD, inherited in an autosomal recessive pattern, results from the deficiency of lysosomal glucocerebrosidase. It is classified into non-neuronopathic (Type 1; MIM 230800) and neuronopathic (Types 2 and 3; MIM 230900 and 231000) forms. While GD3 manifests with impaired horizontal saccades and a spectrum of neurologic symptoms, this child had additional clinical features not associated with GD. The family history of CMT, coupled with the hearing loss and pupillary abnormalities, led to evaluation of the myelin protein zero (MPZ; MIM 159440) gene, which causes CMT2J,1 CMT1B, and other polyneuropathies. Both GBA and MPZ are located about 1Mb apart on chromosome 1q22–23. In the proband, mutational analysis demonstrated homozygosity for the NM_000530.4:c.263C>T (S78L) allele of MPZ and confirmed homozygosity for the GBA mutation. The proband’s father was found to be heterozygous for mutations in both genes, while neither was identified in the mother. Other chromosome 1 microsatellite markers, examined in the proband and both parents, demonstrated complete paternal isodisomy, implicating this mechanism as the etiology of the concurrent disorders (figure).
Figure. Genotype analysis and family pedigree.
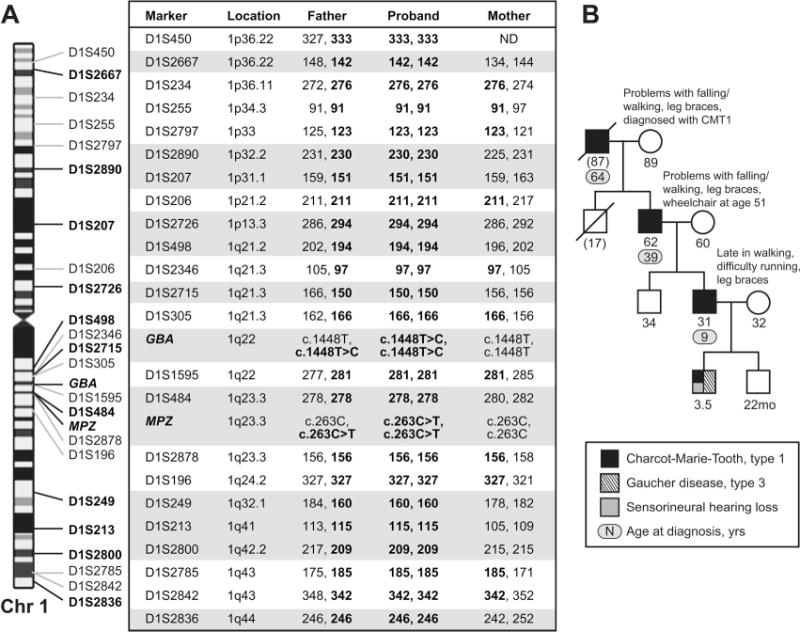
(A) Genotype analysis of the proband and his parents with chromosome 1 markers from the ABI PRISM Linkage Mapping Set, v2.5, confirms complete paternal isodisomy. In addition to homozygosity for the mutant GBA and MPZ alleles, the proband is homozygous for 23 microsatellite markers tested. Eleven of these markers, indicated by shading, are fully informative for the absence of the maternal chromosome 1 in the proband. (B) Family pedigree.
Discussion
UPD occurs when an individual inherits both homologues from one parent.2 This atypical form of inheritance can also have clinical consequences arising from abnormal genomic imprinting, homozygosity of autosomal recessive mutations, or mosaicism. The presence of both homologues from one parent is referred to as heterodisomy, whereas isodisomy describes two copies of one homologue. If the duplicated homologue carries a mutation in a gene, the patient is homozygous for that mutation. The apparent complete paternal isodisomy observed in this patient suggests monosomy rescue as the genetic mechanism (i.e., duplication of a chromosome from a monosomic zygote), but a postfertilization error cannot be excluded. While over 10 cases of paternal UPD of chromosome 1 have been reported,3 this is the first known case of GD or CMT due to UPD.
The CMT manifestations present in our patient matched descriptions of CMT2J (axonal CMT with hearing loss and pupillary abnormalities), except that our patient manifested a demyelinating pattern with slow conduction velocities. Heterozygosity for the MPZ S78L allele is associated with CMT1B4 with variable age at onset, but has been reported in a severely affected family in which the index case presented at 1.5 years of age.5 We hypothesize that the early atypical presentation of our patient is caused by homozygosity for the MPZ mutation, although homozygosity at other loci on chromosome 1 likewise might impact the phenotype.
This patient with both GD and CMT demonstrates that UPD should be considered as an explanation for unusual presentations of well-known diseases. This case also underscores the importance of collecting family pedigrees and parental genotypes.
Acknowledgments
The authors thank the patient and his family; Michael Eblan and Dr. Andre Mignault for genotyping the proband and parents for the GBA and MPZ mutations; and the referring physician, Dr. David B. Flannery.
Footnotes
Disclosure: Dr. Nagan is an employee of Athena Diagnostics, Inc. All other authors report no conflicts of interest.
References
- 1.De Jonghe P, Timmerman V, Ceuterick C, et al. The Thr124Met mutation in the peripheral myelin protein zero (MPZ) gene is associated with a clinically distinct Charcot-Marie-Tooth phenotype. Brain. 1999;122(Pt 2):281–290. doi: 10.1093/brain/122.2.281. [DOI] [PubMed] [Google Scholar]
- 2.Engel E. A new genetic concept: uniparental disomy and its potential effect, isodisomy. Am J Med Genet. 1980;6:137–143. doi: 10.1002/ajmg.1320060207. [DOI] [PubMed] [Google Scholar]
- 3.Kotzot D, Utermann G. Uniparental disomy (UPD) other than 15: phenotypes and bibliography updated. Am J Med Genet A. 2005;136:287–305. doi: 10.1002/ajmg.a.30483. [DOI] [PubMed] [Google Scholar]
- 4.Nelis E, Haites N, Van Broeckhoven C. Mutations in the peripheral myelin genes and associated genes in inherited peripheral neuropathies. Hum Mutat. 1999;13:11–28. doi: 10.1002/(SICI)1098-1004(1999)13:1<11::AID-HUMU2>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 5.Silander K, Meretoja P, Juvonen V, et al. Spectrum of mutations in Finnish patients with Charcot-Marie-Tooth disease and related neuropathies. Hum Mutat. 1998;12:59–68. doi: 10.1002/(SICI)1098-1004(1998)12:1<59::AID-HUMU9>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]