

Supplemental Digital Content is Available in the Text.
The lidocaine metabolite N-ethylglycine specifically reduces GlyT1-dependent glycine uptake and has antinociceptive effects in experimental inflammatory and neuropathic pain, while no adverse effects are observed.
Keywords: Neuropathic pain, Inflammatory pain, N-ethylglycine, Glycine transporter, Glycinergic inhibition, Lidocaine metabolites
Abstract
Glycine transporter 1 (GlyT1) plays a crucial role in regulating extracellular glycine concentrations and might thereby constitute a new drug target for the modulation of glycinergic inhibition in pain signaling. Consistent with this view, inhibition of GlyT1 has been found to induce antinociceptive effects in various animal pain models. We have shown previously that the lidocaine metabolite N-ethylglycine (EG) reduces GlyT1-dependent glycine uptake by functioning as an artificial substrate for this transporter. Here, we show that EG is specific for GlyT1 and that in rodent models of inflammatory and neuropathic pain, systemic treatment with EG results in an efficient amelioration of hyperalgesia and allodynia without affecting acute pain. There was no effect on motor coordination or the development of inflammatory edema. No adverse neurological effects were observed after repeated high-dose application of EG. EG concentrations both in blood and spinal fluid correlated with an increase of glycine concentration in spinal fluid. The time courses of the EG and glycine concentrations corresponded well with the antinociceptive effect. Additionally, we found that EG reduced the increase in neuronal firing of wide-dynamic-range neurons caused by inflammatory pain induction. These findings suggest that systemically applied lidocaine exerts antihyperalgesic effects through its metabolite EG in vivo, by enhancing spinal inhibition of pain processing through GlyT1 modulation and subsequent increase of glycine concentrations at glycinergic inhibitory synapses. EG and other substrates of GlyT1, therefore, may be a useful therapeutic agent in chronic pain states involving spinal disinhibition.
1. Introduction
Persistent inflammatory stimuli or lesions as well as diseases affecting the somatosensory system can lead to distinct and long-lasting alterations in the nociceptive system that result in spontaneous pain and exaggerated responses to innocuous and noxious stimuli. Chronic pain that results from these maladaptive responses loses its warning function and is considered an autonomous disease state.11 Given that roughly 20% of the population suffer from chronic pain,7,22 there is a crucial need for further unraveling the mechanisms of chronic pain states in order to develop new mechanism-oriented treatment strategies.4
The development and maintenance of chronic pain involves profound changes in the neuronal processing of sensory information. In addition to adaptive changes within the peripheral nervous system, there are numerous studies suggesting that pathological alterations in the somatoafferent nervous system in chronic pain conditions result in an imbalance of excitatory and inhibitory spinal neurotransmission predominantly in spinal cord.44 Particularly, the loss of GABAergic and glycinergic synaptic neurotransmission has gained increased interest, since it is believed that a pharmacological restoration of physiological inhibition in the spinal cord might represent a future therapeutic approach.51 Here, targeting of GABAergic neurotransmission has largely failed, in part due to an ubiquitary abundance of GABAA receptors,51 although α1-sparing GABAA receptor agonists have yet to be tested in patients.30 Although there are studies demonstrating that the analgesic effects of cannabinoids result from a facilitation of α3 containing glycine receptors (GlyRs),48 to date, there have been no specific GlyR facilitators/agonists available with suitable pharmacological profiles.
Current recommendations for the management of patients suffering from neuropathic pain include the treatment with tricyclic antidepressants, dual reuptake inhibitors for serotonin and norepinephrine, calcium channel α2δ ligands, opioids, and topical application of lidocaine or capsaicin.2,3,14 In addition to significant adverse effects, all known pharmacological approaches typically show only limited efficacy in the majority of patients.18,45
Systemic long-term application of lidocaine is currently recommended as a promising approach to treat neuropathic pain.9 The principle mode of action of lidocaine is blockade of voltage-gated sodium channels.49 Considering the low concentrations of systemically delivered lidocaine that have proven to be effective in neuropathic pain and its low affinity to voltage-gated sodium channels, this mechanism seems unlikely to be able to explain the antinociceptive effects.12,43 Previously, we have shown that a lidocaine metabolite, N-ethylglycine (EG), is an alternative substrate of the glycine transporter GlyT1, suggesting that EG and not lidocaine itself is responsible for some of the beneficial effects of lidocaine in the treatment of neuropathic pain.46 This hypothesis is supported by studies showing that GlyT1 inhibitors efficiently ameliorated hyperalgesia and allodynia in animal models of chronic pain.8,35,42
Here, we show that the lidocaine metabolite EG is specific for GlyT1 and that it ameliorates hyperalgesia and allodynia in animal models for chronic pain. We observed no adverse effects, in contrast to the application of high-affinity GlyT1 antagonists like ALX5407 or the parental substance lidocaine. Taken together, our findings demonstrate that the use of GlyT1 substrates might be a good alternative for treatment of chronic pain syndromes.
2. Materials and methods
Unless stated otherwise, all reagents were purchased from Sigma-Aldrich (St. Louis, MO). All animal experiments were performed in accordance with animal welfare regulations and approved by local authorities. All animals were housed in a controlled environment (21°C ± 1°C; humidity 60%; lights on from 7 am to 7 pm; food and water available ad libitum).
2.1. Electrophysiological recordings from Xenopus laevis oocytes
Oocytes, isolated from Xenopus laevis, were injected with 1 to 20 ng of the indicated mouse complementary RNA (cRNA), or left noninjected. For coexpression of GlyR β subunits with the respective GlyR α subunit, a 10-fold excess of GlyR β cRNA was injected 24 hours before injecting the respective GlyR α cRNA to ensure that the majority of receptors formed are indeed GlyR heteropentamers. The oocytes were incubated for 2 to 4 days at 18°C in ND96 medium (containing [in mM] NaCl 96, KCl 2, MgCl2 1, CaCl2 1, HEPES/NaOH 5, and 50 μg/mL gentamycin; pH 7.4) to ensure a sufficient expression of the corresponding protein. Ion channel or transporter-mediated currents were analyzed by 2-electrode voltage-clamp recordings. All recordings were performed using a Turbo TEC-03X amplifier and CellWorks (V3.7) data acquisition software (all from NPI Electronics, Göttingen, Germany). For the recordings, the membrane potential was held constant at −50 mV. Typically, substance applications of indicated increasing concentrations were performed for 10 to 30 seconds, followed by a washout step with ND96 superfusion solution (containing [in mM] NaCl 96, KCl 2, MgCl2 1, HEPES/NaOH 5, pH 7.4), or for measurements of N-methyl-d-aspartate receptor (NMDAR)–mediated currents ND96 (-Mg) (containing [in mM] NaCl 96; KCl, 2, CaCl2 0.1, HEPES/NaOH 5, pH 7.4) for at least 30 seconds. The measurements were repeated in at least 3 different populations of X laevis oocytes. Current amplitudes are, if not indicated otherwise, presented as relative substance-induced currents (mean ± SEM, n = 4-6), with the respective maximal glycine-induced current being defined as 100%.
2.2. Animal models of pain and treatment experiments
Inflammatory pain was induced by subcutaneous injection of 20 μL complete Freund's adjuvant (CFA) (1 mg/mL Mycobacterium tuberculosis) solution into the plantar side of the left hind paw in adult male C57BL/6J mice.41 Test drugs were applied 72 hours after injection of CFA and development of reduced withdrawal thresholds in response to mechanical stimulation indicative for mechanical allodynia. To investigate the effect of EG on wide-dynamic-range (WDR) neuron responses in inflammatory pain, carrageenan (1%, 20 μL) was injected into the left hind paw of adult male Wistar rats. When neuronal hyperexcitability had developed 3 hours after the injection of carrageenan, EG at indicated concentrations was applied into the subarachnoid space.
Chronic neuropathic pain was induced through the chronic constriction injury (CCI) model in adult male C57BL/6J mice,6 consisting of 3 loose ligatures around the left sciatic nerve, which were performed under general anesthesia using isoflurane (Baxter, Deerfield, IL). Test drugs were applied 72 hours after surgery. All drugs were injected subcutaneously into the loose skin over the neck. To investigate possible toxic effects of the test substances, the animals were thoroughly observed after high-dose treatment of the respective substance for signs of neurotoxicity (irregular movements, signs of distress) until 360 minutes after subcutaneous injection.
2.3. Behavioral tests
An experimenter who was blinded to the randomized treatment group assignment performed all behavioral tests. Touch perception thresholds were measured using the up–down method for obtaining the 50% threshold using von Frey hairs.10 Behavioral response to heat was measured using the Hargreaves method23 (Plantar Test Apparatus; Ugo Basile, Varese, Italy). Motor coordination behavior was analyzed using the Rota-Rod test (Mouse Rota-Rod; Ugo Basile). Paw thickness was measured using a thickness gauge (Model No. 7313; Mitutoyo Corporation, Kawasaki, Japan). Body weight was recorded before and at the end of the experiments. For determination of general activity and explorative behavior, an open-field experiment was performed. The mice were put in the open-field arena (50 × 50 cm, gray plastic), and their activity was determined by a video tracking system (VideoMot; TSE Systems, Bad Homberg, Germany) for 15 minutes. Total distance and regional preference were determined.
2.4. High-pressure liquid chromatography
To investigate the concentrations of glycine and EG using high-pressure liquid chromatography (HPLC), EG (200 mg/kg) was injected subcutaneously in adult male Wistar rats (n = 24). Blood and cerebrospinal fluid (CSF) samples were taken before, 1, 2, 4, 6, and 24 hours after application (n = 6 each group). Blood samples (≤5 mL) were collected by percutaneous cardiac puncture under deep general anesthesia using isoflurane. Subsequently, CSF samples (≤100 µL) were drawn from the cisterna magna as previously described.40 After collection of whole blood, samples were allowed to clot for 30 minutes at RT and subsequently centrifuged at 2000g for 10 minutes at 4°C in order to carefully acquire the serum as the supernatant. All samples were stored at −20°C before analysis. For calibration, 5 concentrations of glycine and N-ethylglycine (10, 50, 100, 500, and 1000 μM) were prepared in water. Protein-free extracts were prepared by mixing a sample volume of 100 μL with 900 μL methanol containing 20 μM norleucine as internal standard. After centrifugation, methanol was removed from the supernatants using a vacuum evaporation centrifuge for 3 hours at room temperature. Subsequently, 20 μL drying solution containing methanol/1 M sodium acetate/triethylamine (2/2/1 by vol) was added to each sample before lyophilization overnight. Then, samples were treated for 20 minutes at room temperature with 40 μL of a derivatizing buffer containing methanol/triethylamine/phenyl isothiocyanate/water (7/1/1/1 by vol) and lyophilized again overnight. Samples were then dissolved in 100 μL methanol and analyzed using a 150 × 4.6 mm C18 Hypersil column (Synergi 4u Hydro-RP 80A; Phenomenex; Macclesfield, United Kingdom) on an analytical HPLC system (Shimadzu Instruments, Columbia, MD) with ultraviolet detection at 254 nm.
2.5. Spinal cord electrophysiology
Electrophysiological recordings were performed on adult male Wistar rats as previously described.13 General anesthesia was induced in a chamber using 3.0% (vol/vol) isoflurane in 66% (vol/vol) N2O and 33% (vol/vol) O2 and was thereafter maintained at 0.7% (vol/vol) isoflurane in 66% (vol/vol) N2O and 33% (vol/vol) O2. Core body temperature was monitored and maintained at 37°C by a homeothermic heating blanket unit and rectal probe. Laminectomy was performed to expose the L4 and L5 segments of the spinal cord. Using a parylene-coated tungsten electrode (A-M Systems, Carlsborg, WA), neurons in deep laminae (∼500-1000 μm from the dorsal surface of the cord) receiving afferent A-fiber and C-fiber input from the hind paw were sought by periodic light tapping of the glabrous surface of the hind paw. Extracellular recordings made from single neurons were visualized on an oscilloscope and discriminated on a spike amplitude and waveform basis. Electrical stimulation was given through 2 needles inserted into the receptive field, and a train of 16 stimuli was given (2 milliseconds pulse duration, 0.5 Hz at 3 times the C-fiber threshold). Responses evoked by Aβ, Aδ, and C fibers were superimposed and separated according to latency (0-20, 20-90, and 90-350 milliseconds, respectively). A range of natural stimuli, including brush, von Frey filaments (8 and 60g), and heat (40°C and 48°C constant water jet), were applied to the receptive field for 10 seconds per stimulus, and the evoked neuronal firing was quantified. Data were captured and analyzed by a CED 1401 interface coupled to a personal computer with Spike2 software (Cambridge Electronic Design). Stabilization of a neuron's responses to the range of electrical and natural stimuli was confirmed with at least 3 consistent responses (<10%) to all measures. Mean values of these responses were calculated and used as pretreatment controls. Carrageenan (1%, 20 μL) was subsequently injected into the left hind paw to induce an inflammatory response. Evoked responses were recorded at intervals of 20 minutes until hyperexcitability had fully developed at 3 hours after application of carrageenan. Then, EG at concentrations of 10, 30, 100, and 1000 μM was administered onto the dorsal surface of the spinal cord followed by further recordings at 30 minutes after application.
2.6. Statistical analysis
Data are expressed as mean ± SEM. Differences in mean values were tested by 1- or 2-way analysis of variance, with Bonferroni post hoc test where appropriate. Calculations were performed using the GraphPad Prism software version 5.0 (GraphPad Software Inc, La Jolla, CA). Results from behavioral observation regarding neurological side effects were analyzed with the Mantel–Cox log-rank test. P < 0.05 was considered significant.
3. Results
3.1. The lidocaine metabolite EG specifically acts as a substrate for GlyT1
To determine whether the lidocaine metabolite EG acts specifically on GlyT1 or, like sarcosine, also on other key components of the glycinergic system,52,53 we expressed GlyT1, GlyT2, the glycine receptor subunits GlyR α1, α2, and α3, or the NMDAR (NR1/NR2a) in X laevis oocytes. Here, application of increasing concentrations of glycine to the respective GlyT-expressing oocytes resulted in a typical concentration-dependent current response (Fig. 1 and Fig. 2), in line with an electrogenic glycine uptake through both transporters. Consistent with previous studies, we also obtained concentration-dependent currents when GlyT1-expressing oocytes were superfused with sarcosine or EG (Ref. 46; see also Fig. 1). In contrast, no sarcosine- or EG-induced currents were observed in GlyT2-expressing oocytes (Fig. 1). Furthermore, simultaneous application of glycine (20-2000 μM) and EG (2000 μM) onto GlyT2-expressing oocytes did not result in any significant change in the current amplitude as compared with the application of glycine alone (Fig. 1C). These findings clearly indicate that EG is neither an alternative substrate nor a partial inhibitor of GlyT2.
Figure 1.
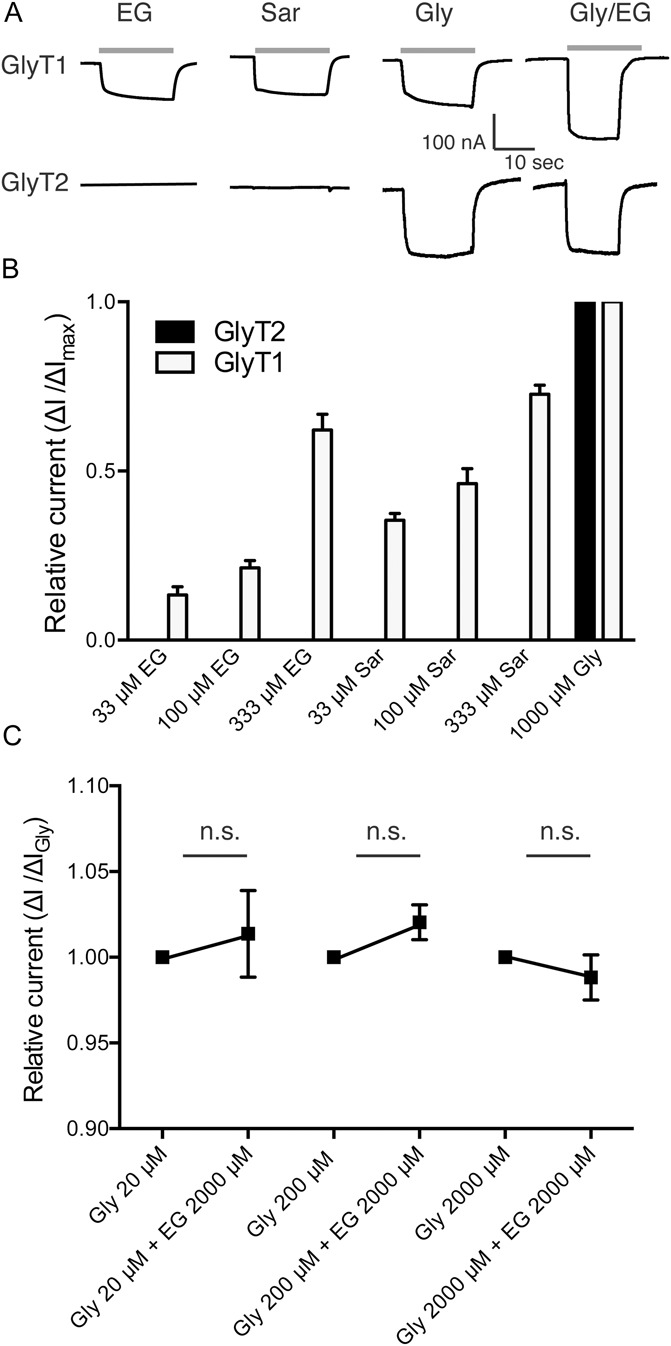
N-ethylglycine (EG) is an artificial substrate for glycine transporter 1 (GlyT1) but has no effect on glycine transporter 2 (GlyT2) function. Xenopus laevis oocytes were injected with complementary RNA encoding for mouse GlyT1 or GlyT2. Substance-induced currents were recorded at a membrane potential held at −50 mV. The respective substances were applied for 20 seconds each followed by a washout of 30 seconds. (A), Representative substance-induced currents for 30 μM glycine (Gly), 1 mM sarcosine (Sar), 1 mM EG, and Gly/EG coapplication were recorded from oocytes expressing GlyT1 or GlyT2, respectively. (B), Average amplitude of the substance-induced currents (EG or sarcosine in the respective concentration) in recordings from GlyT1- or GlyT2-expressing oocytes in relation to the maximal glycine-induced current. Note that EG and sarcosine did not induce currents in GlyT2-expressing oocytes, while glycine led to robust GlyT2-mediated currents. Data are expressed as mean ± SEM (n = 4-6). (C), Comparison of substance-induced currents recorded from GlyT2-expressing oocytes induced by glycine at indicated concentrations or after coapplication of glycine and EG at indicated concentrations. EG did not alter the glycine-induced currents. Data are expressed as mean ± SEM (n = 4-6).
Figure 2.

N-ethylglycine (EG) does not induce significant glycine receptor (GlyR)–mediated currents in Xenopus laevis oocytes. Xenopus laevis oocytes were injected with complementary RNA encoding for mouse GlyR α1, GlyR α2, or GlyR α3 alone or coinjected with complementary RNA encoding for GlyR β. Substance-induced currents were recorded with the membrane potential held at −50 mV. The respective substances were applied for 15 seconds each followed by a washout of 30 seconds. (A), Representative substance-induced currents for 200 μM glycine (Gly), 1 mM sarcosine (Sar), and 1 mM EG were recorded from oocytes expressing the indicated GlyR subunit. (B and C), Concentration/response curve of currents elicited by glycine normalized to maximal glycine inducible currents as determined on oocytes expressing GlyR α1, GlyR α2, or GlyR α3 (depicted in B) or GlyR α1/β, GlyR α2/β, or GlyR α3/β (depicted in C), respectively. Data are expressed as mean ± SEM (n = 4-6). (D-F), Oocytes expressing GlyR α1 (D), GlyR α2 (E), or GlyR α3 (F) or the respective heteropentameric GlyR α/β receptors were superfused with EG- or sarcosine-containing solutions. Substance-induced currents were given in relation to the maximal glycine-induced current. Note that although small sarcosine-induced currents were observed in recordings from oocytes expressing any of the GlyR subunits, no or negligible EG-induced currents were observed. Data are expressed as mean ± SEM (n = 4-6). (G-I), Oocytes expressing the respective GlyR subunit (GlyR α1 in [G], GlyR α2 in [H], or GlyR α3 in [I], depicted with open circles) or the respective heteropentameric GlyR α/β receptors (depicted as filled circles) were superfused with glycine at the indicated concentration alone or in combination with 2 mM EG. Currents were determined as fraction of the current elicited by glycine alone. Data are expressed as mean ± SEM (n = 4-6).
Similarly, no EG-induced currents were observed in recordings from oocytes expressing GlyR α1, α2, or α3 homopentameric receptors, although glycine and even sarcosine-induced currents were readily detectable (Fig. 2A, B, D-F). To analyze whether EG acts as a (partial) antagonist on the glycine receptor, glycine and EG were coapplied to oocytes expressing the respective homopentameric receptor. Here, in none of the recordings, significant effects of EG coapplication on the current amplitude as compared with the currents observed after application of glycine alone were observed, demonstrating that EG does not function as a partial antagonist concerning any of these proteins (Fig. 2G-I). At central nervous system (CNS) synapses, the majority of GlyRs is not present as GlyR α homopentameric receptors but as heteropentamers together with the GlyR β.21 To exclude that EG acts specifically on these heteropentameric receptors, the effects of EG and/or sarcosine were also analyzed in oocytes expressing the respective heteromers. Here, no significant differences to oocytes expressing the respective GlyR α homoreceptor were observed (Fig. 2C-I), confirming that EG acts neither in an agonistic nor antagonistic fashion on GlyRs.
Similar to our experiments on GlyT2 and GlyR receptors, effects of EG on NMDAR (NR1/NR2a) currents were analyzed (Fig. 3). Although NMDAR-mediated currents were readily detectable after application of glycine together with glutamate (Fig. 3A-C, E), no substance-induced currents were observed when glutamate or glycine was applied alone. Application of EG either alone, or in combination with glycine or glutamate, did not produce significant substance-induced currents (Fig. 3A, D). To determine whether EG inhibits NMDAR-mediated currents induced by glutamate and glycine, all 3 substances were applied on NMDAR-expressing oocytes. Similar to our observations in GlyT2- and GlyR-expressing oocytes, the observed current amplitude after application of all 3 substances was indistinguishable from that observed after the application of glutamate and glycine alone (Fig. 3E), indicating that EG does not function as an inhibitor at the NMDAR.
Figure 3.
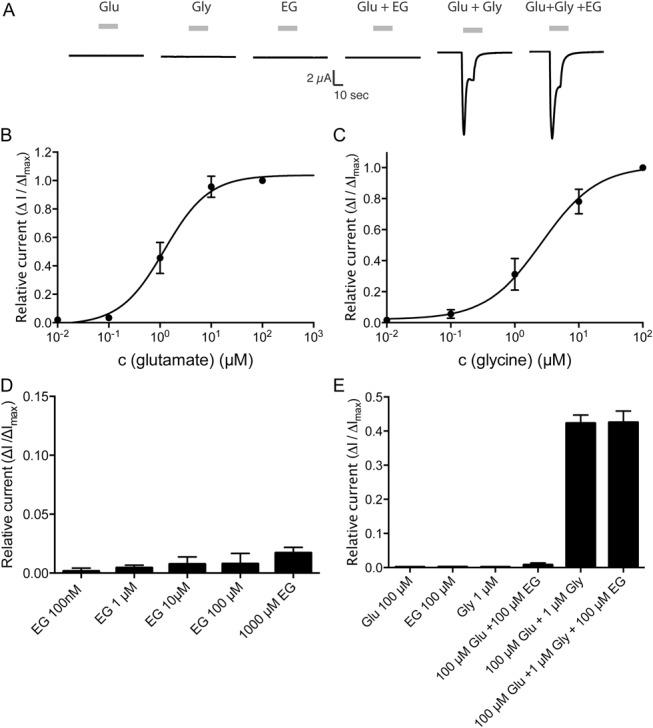
N-ethylglycine (EG) does not influence NMDAR-mediated currents in Xenopus laevis oocytes. Xenopus laevis oocytes were injected with complementary RNA encoding for mouse. Substance-induced currents were recorded with the membrane potential held at −50 mV. The respective substances were applied for 15 seconds each followed by a washout of 30 seconds. (A), Representative substance-induced currents for 1 μM glutamate (Glu), 2 μM glycine (Gly), 100 μM EG, or the indicated coapplications EG were recorded from oocytes expressing the NMDAR (NR1/NR2a). (B), Concentration/response curve of currents elicited by glutamate in the presence of saturating concentrations of glycine (100 μM) normalized to maximal glutamate inducible currents as determined on oocytes expressing the NMDAR (NR1/NR2a). Data are expressed as mean ± SEM (n = 4-6). (C), Concentration/response curve of currents elicited by glycine in the presence of saturating concentrations of glutamate (100 μM) normalized to maximal glycine inducible currents as determined on oocytes expressing the NMDAR (NR1/NR2a). Data are expressed as mean ± SEM (n = 4-6). (D), Oocytes expressing NMDAR superfused with EG at the indicated concentrations in the presence of saturating concentrations of glutamate (100 μM). Substance-induced currents were given in relation to the maximal glycine-induced current (determined after superfusion with 100 μM glutamate and 100 μM glycine). Data are expressed as mean ± SEM (n = 4-6). (E), Oocytes expressing the NMDAR (NR1/NR2a) were superfused with glutamate, glycine, or EG at the indicated concentration alone or in the indicated combinations. Currents were determined as fraction of the current elicited by glycine and glutamate (100 μM each) alone. Data are expressed as mean ± SEM (n = 4-6).
The application of any of the aforementioned substances had no effect on currents in noninjected (wild-type) oocytes (data not shown). Taken together, these data show that EG acts only as a substrate for GlyT1 but has no agonistic or antagonistic function at the other major glycine-responsive proteins within the CNS.
3.2. Systemic application of EG efficiently ameliorates inflammatory hyperalgesia
To test whether the lidocaine metabolite EG can directly exert an antinociceptive effect on inflammatory pain in vivo, CFA was injected into the left hind paw of adult male C57BL/6J mice (n = 8 per group). Testing of mechanical sensitivity using von Frey filaments revealed a profound allodynia that developed in the treated paw 72 hours after injection of CFA, whereas mechanical sensitivity of the untreated paw was not altered. Subcutaneous bolus application of EG (200 mg/kg) or sarcosine (200 mg/kg) increased withdrawal thresholds observed in the CFA-treated paw within 1 hour after application, indicating a reduction of mechanical hyperalgesia. After a plateau of 4 hours, the apparent beneficial effect of EG and sarcosine slowly declined, and after 24 hours, the mechanical sensitivity was back to baseline levels (Fig. 4A). In contrast, mechanical sensitivity of the untreated contralateral hind paw did not change at any point during the experiment (Fig. 4B). The antihyperalgesic effect was fully reproducible when repeating the treatment after 48 hours (Fig. 4A). When repeating EG applications at 12-hour intervals for 3 consecutive days, however, no additional beneficial effects in the sense of a delayed therapeutic response were observed when evaluating 12 hours after injection (data not shown).
Figure 4.
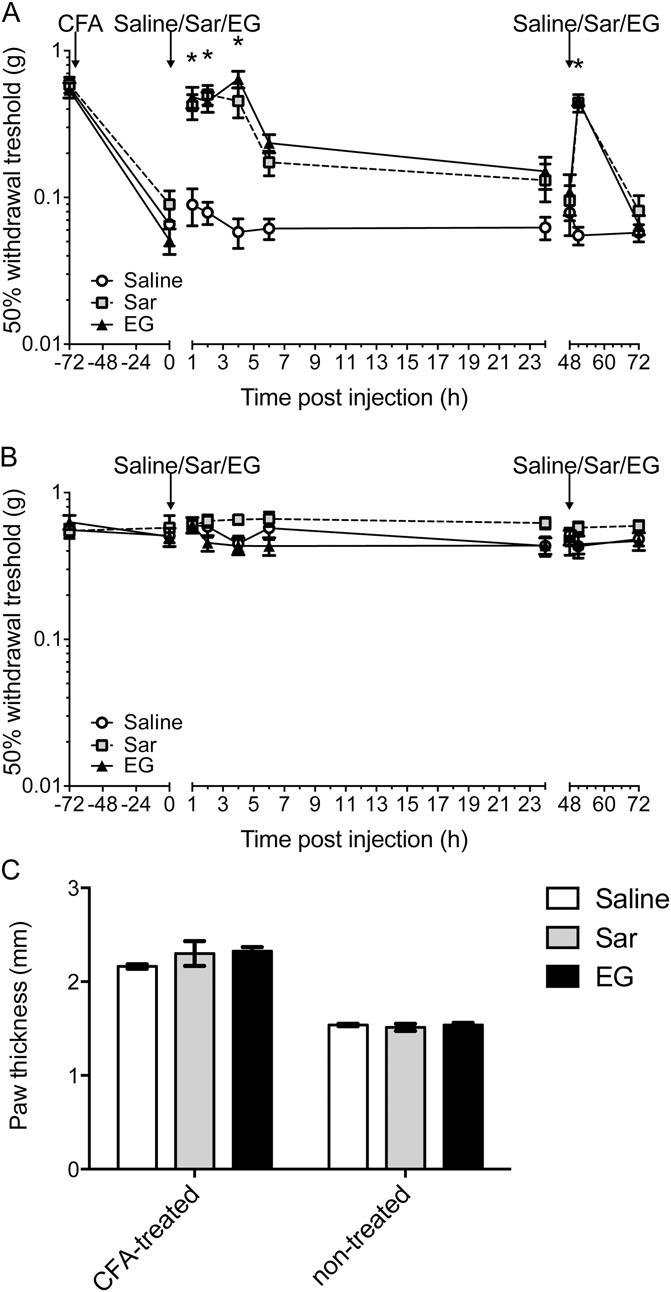
N-ethylglycine (EG) reduces nociceptive behavior in inflammatory pain. (A), Behavioral data in adult mice after induction of inflammatory pain by injection of complete Freund's adjuvant (5 μL) in the left hind paw expressed as 50% withdrawal threshold (in g). EG (200 mg/kg), the known GlyT1 substrate sarcosine (Sar, 200 mg/kg), or saline was applied subcutaneously after allodynia had developed 3 days after induction (marked with arrow). Assessment of mechanical allodynia was performed with von Frey hairs at indicated time points. Please note that the treatment was repeated at 48 hours after initial treatment (marked with arrow) to test whether the effects can be achieved again. Data are expressed as mean ± SEM; n = 8 per group; *P < 0.05 vs control group (saline); 2-way repeated-measures analysis of variance and Bonferroni post hoc test. (B), Corresponding data from complete Freund's adjuvant–untreated right hind paws. (C), Inflammatory edema was assessed by means of paw thickness (mm) 24 hours after first treatment with EG. Data are expressed as mean ± SEM; n = 8 per group; no significant differences were detected between treatment groups; 1-way repeated-measures analysis of variance and Bonferroni post hoc test.
To test whether this antihyperalgesic effect results from an anti-inflammatory effect of EG or sarcosine, the degree of inflammation-induced edema was determined on the basis of paw thickness. Here, thickness of the paw that received the CFA injection (left paw) was approximately 40% increased as compared with the untreated control side, confirming, in addition to the behavioral data, the successful induction of the inflammation. At 24 hours after treatment with sarcosine or EG, however, no change in the inflammation-induced edema was observed, suggesting that, at least in the current experimental setting, neither test drug shows any anti-inflammatory potential (Fig. 4C).
To determine the dose dependency of EG in this model of inflammatory pain, inflammation-induced hyperalgesia was induced by CFA injection in the left hind paw of the animals. After successful verification that a pronounced allodynia had developed, various concentrations of EG were injected subcutaneously and the withdrawal threshold after mechanical stimulation tested 2 hours after injection (n = 8 per group). Half-logarithmic plotting of the data revealed an EC50 of 98 mg/kg (Fig. 5A).
Figure 5.
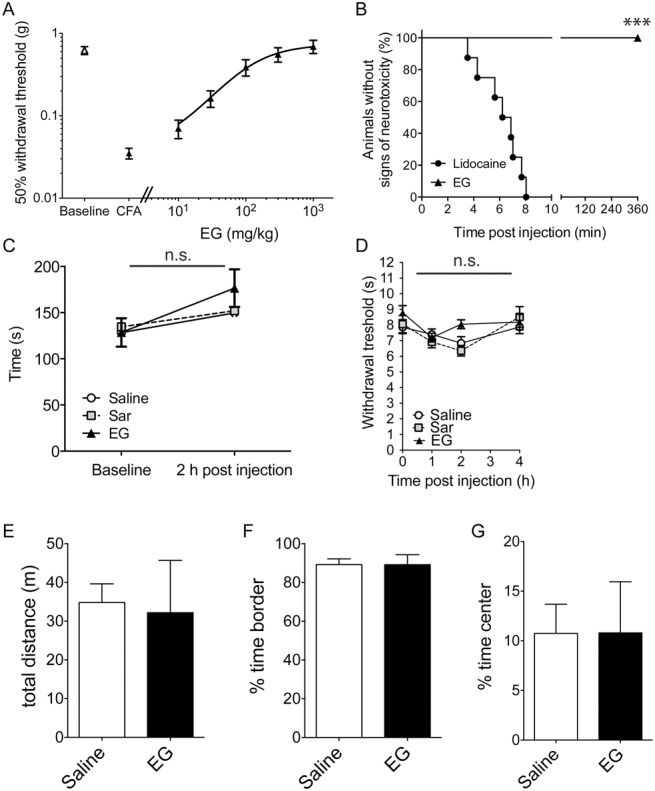
Dose–response relationship of N-ethylglycine (EG) for treatment of inflammatory pain and screening for secondary and acute analgesic effects. (A), Dose–response for the effect of EG on thermal hyperalgesia (withdrawal thresholds using the Hargreaves method) 3 days after the induction of inflammatory pain by complete Freund's adjuvant (5 μL) injection. Data are expressed as mean ± SEM; n = 8 per group. (B), High-dose treatment with EG does not induce adverse neurological side effects; time until occurrence of signs of adverse effects was recorded in adult naive mice after subcutaneous injection of lidocaine (40 mg/kg) or EG (400 mg/kg). Data are expressed as the fraction (in %) of symptom-free animals (no irregular movements or signs of distress); n = 8 per group; ***P < 0.001 between groups; Mantel–Cox log-rank test. (C), EG (200 mg/kg), the known GlyT1 substrate sarcosine (Sar, 200 mg/kg), or saline was applied subcutaneously in adult mice after allodynia had developed 3 days after induction. Motor coordination was tested using the rotarod performance test before and 2 hours after treatment with EG. Riding times (in seconds) were measured. Data are expressed as mean ± SEM; n = 8 per group; no significant differences were detected between treatment groups; 1-way repeated-measures analysis of variance and Bonferroni post hoc test. (D), Behavioral data in naive adult mice in the left hind paw expressed as paw withdrawal threshold (in seconds) after subcutaneous application of EG (200 mg/kg), sarcosine (200 mg/kg), or saline. Assessment of analgesic effects was performed using the Hargreaves method at indicated time points. Data are expressed as mean ± SEM; n = 8 per group; no significant differences vs control group (saline) were detected; 2-way repeated-measures analysis of variance and Bonferroni post hoc test. (E-G), Open-field experiments were performed 2 hours after receiving a single dose of EG (200 mg/kg, subcutaneously). Total distance (E) and the time spent in the periphery (F) or the central quadrant (G) were determined. Data are expressed as mean ± SEM, n = 6 to 7 per group; no significant differences vs control group (saline) were detected (Student t test, P > 0.6).
To test for possible side effects of EG that might result from a general increase of the glycinergic tone or from other routes of action, eg, acute toxic effects, the general activity of mice after treatment with 400 mg/kg EG was analyzed and compared with mice injected with 40 mg/kg of the parent substance lidocaine (n = 8 per group). Although in all lidocaine-treated animals, severe adverse effects like irregular movements or (respiratory) distress were seen within the first 8 minutes after injection, such effects were not observed in any of the mice treated with the lidocaine metabolite EG (Fig. 5B). To determine whether EG treatment causes subtle adverse effects, that result from, for example, a general increase in the glycinergic tone, the motor performance of mice was analyzed with rotarod assay before and 2 hours after EG treatment and compared with that of sarcosine- or saline-treated animals (Fig. 5C; n = 8 per group). Both, before and after EG treatment, the performance of the animals in all 3 groups was indistinguishable, suggesting that short-term treatment with EG does not produce major side effects in mice. Consistent with these findings, no change in the paw withdrawal threshold to heat exposure using the Hargreaves test was observed after application of EG to naive animals at any time point analyzed (Fig. 5D; n = 8 per group), indicating that responses to acute painful stimuli were not altered after EG treatment. The effect of EG application on general activity and explorative behavior was subsequently determined in an open-field experiment. Here, neither the general activity of EG-treated mice (200 mg/kg, subcutaneously) as determined by the total distance traveled nor their explorative behavior as determined by the regional preference for the central quadrant was distinguishable from that of mice that received saline injections only (Fig. 5E-G; n = 6-7 per group). Taken together, these data demonstrate that systemic application of EG, similar to sarcosine, efficiently ameliorates inflammatory hyperalgesia, whereas acute pain sensitivity is not affected. Moreover, these data reveal no major additional effects of acute EG treatment on the general activity of mice.
3.3. Systemic application of EG reduces allodynia and hyperalgesia in mice with neuropathic pain
Subsequently, we analyzed whether EG has an antinociceptive effect only in inflammatory pain or whether this phenomenon can also be observed in an animal model of neuropathic pain. To induce neuropathy, we made use of the CCI model of the sciatic nerve. The establishment of CCI-induced allodynia and hyperalgesia was determined by mechanical and thermal testing of withdrawal thresholds. All animals analyzed (n = 8 per group) developed pronounced mechanical allodynia and thermal hyperalgesia within 72 hours after the operation at the ipsilateral side (Fig. 6), whereas no changes in pain sensitivity were observed at the contralateral side (data not shown). Upon subcutaneous injection of EG, a significant reduction of both CCI-induced mechanical (Fig. 6A) and thermal hyperalgesia (Fig. 6B) was observed within 2 hours after injection. Similar to our findings for inflammatory pain, the observed reduction in hyperalgesia was only transient with a maximal effect observed after 2 to 4 hours. Pain sensitivity was indistinguishable from that of saline-treated mice at 24 hours after injection (Fig. 6).
Figure 6.
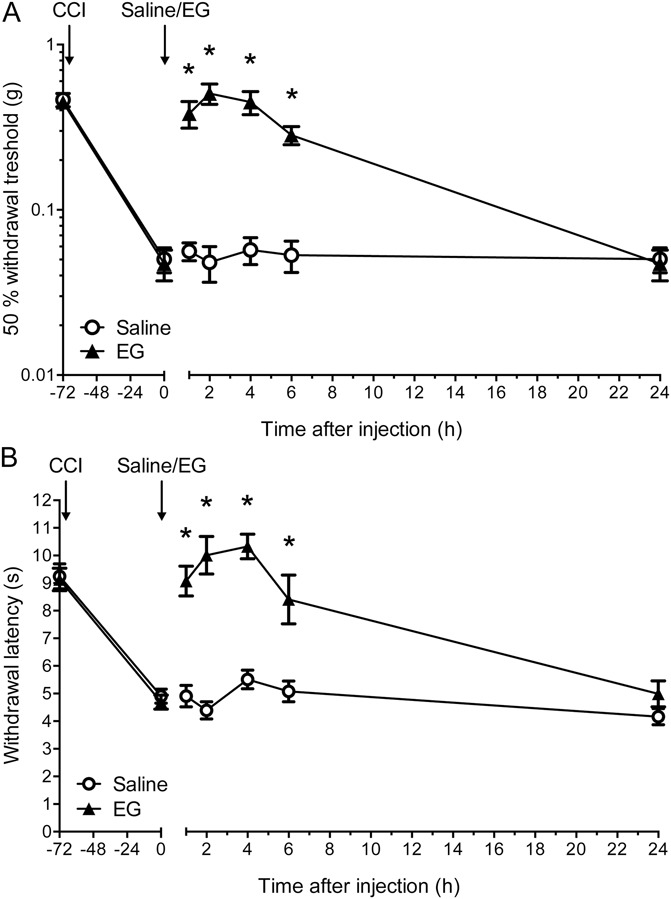
N-ethylglycine (EG) reduces nociceptive behavior in neuropathic pain. (A), Behavioral data in adult mice after induction of neuropathic pain by chronic constriction injury in the left hind paw expressed as 50% withdrawal threshold (in g). EG (200 mg/kg) or saline was applied subcutaneously after mechanical allodynia had developed 3 days after chronic constriction injury. Assessment of mechanical allodynia was performed with von Frey filaments at indicated time points. (B), Thermal hyperalgesia was assessed by means of paw withdrawal thresholds (in seconds) in response to a heat source applied to the left hind paw (Hargreaves method). Data are expressed as mean ± SEM; n = 8 per group; *P < 0.05 vs control group (saline); 2-way repeated-measures analysis of variance and Bonferroni post hoc test.
3.4. Systemic application of EG leads to an increase in serum and cerebrospinal fluid glycine concentrations
Based on its action on GlyT1 as an artificial substrate, we hypothesized that EG might compete with glycine for the uptake into the surrounding tissue of glycinergic synapses and thus cause an increase in the extracellular glycine concentration at least within the CNS. Therefore, we investigated both the concentration of EG and glycine in the serum and CSF in EG-treated animals. As the obtained volume, especially of CSF, was not sufficient for HPLC-based analysis when isolated from mice, we performed these experiments in rats (n = 4 per group). In the serum of untreated rats, a basal glycine concentration of 207 ± 37 μM was observed. Interestingly, EG also was already found in low concentrations (43 ± 6 μM). After subcutaneous injection of EG, a rapid increase in the EG serum concentration was observed that peaked with 384 ± 34 μM only 1 hour after injection. After a 1-hour plateau, a steady decline of the serum EG concentration was observed, and after 24 hours, the serum concentration of EG reached the basal level. During the whole observation period, however, there was no significant change in the serum glycine concentration (Fig. 7A). Based on these measurements, we calculated a volume of distribution for EG of approximately 1.2 L or 4.8 L/kg. Thus, a theoretical dose of 98 mg/kg, estimated as EC50 in our behavioral dose–response experiments (Fig. 5A), would result in a serum concentration of 20.4 mg/L (198 μM) 2 hours after injection.
Figure 7.
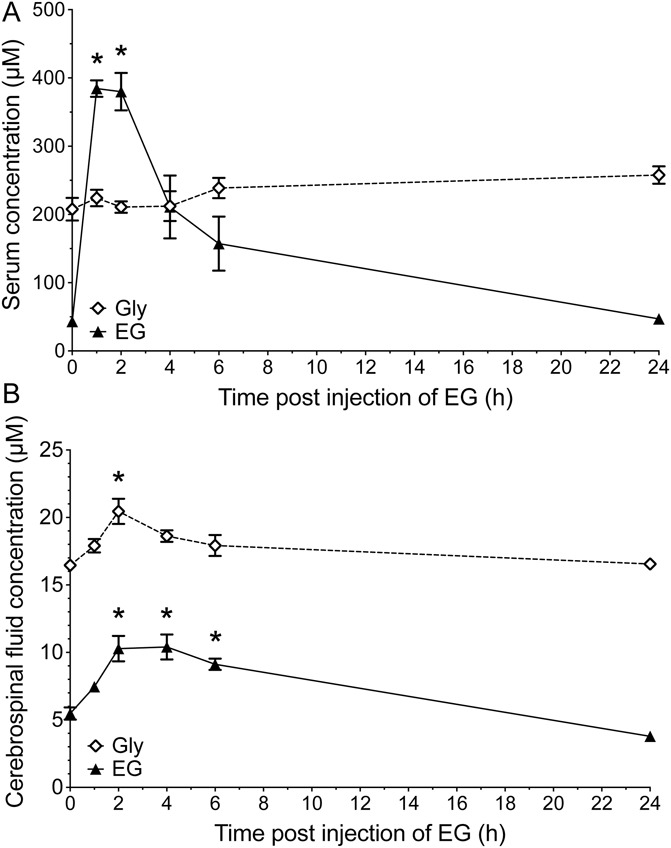
Subcutaneous injection of N-ethylglycine (EG) in rats leads to an increased EG concentration in blood serum and to increased EG and glycine (Gly) concentrations in cerebrospinal fluid. Concentrations of EG and glycine were determined using high-pressure liquid chromatography in (A) blood serum and (B) cerebrospinal fluid samples from adult rats before and at indicated time points after subcutaneous injection of EG (200 mg/kg). Data are expressed as mean ± SEM; n = 4 per group; *P < 0.05 vs before injection of EG; 1-way analysis of variance and Bonferroni post hoc test.
In contrast to our findings on the serum concentrations of EG and glycine, much lower basal concentrations were found in CSF samples of untreated animals (16.5 ± 0.5 μM and 5.5 ± 0.9 μM for glycine and EG, respectively). In samples from animals that received EG injections, the concentration of EG almost doubled within 2 hours after injection, followed by a slow decline. Corroborating our serum concentration findings, the EG CSF levels of rats that received EG injections 24 hours before testing were indistinguishable from those of untreated control animals. Analysis of the CSF glycine concentration revealed that EG injection resulted in a 25% increase in extracellular glycine concentration (to 20.5 ± 1.9 μM). The maximal increase was observed 2 hours after injection of EG. Subsequently, the CSF glycine concentration slowly declined and in rats that received the EG injection 24 hours before CSF sampling, glycine concentrations were comparable with those of untreated animals (Fig. 7B).
Taken together, these findings demonstrate that after systemic application, EG reaches the CSF and has an effect on the extracellular glycine concentration that is consistent with a transient reduction of the GlyT1-mediated glycine transport capacity.
3.5. EG affects the pain processing circuitry by inhibiting the inflammation-induced hyperexcitability of dorsal horn neurons
To investigate the effects of EG on the evoked responses of dorsal horn neurons, we determined the number of action potentials elicited by electrical stimulation in recordings from WDR neurons of the deep laminae in the rat spinal cord dorsal horn. After recording baseline responses after stimulation of the corresponding receptive field at the glabrous surface of the hind paw, an inflammatory response followed by neuronal hyperexcitability was induced by injection of 1% carrageenan solution into the hind paw. Individual action potentials were attributed to the respective fiber types according to their latency (Aβ fibers: 0-20 milliseconds, Aδ fibers: 20-90 milliseconds, and C fibers: 90-350 milliseconds). Here, evaluation of electrical stimulus–evoked responses revealed that the majority of detected action potentials had a stimulus–response latency that corresponded to C fibers (90-350 milliseconds), while neuronal input from Aβ and Aδ fibers was less abundant (see Fig. 8A and Supplemental Digital Content 1 [available online at http://links.lww.com/PAIN/A93], which contains examples of responses of single neurons to electrical stimulation at baseline, after carrageenan injection and subsequent EG treatment). After carrageenan injection, a significant increase in the firing rate in response to Aβ-, Aδ-, and C-fiber activity was observed after 3 hours. Subsequently, EG was applied directly onto the dorsal surface of the spinal cord (n = 4 per group). Further recordings 30 minutes after application revealed a dose-dependent reduction of detected action potentials in response to electrical stimulation reaching complete suppression of neuronal hyperexcitability at concentrations higher than 100 μM (Fig. 8B). As individual neuron responses varied considerably, normalization to maximal responses (3 hours after carrageenan injection) was performed. To determine whether this dose-dependent reduction in the firing rate of dorsal horn neurons was also observed in reaction to physiological stimuli, the respective receptive field at the glabrous surface of the hind paw was stimulated thermally (Fig. 8C; constant water jet 40°C or 48°C) or mechanically (Fig. 8D; brush, 8 or 60g hair). Here, consistent with our findings after electrical stimulation, we observed a strong facilitation of the stimulus-induced firing rate after carrageenan with innocuous stimuli being affected more strongly. After treatment with increasing concentrations of EG, the carrageenan-induced hyperexcitability was dose-dependently reduced. Here, also a stronger effect was observed, when normally innocuous stimuli (8g hair, 40°C water jet) were applied. These findings demonstrate that EG affects the pain processing circuitry by inhibiting the inflammation-induced hyperexcitability of dorsal horn neurons, thus confirming the antihyperalgesic action of EG independently of behavioral approaches.
Figure 8.
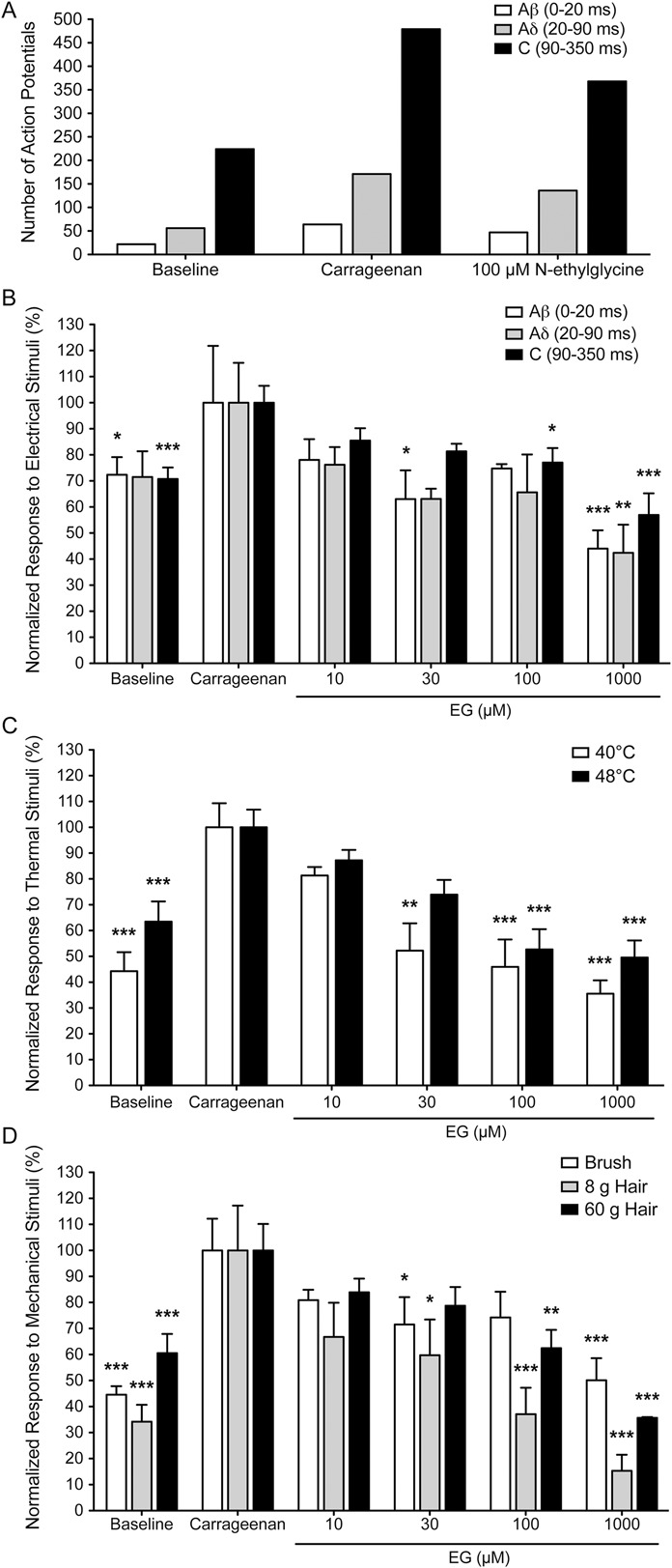
N-ethylglycine (EG) reduces wide-dynamic-range neuron response in inflammatory pain. Action potentials of single neurons in response to electrical stimulation (A/B), thermal stimulation (C) or mechanical stimulation (D) of the receptive field were recorded. Responses evoked by Aβ, Aδ, and C fibers after electrical stimuli were superimposed and separated according to latency (0-20, 20-90, and 90-350 milliseconds, respectively). Representative data from one individual neuron recording (A) demonstrating that the majority of action potentials are elicited by C-fiber input after electrical stimulation. (B-D), Relative number of action potentials elicited by the indicated fiber inputs, with the respective number recorded after carrageenan treatment set to 100%. Thermal stimuli were applied by means of a constant water jet at 40°C and 48°C. Mechanical stimuli were applied with a fine brush or von Frey hairs (8 and 60g force). Inflammatory pain was induced in adult rats by injection of carrageenan (1%, 20 μL) at the left hind paw. EG was applied intrathecally after hyperexcitability had developed 3 hours after carrageenan application. Effect of EG on neuronal response to stimuli was tested 30 minutes after application. (B-D), Data are expressed as mean ± SEM; n = 8 for controls and n = 4 for each concentration of EG; *P < 0.05, **P < 0.01 and ***P < 0.001 vs neuronal response after carrageenan application; 1-way repeated-measures analysis of variance and Bonferroni post hoc test.
4. Discussion
Treatment of chronic pain is still one of the most challenging clinical endeavors. Here, classical analgesics like opioids and nonsteroidal antirheumatics usually have limited efficacy. Despite continuing research, no substantial improvement in therapeutic outcome has emerged.4,29
For more than 50 years, it has been known that systemic long-term application of lidocaine results in a long-lasting general antinociceptive effect,33 although the mechanism mediating these effects remained up to now enigmatic. The relative late onset of the therapeutic benefit, as well as the extremely low levels of the plasma lidocaine concentration required, argue against a direct function of lidocaine on the principal target of this class of substances ie, voltage-gated sodium channels.47 Previous studies have demonstrated that in addition to the general analgesic effect, long-term treatment with lidocaine has an anti-inflammatory potential31 through an up-to-now unknown mechanism, which might contribute to the long-lasting beneficial effects. In this study, we provide evidence that the antinociceptive effect seen after long-term treatment with lidocaine can be mimicked by systemic application of the lidocaine metabolite EG, which acts as an artificial substrate specifically on glycine transporters of the GlyT1 subtype, suggesting a novel additional route of action for systemically applied lidocaine. Systemic application of EG results in a significant elevation of the extracellular glycine concentration within the CSF and thereby inhibition of inflammation-induced hyperexcitability of WDR neurons in the deep lamina of the dorsal horn.
In the mature nervous system, the extracellular glycine concentration is synergistically regulated by GlyT1 and GlyT215 with GlyT2 being essential for replenishing the presynaptic glycine pool. Loss of the GlyT2 activity results in a pronounced disinhibition of motor neurons and consequently leads to severe neuromotor deficits in both rodents and humans.16,20,38 In contrast, the major function of glial GlyT1 is the maintenance of low extracellular glycine concentrations.19 Inactivation of GlyT1 expression in glial cells of neonatal animals results in severe hypotonia, respiratory depression and premature death. At later developmental stages, however, a loss of glial GlyT1 seems to be well tolerated.17 Additionally, neuronal expression of GlyT1 has been shown to be an important modulator of glutamatergic neurotransmission through glutamate receptors of the NMDAR subtype.50 Therefore, GlyT1 inhibitors are currently in phase 3 trials for the treatments of diseases associated with NMDAR dysfunction like schizophrenia (see, eg, ClinicalTrials.gov Identifier: NCT01192867).
There is an increasing body of evidence, in addition to this study, that suggests both inhibition of GlyT1 and GlyT2 are suitable strategies for the treatment of chronic pain conditions including inflammatory and neuropathic pain.25 The interpretation of these findings has in many cases been hampered by the fact that the currently available inhibitors are poorly characterized and/or are proven to cross-react with other components of glycine-dependent neurotransmission. For example, the GlyT2 inhibitor ALX1393 also inhibits GlyT1 at low micromolar concentrations,34 while the GlyT1 substrate sarcosine was shown to act also as a partial agonist on GlyR53 and NMDAR.52 In contrast, we found EG to be specific for GlyT1 without any effects on GlyT2, GlyRs, or NMDARs in vitro. This high specificity for GlyT1 might facilitate better predictability of the expected effects in vivo and therefore prevent undesired secondary effects.
Our behavioral experiments show that systemic EG is dose-dependently antinociceptive in chronic but not acute pain. Interestingly, EG diminished pain states induced by either inflammation or CCI in the same manner, although the spinal cord pathophysiology has been shown to be different. In contrast to neuropathic pain,26,28 inflammation-induced pain requires an increase in the production of the pronociceptive prostaglandin PGE2 in the spinal cord, which diminishes glycinergic inhibition in the dorsal horn by phosphorylation of α3 subunit containing GlyR.1,24 Thus, it appears that the reduction of the glycine uptake capacity of GlyT1 might constitute a general treatment strategy for diseases associated with a diminished inhibitory neurotransmission in caudal regions of the CNS like chronic pain with central sensitization.
The time course of the antinociceptive effect elicited by EG was similar in CFA- and CCI-induced pain; withdrawal thresholds were already significantly increased within 1 hour after application, and this increase lasted for 4 to 6 hours. Thus, onset of antinociceptive effects was significantly faster than what was observed with GlyT1 inhibitors previously.5,35 Whether this discrepancy in the kinetics of action results from differences in the pharmacokinetics or from a different mechanism (eg, inhibitor vs artificial substrate) is not clear at present.
Measurements of serum concentrations revealed that after subcutaneous injection, serum EG concentrations increased quickly and remained elevated for 2 hours at levels slightly below 400 μM while serum glycine concentration remained unchanged. These findings support the idea that EG specifically acts on GlyT1 because the plasma concentration of glycine has been suggested to be controlled by other (low affinity) transporters such as System A.32 Interestingly, EG concentrations found after subcutaneous EG injection were similar to those reported in the plasma of patients receiving continuous systemic or epidural applications of lidocaine (range: 3-65 mg/L, ie, 30-650 μM).39 By measuring the spinal fluid EG and glycine levels, we could show that EG crosses the blood–brain barrier and leads to a short-term increase in spinal fluid glycine concentration. This further supports the hypothesis that EG functions through a reduction of the glycine transport capacity of GlyT1. It has to be noted, however, that the concentration measurements performed here only allow for an evaluation of extrasynaptic glycine levels that may differ from those at synaptic sites. Taken together, these data suggest that after continuous application of lidocaine, EG can accumulate and reach plasma concentrations that are sufficient for a significant glycine transport inhibition in vitro and antinociceptive effects in vivo. These results strongly suggest that the antinociceptive effects of systemic lidocaine are caused by EG accumulation that results in elevated glycine levels at synapses of spinal inhibitory circuits.
In previous studies, the beneficial effect of GlyT inhibitors was observed only when a fraction of the total GlyT activity was blocked. Strong inhibition of GlyT1 activity caused severe adverse effects like pronociceptive effects, neuromotor disturbances, or respiratory depression.27,35,37 Such negative consequences were not observed in animals treated with even twice the antinociceptive dose of EG. This possibly could be explained by EG's only moderate affinity for GlyT1 and the fact that EG, in contrast to previously evaluated substances, constitutes an artificial GlyT1 substrate and does not act as an inhibitor of GlyT1.
Interestingly, previous studies have shown that long-term blockade of GlyT1 by the irreversible GlyT1 inhibitor ALX5407 appears not only to ameliorate pathological pain symptoms but also to reverse some of the neuroplastic changes involved in the manifestation of neuropathic pain such as an increase of NMDAR subunit expression NR1 within the spinal cord.5 This might result from the previously described internalization of NMDAR after sustained saturation of the NMDAR glycine binding.36 These effects might facilitate the beneficial effects of GlyT1 in the treatment of pathological pain and explain some of the long-lasting effects of even a single dose application of GlyT1 inhibitors.35 In this study, using the GlyT1 substrate EG, no long-lasting antihyperalgesic effect was observed, most likely due to the relative short treatment period and the fact that here we used not a GlyT1 inhibitor but an artificial substrate. Whether injection of higher doses or continuous application of EG might result in sustained or long-lasting effects on nociception remains to be elucidated in further experiments.
Using in vivo extracellular recordings at dorsal horn neurons in the deep laminae of the rat lumbar spinal cord, we identified strong inhibitory effects of EG on inflammation-induced neuronal hyperexcitability in WDR neurons. Here, only intrathecal concentrations of more than 100 μM EG led to complete suppression of neuronal hyperexcitability, although significant effects were observed using 30 μM EG. This finding most likely results from inhibition of glycine uptake capacity and the subsequent accumulation of extracellular glycine. Support for the hypothesis that this is the most likely mode of action also comes from our HPLC experiments, which show that CSF concentrations of up to 20 μM EG can be reached by systemic application of EG at a dose that has been shown to be effective in behavioral experiments (200 mg/kg). However, it has to be considered that all in vivo electrophysiological experiments were performed under general anesthesia and thus a direct correlation with behavioral data is difficult.
In conclusion, our findings suggest that EG, as a substrate of GlyT1, mediates at least in part the antinociceptive effect of systemically applied lidocaine. Furthermore, our results demonstrate that competitive inhibition of GlyT1 by application of GlyT1 substrates like EG is a promising strategy for the therapy of chronic pain conditions. Future investigations are needed to determine the precise molecular mechanisms behind the antinociceptive action, eg, to what extent altered spinal and supraspinal glycinergic and glutamatergic neurotransmission contributes to this phenomenon or whether low-affinity glycine transport by systems A, ASC, etc., is sensitive to EG. Finally, whether these effects can be confirmed in clinical studies remains to be elucidated.
Conflict of interest statement
The authors have no conflicts of interest to declare.
This work was supported by the German Research Foundation (DFG We4860/1-1 to R.W. and EU110/3-1 to V.E.), the Wellcome Trust funded London Pain Consortium (to J.N.W.), and the Interdisciplinary Center for Clinical Research (IZKF) of the University Hospital Erlangen (TP E15 to V.E.).
Supplementary Material
Acknowledgements
The authors would like to gratefully acknowledge Dr Carolyn Hyde (Bioanalysis Manager, University College London, United Kingdom) for technical guidance in high-pressure liquid chromatography and Dr Christoph Korbmacher (Institute for Physiology, University of Erlangen-Nürnberg) for providing Xenopus laevis oocytes. The excellent technical assistance of Marina Wenzel (Institute of Biochemistry, University of Erlangen-Nürnberg) is gratefully acknowledged.
Appendix A. Supplemental Digital Content
Supplemental Digital Content associated with this article can be found online at http://links.lww.com/PAIN/A93.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painjournalonline.com).
H. Hermanns and V. Eulenburg share senior authorship.
References
- [1].Ahmadi S, Lippross S, Neuhuber WL, Zeilhofer HU. PGE(2) selectively blocks inhibitory glycinergic neurotransmission onto rat superficial dorsal horn neurons. Nat Neurosci 2002;5:34–40. [DOI] [PubMed] [Google Scholar]
- [2].Attal N, Cruccu G, Baron R, Haanpaa M, Hansson P, Jensen TS, Nurmikko T; European Federation of Neurological S. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur J Neurol 2010;17:1113–e88. [DOI] [PubMed] [Google Scholar]
- [3].Backonja MM. Neuropathic pain therapy: from bench to bedside. Semin Neurol 2012;32:264–8. [DOI] [PubMed] [Google Scholar]
- [4].Baron R, Binder A, Wasner G. Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol 2010;9:807–19. [DOI] [PubMed] [Google Scholar]
- [5].Barthel F, Urban A, Schlosser L, Eulenburg V, Werdehausen R, Brandenburger T, Aragon C, Bauer I, Hermanns H. Long-term application of glycine transporter inhibitors acts antineuropathic and modulates spinal N-methyl-D-aspartate receptor subunit NR-1 expression in rats. Anesthesiology 2014;121:160–9. [DOI] [PubMed] [Google Scholar]
- [6].Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. PAIN 1988;33:87–107. [DOI] [PubMed] [Google Scholar]
- [7].Breivik H, Collett B, Ventafridda V, Cohen R, Gallacher D. Survey of chronic pain in Europe: prevalence, impact on daily life, and treatment. Eur J Pain 2006;10:287–333. [DOI] [PubMed] [Google Scholar]
- [8].Centeno MV, Mutso A, Millecamps M, Apkarian AV. Prefrontal cortex and spinal cord mediated anti-neuropathy and analgesia induced by sarcosine, a glycine-T1 transporter inhibitor. PAIN 2009;145:176–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Challapalli V, Tremont-Lukats IW, McNicol ED, Lau J, Carr DB. Systemic administration of local anesthetic agents to relieve neuropathic pain. Cochrane Database Syst Rev 2005;4:CD003345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 1994;53:55–63. [DOI] [PubMed] [Google Scholar]
- [11].Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci 2009;32:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Devor M, Wall PD, Catalan N. Systemic lidocaine silences ectopic neuroma and DRG discharge without blocking nerve conduction. PAIN 1992;48:261–8. [DOI] [PubMed] [Google Scholar]
- [13].Dickenson AH, Sullivan AF. Electrophysiological studies on the effects of intrathecal morphine on nociceptive neurones in the rat dorsal horn. PAIN 1986;24:211–22. [DOI] [PubMed] [Google Scholar]
- [14].Dworkin RH, O'Connor AB, Audette J, Baron R, Gourlay GK, Haanpaa ML, Kent JL, Krane EJ, Lebel AA, Levy RM, Mackey SC, Mayer J, Miaskowski C, Raja SN, Rice AS, Schmader KE, Stacey B, Stanos S, Treede RD, Turk DC, Walco GA, Wells CD. Recommendations for the pharmacological management of neuropathic pain: an overview and literature update. Mayo Clin Proc 2010;85(3 suppl):S3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Eulenburg V, Armsen W, Betz H, Gomeza J. Glycine transporters: essential regulators of neurotransmission. Trends Biochem Sci 2005;30:325–33. [DOI] [PubMed] [Google Scholar]
- [16].Eulenburg V, Becker K, Gomeza J, Schmitt B, Becker CM, Betz H. Mutations within the human GLYT2 (SLC6A5) gene associated with hyperekplexia. Biochem Biophys Res Commun 2006;348:400–5. [DOI] [PubMed] [Google Scholar]
- [17].Eulenburg V, Retiounskaia M, Papadopoulos T, Gomeza J, Betz H. Glial glycine transporter 1 function is essential for early postnatal survival but dispensable in adult mice. Glia 2010;58:1066–73. [DOI] [PubMed] [Google Scholar]
- [18].Finnerup NB, Sindrup SH, Jensen TS. The evidence for pharmacological treatment of neuropathic pain. PAIN 2010;150:573–81. [DOI] [PubMed] [Google Scholar]
- [19].Gomeza J, Hulsmann S, Ohno K, Eulenburg V, Szoke K, Richter D, Betz H. Inactivation of the glycine transporter 1 gene discloses vital role of glial glycine uptake in glycinergic inhibition. Neuron 2003;40:785–96. [DOI] [PubMed] [Google Scholar]
- [20].Gomeza J, Ohno K, Hulsmann S, Armsen W, Eulenburg V, Richter DW, Laube B, Betz H. Deletion of the mouse glycine transporter 2 results in a hyperekplexia phenotype and postnatal lethality. Neuron 2003;40:797–806. [DOI] [PubMed] [Google Scholar]
- [21].Grudzinska J, Schemm R, Haeger S, Nicke A, Schmalzing G, Betz H, Laube B. The beta subunit determines the ligand binding properties of synaptic glycine receptors. Neuron 2005;45:727–39. [DOI] [PubMed] [Google Scholar]
- [22].Gureje O, Von Korff M, Simon GE, Gater R. Persistent pain and well-being: a World Health Organization study in primary care. JAMA 1998;280:147–51. [DOI] [PubMed] [Google Scholar]
- [23].Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. PAIN 1988;32:77–88. [DOI] [PubMed] [Google Scholar]
- [24].Harvey RJ, Depner UB, Wassle H, Ahmadi S, Heindl C, Reinold H, Smart TG, Harvey K, Schutz B, Abo-Salem OM, Zimmer A, Poisbeau P, Welzl H, Wolfer DP, Betz H, Zeilhofer HU, Muller U. GlyR alpha3: an essential target for spinal PGE2-mediated inflammatory pain sensitization. Science 2004;304:884–7. [DOI] [PubMed] [Google Scholar]
- [25].Harvey RJ, Yee BK. Glycine transporters as novel therapeutic targets in schizophrenia, alcohol dependence and pain. Nat Rev Drug Discov 2013;12:866–85. [DOI] [PubMed] [Google Scholar]
- [26].Harvey VL, Caley A, Muller UC, Harvey RJ, Dickenson AH. A selective role for alpha3 subunit glycine receptors in inflammatory pain. Front Mol Neurosci 2009;2:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hermanns H, Muth-Selbach U, Williams R, Krug S, Lipfert P, Werdehausen R, Braun S, Bauer I. Differential effects of spinally applied glycine transporter inhibitors on nociception in a rat model of neuropathic pain. Neurosci Lett 2008;445:214–19. [DOI] [PubMed] [Google Scholar]
- [28].Hosl K, Reinold H, Harvey RJ, Muller U, Narumiya S, Zeilhofer HU. Spinal prostaglandin E receptors of the EP2 subtype and the glycine receptor alpha3 subunit, which mediate central inflammatory hyperalgesia, do not contribute to pain after peripheral nerve injury or formalin injection. PAIN 2006;126:46–53. [DOI] [PubMed] [Google Scholar]
- [29].Kissin I. The development of new analgesics over the past 50 years: a lack of real breakthrough drugs. Anesth Analg 2010;110:780–9. [DOI] [PubMed] [Google Scholar]
- [30].Knabl J, Witschi R, Hosl K, Reinold H, Zeilhofer UB, Ahmadi S, Brockhaus J, Sergejeva M, Hess A, Brune K, Fritschy JM, Rudolph U, Mohler H, Zeilhofer HU. Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature 2008;451:330–4. [DOI] [PubMed] [Google Scholar]
- [31].Lirk P, Picardi S, Hollmann MW. Local anaesthetics: 10 essentials. Eur J Anaesthesiol 2014;31:575–85. [DOI] [PubMed] [Google Scholar]
- [32].Mackenzie B, Erickson JD. Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Pflugers Arch 2004;447:784–95. [DOI] [PubMed] [Google Scholar]
- [33].Mao J, Chen LL. Systemic lidocaine for neuropathic pain relief. PAIN 2000;87:7–17. [DOI] [PubMed] [Google Scholar]
- [34].Mingorance-Le Meur A, Ghisdal P, Mullier B, De Ron P, Downey P, Van Der Perren C, Declercq V, Cornelis S, Famelart M, Van Asperen J, Jnoff E, Courade JP. Reversible inhibition of the glycine transporter GlyT2 circumvents acute toxicity while preserving efficacy in the treatment of pain. Br J Pharmacol 2013;170:1053–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Morita K, Motoyama N, Kitayama T, Morioka N, Kifune K, Dohi T. Spinal antiallodynia action of glycine transporter inhibitors in neuropathic pain models in mice. J Pharmacol Exp Ther 2008;326:633–45. [DOI] [PubMed] [Google Scholar]
- [36].Nong Y, Huang YQ, Ju W, Kalia LV, Ahmadian G, Wang YT, Salter MW. Glycine binding primes NMDA receptor internalization. Nature 2003;422:302–7. [DOI] [PubMed] [Google Scholar]
- [37].Perry KW, Falcone JF, Fell MJ, Ryder JW, Yu H, Love PL, Katner J, Gordon KD, Wade MR, Man T, Nomikos GG, Phebus LA, Cauvin AJ, Johnson KW, Jones CK, Hoffmann BJ, Sandusky GE, Walter MW, Porter WJ, Yang L, Merchant KM, Shannon HE, Svensson KA. Neurochemical and behavioral profiling of the selective GlyT1 inhibitors ALX5407 and LY2365109 indicate a preferential action in caudal vs. cortical brain areas. Neuropharmacology 2008;55:743–54. [DOI] [PubMed] [Google Scholar]
- [38].Rees MI, Harvey K, Pearce BR, Chung SK, Duguid IC, Thomas P, Beatty S, Graham GE, Armstrong L, Shiang R, Abbott KJ, Zuberi SM, Stephenson JB, Owen MJ, Tijssen MA, van den Maagdenberg AM, Smart TG, Supplisson S, Harvey RJ. Mutations in the gene encoding GlyT2 (SLC6A5) define a presynaptic component of human startle disease. Nat Genet 2006;38:801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Roberts RT, Alexander NM, Kelner MJ. Definitive liquid-chromatographic demonstration that N-ethylglycine is the metabolite of lidocaine that interferes in the Kodak sarcosine oxidase-coupled method for creatinine. Clin Chem 1988;34:2569–72. [PubMed] [Google Scholar]
- [40].Sharma AK, Schultze AE, Cooper DM, Reams RY, Jordan WH, Snyder PW. Development of a percutaneous cerebrospinal fluid collection technique in F-344 rats and evaluation of cell counts and total protein concentrations. Toxicol Pathol 2006;34:393–5. [DOI] [PubMed] [Google Scholar]
- [41].Stein C, Millan MJ, Herz A. Unilateral inflammation of the hindpaw in rats as a model of prolonged noxious stimulation: alterations in behavior and nociceptive thresholds. Pharmacol Biochem Behav 1988;31:445–51. [DOI] [PubMed] [Google Scholar]
- [42].Tanabe M, Takasu K, Yamaguchi S, Kodama D, Ono H. Glycine transporter inhibitors as a potential therapeutic strategy for chronic pain with memory impairment. Anesthesiology 2008;108:929–37. [DOI] [PubMed] [Google Scholar]
- [43].Tanelian DL, Brose WG. Neuropathic pain can be relieved by drugs that are use-dependent sodium channel blockers: lidocaine, carbamazepine, and mexiletine. Anesthesiology 1991;74:949–51. [DOI] [PubMed] [Google Scholar]
- [44].Todd AJ. Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci 2010;11:823–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Turk DC, Wilson HD, Cahana A. Treatment of chronic non-cancer pain. Lancet 2011;377:2226–35. [DOI] [PubMed] [Google Scholar]
- [46].Werdehausen R, Kremer D, Brandenburger T, Schlosser L, Jadasz J, Kury P, Bauer I, Aragon C, Eulenburg V, Hermanns H. Lidocaine metabolites inhibit glycine transporter 1: a novel mechanism for the analgesic action of systemic lidocaine? Anesthesiology 2012;116:147–58. [DOI] [PubMed] [Google Scholar]
- [47].Xiao WH, Bennett GJ. C-fiber spontaneous discharge evoked by chronic inflammation is suppressed by a long-term infusion of lidocaine yielding nanogram per milliliter plasma levels. PAIN 2008;137:218–28. [DOI] [PubMed] [Google Scholar]
- [48].Xiong W, Cui T, Cheng K, Yang F, Chen SR, Willenbring D, Guan Y, Pan HL, Ren K, Xu Y, Zhang L. Cannabinoids suppress inflammatory and neuropathic pain by targeting α3 glycine receptors. J Exp Med 2012;209:1121–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yanagidate F, Strichartz GR. Local anesthetics. Handb Exp Pharmacol 2007;177:95–127. [DOI] [PubMed] [Google Scholar]
- [50].Yee BK, Balic E, Singer P, Schwerdel C, Grampp T, Gabernet L, Knuesel I, Benke D, Feldon J, Mohler H, Boison D. Disruption of glycine transporter 1 restricted to forebrain neurons is associated with a procognitive and antipsychotic phenotypic profile. J Neurosci 2006;26:3169–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zeilhofer HU, Benke D, Yevenes GE. Chronic pain states: pharmacological strategies to restore diminished inhibitory spinal pain control. Annu Rev Pharmacol Toxicol 2012;52:111–33. [DOI] [PubMed] [Google Scholar]
- [52].Zhang HX, Hyrc K, Thio LL. The glycine transport inhibitor sarcosine is an NMDA receptor co-agonist that differs from glycine. J Physiol 2009;587:3207–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zhang HX, Lyons-Warren A, Thio LL. The glycine transport inhibitor sarcosine is an inhibitory glycine receptor agonist. Neuropharmacology 2009;57:551–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.