

Abstract
Purpose of review
Randomized clinical trials provide strong evidence that pharmacological elevation of HDL-cholesterol (HDL-C) fails to reduce cardiovascular disease risk in statin-treated humans. It is thus critical to identify new metrics that capture HDL's cardioprotective effects.
Recent findings
We review recent evidence that HDL's cholesterol efflux capacity is a strong inverse predictor of incident and prevalent cardiovascular disease in humans. In light of those findings, we assess the proposal that impaired macrophage cholesterol efflux to HDL contributes to disease risk. We also discuss recent studies implicating small HDL particles in cholesterol efflux from macrophages.
Summary
These observations lay the foundation for a new approach to understanding mechanistically how HDL's functional properties help reduce CVD risk.
Keywords: high density lipoprotein (HDL), cholesterol homeostasis, atherosclerosis
Introduction
Epidemiological and clinical studies demonstrate a robust positive correlation between low density lipoprotein-cholesterol (LDL-C) and cardiovascular disease (CVD) risk. In contrast, high density lipoprotein-cholesterol (HDL-C) has a strong inverse association with CVD[1]. Indeed, these associations between lipid levels and vascular health led to the ubiquitous terms “bad cholesterol” for LDL-C and “good cholesterol” for HDL-C, signaling near universal acceptance of the lipoproteins' roles in vascular health.
With the introduction of HMG-CoA-reductase inhibitors in the late 1980's—and the resulting reduction in CVD observed in multiple randomized clinical trials—it became clear that LDL-C is indeed “bad” and that it resides in the causal pathway of atherosclerotic vascular disease. This conclusion is strongly supported from a mechanistic point of view because the bulk of cholesteryl ester found inside vascular lesions appears to originate directly from cholesteryl ester-rich LDL particles. The complex interactions between atherogenic LDL and the artery wall are reviewed elsewhere in this issue.
Because of the strong inverse correlation between HDL-C and CVD, and because of the dramatic success of approaches that lower LDL-C, it was anticipated that therapies designed to raise “good cholesterol” would further reduce CVD risk. However, the recent failure of several different HDL-C-raising agents, such as niacin, to reduce CVD risk in statin-treated subjects cast doubt on the cardioprotective nature of HDL [2,3]. These observations further suggest that increasing HDL-C levels does not directly confer atheroprotection, at least in the context of statin treatment. However, HDL is a heterogeneous collection of noncovalent particles comprising amphipathic lipids, neutral lipids, and a wide variety of proteins [4]. Therefore, measurements of HDL cholesterol content does not necessarily reflect either the overall abundance of HDL particles or the distribution of HDL subspecies [5]. Furthermore, HDL-C levels only explain a fraction (∼1/3) of the variance in functional assays such as sterol efflux capacity [6]. The potential role of HDL particle size and cholesterol content in modulating HDL particle number is summarized in Figure 1. Thus, modulating HDL cholesterol content might have no impact on the population of HDL particles responsible for atheroprotection. To successfully implement CVD therapies that target HDL, it will be critical to ascertain which HDL properties are responsible for cardioprotection and to identify the means to monitor and ultimately modulate those factors.
Figure 1. Potential mechanisms for discordance between HDL-C, HDL particle number, and HDL function.
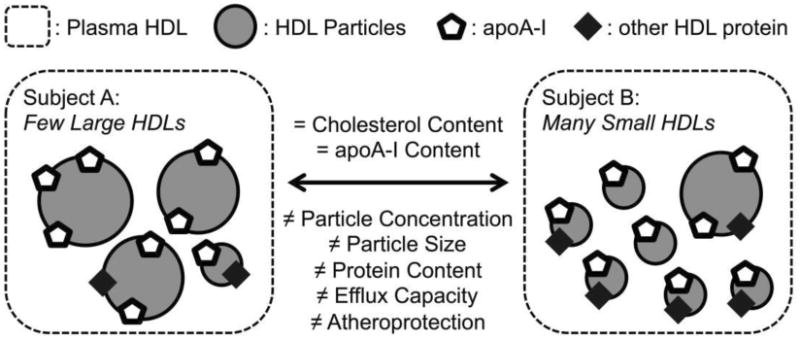
HDL particles are heterogeneous and the concentrations of individual subpopulations can vary markedly between individuals with the same levels of HDL-C and apoA-I. Moreover, the content of proteins with different biological activities can also vary widely, in part because of differences in the protein cargo carried by different HDL subspecies. In this hypothetical example, subjects A and B have equal HDL-C (the traditional HDL metrics) but unequal particle concentrations, particle size distributions, protein contents, and functional capacities.
Many lines of evidence suggest that HDL's anti-atherogenic activities include fighting inflammation, combatting oxidation, and promoting endothelial repair. However, HDL's best-studied cardioprotective property is its ability to accept cholesterol from macrophages in the artery wall—the first step of reverse cholesterol transport (RTC). Indeed the HDL hypothesis proposes that HDL mediated RTC is responsible for the inverse correlation of HDL-C and CVD risk; thus the hypothesis has always related to HDL function, not HDL cholesterol content [7,8]. Enhanced transport of cellular cholesterol from macrophages to the liver by HDL retards plaque progression in animal models [9]. Recent human studies indicate that the ability of serum HDL (serum depleted of LDL and other apoB-containing lipoproteins) to promote cholesterol efflux from cultured macrophages associates strongly and inversely with prevalent and incident CVD, supporting HDL's proposed role in reverse cholesterol transport and atherogenesis [10,11].
This review will focus on HDL/macrophage interactions that initiate reverse cholesterol transport, their impact on cholesterol mobilization from the artery wall, and how these pathways can inform HDL biology and future drug development.
Macrophages and reverse cholesterol transport
When macrophages take up and degrade more lipoprotein-derived cholesterol than they can excrete, they convert free cholesterol to cholesteryl ester. Macrophages laden with lipid droplets of cholesteryl ester, called foam cells, are the hallmark of early atherosclerotic lesions [12]. By acting as an acceptor for free cholesterol, HDL can remove cholesterol from macrophages and retard and/or reverse foam cell formation.
Multiple pathways contribute to macrophage cholesterol efflux, including the ATP-binding cassette transporters ABCA1 and ABCG1, aqueous diffusion, and scavengerreceptor class B member 1 (SR-B1) [13]. ABCA1 is defective in Tangier's disease, a disorder associated with low HDL-C levels and early onset atherosclerosis [14,15]. Because it is clearly relevant to macrophage cholesterol homeostasis, while the physiological significance of the other pathways in humans is unclear, this review will focus on the role of that transporter.
It has long been thought that ABCA1 interacts only with lipid-free or poorly lipidated apolipoproteins (apos)—such as apoA-I, A-II, E, CI/II/III or A-IV—to form nascent HDL particles [16]. The most important of these is apoA-I, HDL's main structural protein, which accounts for ∼70% of all HDL protein mass and is found on most HDL particles. The immediate product of the interaction between ABCA1 and apoA-I is a nascent HDL particle likely containing 2 molecules of apoA-I, and ∼1 mass equivalent (∼60 kDa) of phospholipid (PL) and free cholesterol (FC) [17]. No neutral lipids (cholesteryl ester or triglyceride) are incorporated. Strong evidence supports the notion that ABCA1 acts efficiently to transform lipid-free apoA-I to lipidated HDL particles. The most compelling observation is that plasma from Tangier's disease humans contains essentially no lipidated HDLs; only small amounts of lipid-free apoA-I can be detected [18]. It should be noted that recent evidence suggests that ABCA1 might also interact with more mature HDL [19].
The combined results of many investigators have produced the following model for ABCA1's role in promoting cholesterol efflux from macrophages [13,16, 20]. ABCA1 is a phospholipid translocase; the preferred substrate is phosphatidylcholine. ATP-dependent translocase activity accumulates phospholipid substrate at a membrane site adjacent to ABCA1 referred to as the activated lipid domain. ApoA-I binds the activated lipid domain created by ABCA1 and to the enzyme itself during the formation of nascent HDL. Recent single-molecule observations suggest that only ABCA1 dimers interact with apoA-I, raising the possibility the translocase is directly involved in the formation of nascent HDL [21]. ABCA1 lipid translocalization, combined with the presence of apoA-I, leads to the simultaneous release of both phospholipid and free cholesterol to lipid-free apoA-I and the release of very small immature HDL particles with no neutral lipid core. The free cholesterol is derived from both plasma membrane and intracellular membrane compartments.
Newly synthesized ABCA1 has a short half-life; the protein recycles between the plasma membrane and late endocytic vesicles [13,20]. Binding of apoA-I to the transporter stabilizes the protein by protecting it from calpain-mediated intracellular proteolysis. The phospholipid translocase activity of ABCA1 causes reorganization of lipid domains in the plasma membrane that in turn promote export of phospholipid and free cholesterol to apoA-I and other plasma apolipoproteins.
In lipid-laden macrophage foam cells, transport of esterified cholesterol—contained within lipid droplets—must be preceded by hydrolysis and release of free cholesterol. This process, though incompletely understood, possibly involves several neutral cholesteryl ester hydrolases [22]. Some studies suggest that enzymatic oxidation of cholesteryl ester acyl chain moieties potentiate this process by increasing hydrolase affinity, possibly contributing to the array of oxidized lipids observed in human atheromata [23–26].
The overall net transfer of cholesterol from macrophages (and perhaps other arterial cells) to extracellular acceptors (e.g., apoA-I) at the artery wall ultimately reduces cellular cholesterol content and thus attenuates atherogenesis.
Cholesterol efflux capacity of serum HDL and human CVD
Using J744 macrophages as a model system, Rothblat and colleagues have extensively characterized the ability of HDL and serum HDL (HDL depleted of LDL and other apoB-containing lipoproteins) to accept cholesterol [6]. Prior to incubation, the macrophages are incubated with radiolabeled cholesterol in the presence of an ACAT inhibitor to limit esterification of cellular free cholesterol. This process increases free cholesterol levels; it also prevents esterification of radiolabeled cholesterol, thereby maximizing localization of the sterol in the plasma membrane. Additionally, ABCA1 expression is upregulated, using stable cAMP analogs. Following incubation, the medium of the cells is collected, cellular debris removed, and cholesterol efflux is quantified by scintillation counting of radiolabeled cholesterol. Cholesterol efflux capacity is calculated as the percentage of radiolabel in the medium relative to that in the total system (medium + cells). In this model system, studies with inhibitors suggest that ABCA1, ABCG1 and aqueous diffusion, and SR-B1 each account for ∼ ⅓ of cholesterol efflux [27].
A key early study demonstrated that serum HDL (serum depleted of apoB-containing lipoproteins) from different humans have very different efflux capacities in this system, despite their similar levels of HDL cholesterol [6]. This finding strongly suggests that HDL cholesterol content is not the major determinant of cholesterol efflux capacity in this system. The potential clinical relevance of this observation was demonstrated by analyzing samples from two large cohorts [10]. Impaired efflux capacity of HDL was a superior predictor of cardiovascular disease status than traditional CVD risk factors. Indeed, efflux capacity strongly and negatively associated with CVD status after correction for HDL-C levels. Moreover, apoA-I explained only 40% of the variance in efflux capacity, while HDL-C accounted for only 34%. These observations strongly support the proposal that HDL's ability to remove cholesterol from macrophages is important for human cardioprotection. They also indicate that HDL-C is not a major determinant of efflux capacity in this model of macrophage reverse cholesterol transport.
The efflux capacity of J774 macrophages can also be quantified with a BODIPY cholesterol (a fluorescently labeled form of free cholesterol). Using this model, Rothblat and colleagues found that efflux of BODIPY cholesterol is mediated mainly by the ABCA1 pathway [28]. In the Dallas Heart Study, impaired cholesterol efflux capacity measured with BODIPY predicted future cardiovascular events [11]. Indeed, it was the strongest predictor of incident CVD events. Importantly, the Dallas cohort was multiethnic and included a large number of relatively young, apparently healthy subjects. In a logistic model that adjusted for traditional lipid factors, including HDL-C and LDL-C, impaired sterol efflux capacity remained a strong predictor of future events, suggesting that this metric provides clinically valuable information that is independent of traditional lipid risk factors. Collectively, these studies indicate that the cholesterol efflux capacity of serum HDL, as measured by these in vitro functional assays, is biologically relevant and—importantly—is not reliably reflected by common measures of HDL such HDL-C or apoA-I.
Therapies that target HDL-C and atheroprotective HDL
To date, therapies directed at HDL have specifically targeted HDL-C, owing to its robust inverse association with CVD risk and its facile measurement. Inhibition of cholesteryl ester transport protein (CETP) perhaps represents the most targeted approach in terms of mechanism. CETP enables the passive movement of neutral lipid between HDL particles and LDL/VLDL particles in a 1:1 molecular exchange of cholesteryl ester for triacylglyceride. Because of the relative concentrations of these constituents, CETP typically facilitates a net flux of cholesteryl ester from HDL to LDL and VLDL [29]. Thus, inhibiting CETP causes cholesteryl ester to accumulate in HDL particles and triacylglyceride to accumulate in LDL and VLDL particles. However, two CETP inhibitor trials in statin-treated subjects —ILLUMINATE (Investigation of Lipid Level Management to Understand Its Impact in Atherosclerotic Events) and the Dal-OUTCOMES study —were recently halted because of adverse results and predicted futility, respectively [30,31].
Another HDL-C targeted intervention, niacin, also failed to reduce clinical events in statin-treated subjects [3]. Taken together, these observations indicate that increasing the cholesterol content of HDL particles does not necessarily reduce CVD events. This conclusion argues strongly against the hypothesis that increasing HDL-C is atheroprotective in statin-treated subjects.
In contrast, the power of impaired efflux capacity of serum HDL to predict both incident and prevalent CVD strongly suggests that this metric reflects the cardioprotective actions of HDL. However, the factors that control the efflux capacity of human serum HDL are poorly understood. It is noteworthy that the Dallas Heart study focused on future CVD events in healthy subjects, and that the investigators used the BODIPY assay, which predominantly measures cholesterol efflux by the ABCA1 pathway [11]. Moreover, this pathway is defective in humans with Tangier's disease, and these subjects accumulate cholesterol-laden macrophages in many different tissues [18], strongly supporting the idea that ABCA1 plays a critical role in mediating cholesterol efflux from macrophages in humans.
One key unresolved issue is the primary ligand for ABCA1. Although current dogma favors poorly lipidated or lipid-free apoA-I, this species is only a minor component of total apoA-I in blood. Moreover, levels of poorly lipidated apoA-I are elevated in subjects with established CVD, suggesting it is unlikely to be cardioprotective [32,33].
Jessup and colleagues recently provided strong evidence that small HDLs, but not large HDLs could also accept sterol from cells by the ABCA1 pathway [19]. Importantly, small HDLs carry much less cholesterol per particle, indicating that HDL-C levels do not necessarily provide useful information on the size and concentration of this HDL subspecies. Moreover, both niacin and CETP inhibitors decrease the catabolism of HDL, thereby increasing the size of the HDL particle and raising HDL-C. The impact of these therapies on small and medium-sized HDL particles is less well understood.
These observations indicate that another key metric of HDL's cardioprotective effects, in addition to the efflux capacity of HDL, may be the concentrations of the various HDL subspecies. However, there is no agreement on how to accurately quantify the size and concentration of HDL, and two widely used methods—NMR and non-calibrated ion mobility—give substantially different concentration values.
We recently converted the ion mobility method of Kraus and colleagues to a quantitative metric of HDL size and concentration [5]., which we call ‘calibrated ion mobility analysis.’ Importantly, we demonstrated that our calibrated approach accurately quantifies the size and concentration of reconstituted HDL particles as well as gold nanoparticles, providing strong evidence that it can accurately quantify HDL from biological samples. In a small study, the concentration of medium-sized HDL particles was selectively lower in subjects with advanced carotid atherosclerotic disease, and this association remained highly significant after correction for HDL-C [5]. Another study of a cohort from the same study population concluded that HDL3 (the small and dense HDL fraction) was a strong and independent predictor of carotid disease, bolstering the notion that HDL's carrying less cholesterol may impact atheroprotection [34].
Conclusions
It is clearly time to end the clinical focus on HDL-C and to understand mechanistically how HDL's functional properties contribute to CVD risk. It will also be important to link variations in HDL's size and function to HDL-targeted therapies, genetics, and populations at high risk for CVD, such as diabetics. The development of new metrics for quantifying HDL function, based on a better understanding of macrophage reverse cholesterol transport, is critical for achieving these goals.
Key points.
Pharmacological elevation of HDL-C using multiple agents with different mechanisms of action has failed to reduce cardiovascular risk in statin treated subjects.
Impaired cholesterol efflux capacity of HDL is a better predictor than HDL-C of incident and prevalent cardiovascular disease; the association is independent of HDL-C levels.
HDL-C levels account for only ∼1/3 of the variance in HDL's cholesterol efflux capacity.
In future studies, it will be critical to understand the mechanistic basis for impaired cholesterol efflux capacity. We will also need to integrate this information with the concentrations and sizes of HDL subspecies derived by reliable quantitative methods.
Acknowledgments
None
Financial Support and Sponsorship: This work was supported by awards from the National Institutes of Health (HL112625, HL108897, P30 DK17047, P01 HL092969, T32HL007828).
Footnotes
Disclosures: Dr. Heinecke is named as a coinventor on patents from the US Patent Office on the use of HDL markers to predict the risk of cardiovascular disease. Dr. Heinecke has served as a consultant for Merck, Amgen, Bristol Meyer Squibb, GSK and Pacific Biomarkers.
References
- 1.Gordon DJ, Rifkind BM. High-density lipoprotein--the clinical implications of recent studies. N Engl J Med. 1989;321:1311–1316. doi: 10.1056/NEJM198911093211907. [DOI] [PubMed] [Google Scholar]
- 2.Kingwell BA, Chapman MJ, Kontush A, Miller NE. HDL-targeted therapies: progress, failures and future. Nat Rev Drug Discov. 2014;13:445–464. doi: 10.1038/nrd4279. [DOI] [PubMed] [Google Scholar]
- 3.AIM-HIGH Investigators. Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 4.Shah AS, Tan L, Long JL, Davidson WS. Proteomic diversity of high density lipoproteins: our emerging understanding of its importance in lipid transport and beyond. J Lipid Res. 2013;54:2575–2585. doi: 10.1194/jlr.R035725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5*.Hutchins PM, Ronsein GE, Monette JS, Pamir N, Wimberger J, He Y, Anantharamaiah GM, Kim DS, Ranchalis JE, Jarvik GP, et al. Quantification of HDL Particle Concentration by Calibrated Ion Mobility Analysis. Clin Chem. 2014 doi: 10.1373/clinchem.2014.228114. First validated description of a method to quantify HDL particle size and concentration accurately. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De la Llera-Moya M, Drazul-Schrader D, Asztalos BF, Cuchel M, Rader DJ, Rothblat GH. The ability to promote efflux via ABCA1 determines the capacity of serum specimens with similar high-density lipoprotein cholesterol to remove cholesterol from macrophages. Arterioscler Thromb Vasc Biol. 2010;30:796–801. doi: 10.1161/ATVBAHA.109.199158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller GJ, Miller NE. Plasma-high-density-lipoprotein concentration and development of ischaemic heart-disease. Lancet. 1975;1:16–19. doi: 10.1016/s0140-6736(75)92376-4. [DOI] [PubMed] [Google Scholar]
- 8.Rosenson RS, Brewer HB, Jr, Davidson WS, Fayad ZA, Fuster V, Goldstein J, Hellerstein M, Jiang XC, Phillips MC, Rader DJ, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125:1905–1919. doi: 10.1161/CIRCULATIONAHA.111.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rader DJ, Tall AR. The not-so-simple HDL story: Is it time to revise the HDL cholesterol hypothesis? Nat Med. 2012;18:1344–1346. doi: 10.1038/nm.2937. [DOI] [PubMed] [Google Scholar]
- 10.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11**.Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, Neeland IJ, Yuhanna IS, Rader DR, de Lemos JA, et al. HDL Cholesterol Efflux Capacity and Incident Cardiovascular Events. N Engl J Med. 2014;371:2383–2393. doi: 10.1056/NEJMoa1409065. First demonstration that impaired cholesterol efflux from macrophages predicts incident CVD events in a large multiethnic clinical population. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heinecke JW. Oxidants and antioxidants in the pathogenesis of atherosclerosis: implications for the oxidized low density lipoprotein hypothesis. Atherosclerosis. 1998;141:1–15. doi: 10.1016/s0021-9150(98)00173-7. [DOI] [PubMed] [Google Scholar]
- 13.Phillips MC. Molecular Mechanisms of Cellular Cholesterol Efflux. J Biol Chem. 2014;289:24020–24029. doi: 10.1074/jbc.R114.583658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rust S, Rosier M, Funke H, Real J, Amoura Z, Piette JC, Deleuze JF, Brewer HB, Duverger N, Denèfle P, et al. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat Genet. 1999;22:352–355. doi: 10.1038/11921. [DOI] [PubMed] [Google Scholar]
- 15.Santos RD, Asztalos BF, Martinez LRC, Miname MH, Polisecki E, Schaefer EJ. Clinical presentation, laboratory values, and coronary heart disease risk in marked high-density lipoprotein–deficiency states. J Clin Lipidol. 2008;2:237–247. doi: 10.1016/j.jacl.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oram JF, Heinecke JW. ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol Rev. 2005;85:1343–1372. doi: 10.1152/physrev.00005.2005. [DOI] [PubMed] [Google Scholar]
- 17.Ayyobi AF, Lacko AG, Murray K, Nair M, Li M, Molhuizen HO, Pritchard PH. Biochemical and compositional analyses of recombinant lecithin:cholesterol acyltransferase (LCAT) obtained from a hepatic source. Biochim Biophys Acta. 2000;1484:1–13. doi: 10.1016/s1388-1981(99)00199-7. [DOI] [PubMed] [Google Scholar]
- 18.Assmann G. Familial Analphalipoproteinemia: Tangier Disease [Internet] In: von Eckardstein A, editor. The Online Metabolic and Molecular Bases of Inherited Disease. 2013. [Google Scholar]
- 19**.Du X, Kim MJ, Hou L, Le Goff W, Chapman MJ, van Eck M, Curtiss LK, Burnett J, Cartland SP, Quinn CM, et al. HDL Particle Size is a Critical Determinant of ABCA1-Mediated Macrophage Cellular Cholesterol Export [Internet] Circ Res. 2015 doi: 10.1161/CIRCRESAHA.116.305485. Strong evidence that small discoidal reconstituted HDL and small, dense HDL particles isolated from human blood are good substrates for promoting cholesterol efflux from human and mouse macrophages by the ABCA1 pathway. [DOI] [PubMed] [Google Scholar]
- 20.Westerterp M, Bochem AE, Yvan-Charvet L, Murphy AJ, Wang N, Tall AR. ATP-Binding Cassette Transporters, Atherosclerosis, and Inflammation. Circ Res. 2014;114:157–170. doi: 10.1161/CIRCRESAHA.114.300738. [DOI] [PubMed] [Google Scholar]
- 21.Nagata KO, Nakada C, Kasai RS, Kusumi A, Ueda K. ABCA1 dimer-monomer interconversion during HDL generation revealed by single-molecule imaging. Proc Natl Acad Sci. 2013;110:5034–5039. doi: 10.1073/pnas.1220703110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakai K, Igarashi M, Yamamuro D, Ohshiro T, Nagashima S, Takahashi M, Enkhtuvshin B, Sekiya M, Okazaki H, Osuga J, et al. Critical role of neutral cholesteryl ester hydrolase 1 in cholesteryl ester hydrolysis in murine macrophages. J Lipid Res. 2014;55:2033–2040. doi: 10.1194/jlr.M047787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belkner J, Wiesner R, Rathman J, Barnett J, Sigal E, Kühn H. Oxygenation of lipoproteins by mammalian lipoxygenases. Eur J Biochem FEBS. 1993;213:251–261. doi: 10.1111/j.1432-1033.1993.tb17755.x. [DOI] [PubMed] [Google Scholar]
- 24.Belkner J, Stender H, Holzhütter HG, Holm C, Kühn H. Macrophage cholesteryl ester hydrolases and hormone-sensitive lipase prefer specifically oxidized cholesteryl esters as substrates over their non-oxidized counterparts. Biochem J. 2000;352 Pt 1:125–133. [PMC free article] [PubMed] [Google Scholar]
- 25.Hutchins PM, Moore EE, Murphy RC. Electrospray MS/MS reveals extensive and nonspecific oxidation of cholesterol esters in human peripheral vascular lesions. J Lipid Res. 2011;52:2070–2083. doi: 10.1194/jlr.M019174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hutchins PM, Murphy RC. Cholesteryl ester acyl oxidation and remodeling in murine macrophages: formation of oxidized phosphatidylcholine. J Lipid Res. 2012;53:1588–1597. doi: 10.1194/jlr.M026799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adorni MP, Zimetti F, Billheimer JT, Wang N, Rader DJ, Phillips MC, Rothblat GH. The roles of different pathways in the release of cholesterol from macrophages. J Lipid Res. 2007;48:2453–2462. doi: 10.1194/jlr.M700274-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Sankaranarayanan S, Kellner-Weibel G, de la Llera-Moya M, Phillips MC, Asztalos BF, Bittman R, Rothblat GH. A sensitive assay for ABCA1-mediated cholesterol efflux using BODIPY-cholesterol. J Lipid Res. 2011;52:2332–2340. doi: 10.1194/jlr.D018051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charles MA, Kane JP. New molecular insights into CETP structure and function: a review. J Lipid Res. 2012;53:1451–1458. doi: 10.1194/jlr.R027011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwartz GG, Olsson AG, Ballantyne CM, Barter PJ, Holme IM, Kallend D, Leiter LA, Leitersdorf E, McMurray JJV, Shah PK, et al. Rationale and design of the dal-OUTCOMES trial: efficacy and safety of dalcetrapib in patients with recent acute coronary syndrome. Am Heart J. 2009;158:896–901.e3. doi: 10.1016/j.ahj.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 31.McKenney JM, Davidson MH, Shear CL, Revkin JH. Efficacy and safety of torcetrapib, a novel cholesteryl ester transfer protein inhibitor, in individuals with below-average high-density lipoprotein cholesterol levels on a background of atorvastatin. J Am Coll Cardiol. 2006;48:1782–1790. doi: 10.1016/j.jacc.2006.06.066. [DOI] [PubMed] [Google Scholar]
- 32.Asztalos BF, Cupples LA, Demissie S, Horvath KV, Cox CE, Batista MC, Schaefer EJ. High-density lipoprotein subpopulation profile and coronary heart disease prevalence in male participants of the Framingham Offspring Study. Arterioscler Thromb Vasc Biol. 2004;24:2181–2187. doi: 10.1161/01.ATV.0000146325.93749.a8. [DOI] [PubMed] [Google Scholar]
- 33.Guey LT, Pullinger CR, Ishida BY, O'Connor PM, Zellner C, Francone OL, Laramie JM, Naya-Vigne JM, Siradze KA, Deedwania P, et al. Relation of increased prebeta-1 high-density lipoprotein levels to risk of coronary heart disease. Am J Cardiol. 2011;108:360–366. doi: 10.1016/j.amjcard.2011.03.054. [DOI] [PubMed] [Google Scholar]
- 34.Jarvik GP, Rozek LS, Brophy VH, Hatsukami TS, Richter RJ, Schellenberg GD, Furlong CE. Paraoxonase (PON1) phenotype is a better predictor of vascular disease than is PON1(192) or PON1(55) genotype. Arterioscler Thromb Vasc Biol. 2000;20:2441–2447. doi: 10.1161/01.atv.20.11.2441. [DOI] [PubMed] [Google Scholar]