

Abstract
Background
Pleomorphic xanthoastrocytomas with anaplastic features (PXA-As) are rare tumors about which little is known regarding clinicopathological and molecular features. Several studies have identified BRAF V600E mutations in PXA-As, but the percentage with mutation may differ between adult and pediatric examples, and limited information exists about immunohistochemistry for IDH1 (isocitrate dehydrogenase 1). Design: 10 cases of adult PXA-As seen at our institution since 2000 were assessed for BRAF V600E mutation by polymerase chain reaction testing (PCR) and IDH1 by immunohistochemistry.
Results
Patients ranged in age from 18-68 years; 4 PXA-As affected temporal lobe and 2 were cystic. Four patients underwent gross total resection; and 9/10 received cranial irradiation and/or adjuvant chemotherapy. Five survived less than 5 years, although 2/5 died from non-tumor causes. Four long-term survivors are alive at 7.5, 9.8, 11.4, and 11.9 years post-diagnosis. 2 of 4 long term survivors had BRAF V600E mutation: patients were ages 18 and 28 years. A 48-year old male without BRAF mutation survives at 9.8 years, even with thalamic location; conversely a 68-year-old female with temporal lobe tumor and BRAF mutation survived 1.9 years after diagnosis. All tumors were IDH1 immunonegative.
Conclusion
This case series details clinicopathological features of a subset of rare PXA-As in adults. BRAF V600E mutation was identified in 50% of these cases.
Keywords: pleomorphic xanthoastrocytoma, PXA, anaplastic, BRAF, IDH1, clinical outcome
Introduction
Pleomorphic xanthoastrocytomas (PXAs) are rare WHO grade II tumors, first fully characterized by Kepes et al. [1], which demonstrate a relatively favorable clinical course [1-3]. PXA with anaplastic features (PXA-A) designates a subset with more aggressive histopathologic features and more adverse prognosis, and has yet to be assigned a formal WHO grade [2]. PXAs and PXA-As collectively constitute no more than 1% of astrocytic tumors and are usually seen in children and young adults [1-3], with a mean age of 26 years [3]. Nearly 2/3 of examples occur in patients less than age 25 years [3].
Adult examples are thus uncommon, although well-documented cases have been seen in patients 40 years or older [4-9]. Some authors have suggested a worse prognosis is associated with older patient age [9]. PXA-As may arise de novo [10, 11], or may develop anaplastic features, including necrosis and/or increased mitotic index [3] at recurrence of a previous WHO grade II PXA [3, 5, 7, 10-16]. Recent reviews, by Tekkok et al. [14], Okazaki et. al. [15] and Vu et al. [16], emphasize the fact that for either de novo PXA-As or for those that acquire anaplastic features at recurrence, the prognosis is often, but not invariably, poor.
Which histological feature(s) correlates best with prognosis in individual patients remains somewhat unclear. A seminal study of 71 cases and review of an additional 121 published cases of PXA by Giannini et al. in 1999 [3], lead the World Health Organization (WHO) in 2007 to adopt her findings that mitotic activity of greater than or equal to 5 mitoses per 10 high power fields (HPFs) is the sole independent predictor of overall survival by multivariate analysis [2]. A “significant association of necrosis with survival” was also acknowledged, although this did not reach statistical significance on multivariate analysis [2]. On univariate analysis, three categories of patients with differing recurrence-free survival and overall survival rates could be identified: those with 0 mitoses, those with 1-4 and those with >/= 5 mitoses [3]. Korshunov et al. in their 2001 series of 34 patients [17] were more clearly able to identify three subsets of PXAs: typical: tumors without mitoses per 20 HPFs; proliferating: tumors with mitoses but without necrosis; and malignant: tumors with elevated mitotic index and necrotic foci. There were no recurrences or death amongst typical tumors. In contrast, 36% of proliferating tumors recurred and 1 patient died, and 5/5 malignant tumors rapidly recurred and 4/5 succumbed. Other authors have placed emphasis on necrosis [18, 19] and MIB-1/Ki-67 cell cycling rate [10-12].
Recently, mutation in BRAF V600E at position 600, specifically V600E (NM_004333.4 c.1799T>A, hereafter referred to as BRAF V600E), has been identified as a common finding in certain central nervous system (CNS) tumors, most commonly in PXAs, (WHO grade II) and PXA-As. The same mutation has also been found in ganglioglioma and extra-cerebellar pilocytic astrocytoma [20-22]. Amongst CNS tumors, the presence of BRAF V600E may be helpful in distinguishing PXAs from diffuse astrocytic tumors WHO grades II, III, and IV [22], since the latter almost always lack this finding [20].
In contrast, a common mutation in isocitrate dehydrogenase 1 (IDH1) is typical of the majority of diffuse astrocytomas, oligodendrogliomas, and mixed oligoastrocytomas of WHO grades II and III [23]. Immunohistochemical (IHC) staining for IDH1 correlates strongly with the IDH1 mutational status as assessed by polymerase chain reaction testing [24] and thus can serve as a cost- and time-effective substitute.
Relatively few examples of adult PXA-As have been studied for BRAF mutational status. Studies by Schindler et al. hint that there may be age-related differences in BRAF V600E mutational status, based on the fact that, in their study, 38% of adult PXA-As showed this mutation (5/13 adult cases), compared to 100% of pediatric PXA-As (10/10 pediatric cases) [20].
Finally, although IDH1 immunoreactivity appears to be a signature of diffuse astrocytomas and not PXAs, large numbers of adult PXA-A cases have also not been assessed [23]. We undertook this study of PXA-As to address these issues. We confined our work to a series of adult patients well-known within our system for which treatment data and long term clinical followup could be obtained.
Materials and Methods
Case accrual
Institutional research review board approval was obtained for this study. Cases were identified by diagnosis from our pathology department databank and/or personal files of the authors for the years 2000-2011, inclusive. Medical records were reviewed by the neuro-oncologist (DMD) to obtain neuroimaging features, treatment regimens, and survival data. Followup data was sought for all patients, and patients recorded as alive or dead. Those who died were recorded as succumbing either to disease (ie., their PXA-A) or from other causes. For those patients still living, survival data was calculated from time of PXA-A diagnosis to 3/2012, the closure of study.
All slides were re-reviewed for diagnosis confirmation (BKD). For diagnosis of PXA-A, criteria identified by Giannini et al. [2, 3] were utilized. All cases in this study contained the major histological findings previously identified as essential to diagnosis, including multinucleated pleomorphic large cells (seen in 92% of PXA cases in her study [3]), GFAP immunoreactivity (98% of cases), granular bodies (93%: eosinophilic 81%/pale 71%), lymphocytic collections (83% of cases), foci of increased reticulin (81%), and xanthic cells (66% of cases). Most cases were quite stereotypic and similar in features. All cases had pleomorphic cells and GFAP immunoreactivity. Granular bodies, especially eosinophilic, refractile, droplet-like, eosinophilic, PAS-positive (Periodic acid Schiff-positive) granular bodies were specifically sought and tabulated (see Table). As noted by Giannini et al., “although it was not stressed in the original description of PXA or in the WHO monograph, granular bodies of varying size, texture, and eosinophilia are a regular feature of this tumor” [3]. In our own experience, the presence of granular bodies has been the single most distinctive and helpful clue since they are usually not identified in glioblastomas (lipidized, giant cell or ordinary), the main tumor type in the differential diagnosis for PXA-A.
Methods for histology
For light microscopy, tumor sections were cut at 4 microns and stained with hematoxylin and eosin (H&E), reticulin (Gordon Sweet reticulum II), and periodic acid Schiff in selected cases where eosinophilic granular bodies were rare on H&E. Immunohistochemistry for glial fibrillary acidic protein (GFAP, Dako #M761, Carpinteria, CA, monoclonal, 1:100 dilution) and synaptophysin (Ventana 790-4286, Tucson, AZ, rabbit monoclonal, Pre Dilute) was additionally utilized for diagnosis.
Immunohistochemistry for IDH1 (Dianova HistoBio, Miami Beach, Florida, clone H09, monoclonal, 1:40 dilution) and MIB-1 cell cycle labeling (Ventana 790-4286, Tucson, AZ, rabbit monoclonal, Pre Dilute) was conducted on all cases. All assessments for IHC were performed on the first resection at which PXA-A diagnosis was made.
Mitotic figures were counted at high power objective (40×), with ten consecutive fields counted after identifying a mitosis. Considerable variation in mitotic rates from one region to the next was encountered in all cases, a feature previously well described by others in the literature [3, 27]. The presence or absence of eosinophilic granular bodies was assessed and recorded as present, rare, single, or absent on H&E; in select cases, on PAS, was performed. Necrosis and microvascular proliferation were also assessed and recorded, detailed in the Table. MIB-1 was assessed as percentage of immuno-positive tumor nuclei on 1000 cell counts, avoiding non-tumoral cells such as lymphocytes in the counts.
Methods for V600E BRAF mutational analyses
Blocks (or in one case unstained slides with scrape of tissue) with PXA morphology were specifically targeted for assessment. In the case of the scraped slides, paraffin sections were thoroughly deparaffinized in xylene, hydrated through graded alcohols to water and stained with Gill's hematoxylin. Slides were covered with glycerol to prevent cell dispersion and isolated under dissecting microscope using a scalpel point or hollow borosilicate glass pipette. The scraped material was washed in PBS and digested in proteinase K overnight at 37°C in ATL Buffer (Qiagen Inc.). DNA was then isolated using QIAamp DNA FFPE extraction kit (cat # 56404) according to manufacturer instruction.
In the case of paraffin tissue blocks (remaining cases), paraffin scrolls were deparaffinized in xylene as above, followed by reconstitution in graded alcohols and PBS and treated in parallel fashion. DNA yields were then quantified using a Nanodrop spectrophotometer ND-1000 (Thermo Fisher Scientific Inc., Waltham, MA).
For direct sequencing, approximately 10 ng of template DNA was PCR amplified using 5 pmol each of forward (5′TGCTTGCTCTGATAGGAAAAT3′) and reverse (5′TCAGGGCCAAAAATTTAATCA3′) BRAF exon 15 primers KAPA2G™ Robust HotStart Enzyme and PCR master mix with KAPA™ dNTP mix (KAPA Biosystems cat# KK5525 and KK1017) in a 25μl reaction. PCR was performed on an ABI 9700 thermocycler with an initial denaturing step at 95°C, followed by 20 cycles of touchdown PCR (starting annealing temperature of 65°C, decremented 0.5°C per cycle) and 25 cycles at 94°C denaturation, 55°C annealing and 72°C extension finished by a 10 minute 72°C final extension. The resultant PCR products were purified with the QIAquick 96 well PCR cleanup kit (Qiagen cat# 28106). The purified PCR products were sequenced in forward and reverse directions using an ABI 3730 automated sequencer that utilizes BigDye Terminator Version 1.1 (Applied Biosystems). Each chromatogram was visually inspected for any abnormalities, using NM_004333.4 as a reference sequence, with particular attention directed to codon 600. Sequences were also evaluated using Mutation Surveyor software (Soft Genetics, State College, PA). Mutations were determined to be present when peaks reached a threshold value above baseline calculated from background level, combined with visual inspection of the chromatogram.
Results
Demographic, clinical, treatment, and survival data is listed in Table 1, with histological and genetic features detailed in Table 2. Ten patients were identified (5 females, 5 males). Patients ranged in age from 18-68 years; 4 PXA-As affected temporal lobe and 2 tumors were cystic. Patients had been treated as high grade gliomas with surgery, external beam irradiation, and chemotherapy, except for one patient who refused treatment beyond subtotal resection (patient #7, see Table 1), and another who could not tolerate a full course of radiotherapy (patient #9). Data was not available for 2 cases (see Table 1). Most patients receiving chemotherapy had been treated with standard temozolomide and 4 had undergone a second surgical resection after the PXA-A diagnosis (see Table). One patient had a past medical history of neurofibromatosis type I (patient #1), one had colon cancer (patient #2), and one had a previous diagnosis of a high grade glioma antecedent to their PXA-A diagnosis at our institution (patient #8). Patient #8 had been diagnosed with anaplastic astrocytoma 3 years before. Slides from this surgical procedure performed at an outside facility were not available for review. The remaining 9 patients with PXA-As occurred de novo.
Table 1. PXA-As: Clinical, pathologic, and survival data.
| # | Age | Sex | Significant Clinical History | Location | Cystic lesion | Extent of surgical resection | Initial therapy | Additional Treatment | Alive vs dead | Survival (years) calculated as of study closure |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 27 | F | Neurofibromatosis, type I; mother with NF1 | L frontal | N | GTR | RT TMZ |
Re-resection 5 years later | Alive | 11 years, 4 months |
| 2 | 52 | F | Colon cancer 5 years prior | L frontal | N | Unknown | Unknown | None known | Died of other causes | 1 year, 1 month |
| 3 | 68 | F | Initially biopsied: diagnosed as PXA-A (Mayo); resection soon thereafter | R temporal | N | GTR | RT TMZ |
None known | Died of disease | 1 year, 9 months |
| 4 | 51 | M | No known relevant history | L temporal | N | Subtotal resection | RT TMZ |
Re-resection at outside hospital 2 years after first surgery: pathology “identical to first resection material” | Died of other causes | 1 year, 6 months |
| 5 | 28 | M | No known relevant history | L fronto-parietal | N | Subtotal resection | RT BCNU |
SRS Re-resection 5 years after first surgery: extensive radionecrosis + some tumor | Alive | 11 years, 9 months |
| 6 | 48 | M | No known relevant history | R thalamus | Y | Subtotal resection | RT TMZ |
TMZ Re-resection 9 years after first surgery: residual tumor histologically similar to original tumor | Alive | 9 years, 8 months |
| 7 | 22 | M | No known relevant history | R temporal | N | Subtotal resection | None | None known | Died of disease | 1 year, 7 months |
| 8 | 26 | F | Diagnosed as anaplastic astrocytoma 3 years prior elsewhere (2003); treated with RT, re-presented after delivery of child | R frontal | N | Unknown | Unknown | None known | Lost to follow-up after 2 years | 2+ years |
| 9 | 18 | F | No known relevant history | L temporal | Y | GTR | Gliadel wafer placement Partial RT (ceased due to intolerance) TMZ |
Additional TMZ | Alive | 7 years, 5 months |
| 10 | 38 | M | History of many years migraines, recently worsened | R frontal | N | GTR | RT Avastin TMZ |
None | Alive but <2 years from surgery | 7 months |
Patient survival assessed as of study closure date of 3/2012
Key: GTR=gross total resection; RT:=radiation therapy; SRS:=stereotactic radiosurgery; TMZ:=temozolomide; BCNU:=carmustine, bis-chloroethylnitrosourea
Table 2. PXA-As: Histological and genetic features.
| # | Age | Sex | Histologic features | Mitotic count | MIB-1 rate | IDH1 by IHC | BRAF V600E mutation status |
|---|---|---|---|---|---|---|---|
| 1 | 27 | Female | EGBs Necrosis MVP |
5/10 HPFs | 1.9% | Negative | No mutation detected |
| 2 | 52 | Female | Rare EGBs Necrosis MVP |
7/10 HPFs | 23% | Negative | No mutation detected |
| 3 | 68 | Female | EGBs Necrosis MVP |
15/10 HPFs | 22% | Negative | V600E mutation present |
| 4 | 51 | Male | Single EGB No necrosis No MVP |
6/10 HPFs | 8.5% | Negative | No mutation detected |
| 5 | 28 | Male | Rare EGB Necrosis No MVP |
9/10 HPFs | 9% | Negative | V600E mutation present |
| 6 | 48 | Male | EGBs No necrosis Focal MVP |
7/10 HPFs | 8.5% | Negative | No mutation detected |
| 7 | 22 | Male | EGBs Focal remote Hemorrhagic necrosis No MVP |
8/10 HPFs | 5.5% | Negative | V600E mutation present |
| 8 | 26 | Female | EGBs Necrosis MVP |
15/10 HPFs | 16% | Negative | V600E mutation present |
| 9 | 18 | Female | EGBs Necrosis MVP |
5/10 HPFs | 5.5% | Negative | V600E mutation present |
| 10 | 38 | Male | EGBs Necrosis MVP |
34/10 HPFs | 49.5% | Negative | No mutation detected |
Key: EGBs=eosinophilic granular bodies; MVP=microvascular proliferation; HPFs=high power microscopic fields; IDH-1=isocitrate dehydrogenase-1; IHC= immunohistochemistry
Tumors predominantly affected frontal or temporal lobes (see Table 1), with a single case involving subcortical gray matter (patient # 6). The range of neuroimaging features is illustrated in Figure 1. Since a significant number of our cases had first been diagnosed with PXA-As in the early 2000's, 4/5 of the survivors have been followed for >7 years (range: 7 years, 5 months to 11 years, 9 months). The final patient was diagnosed more recently and, as of the study closure time point of 3/2012, had a clinical followup time of 7 months (see Table 1). Of the 5 survivors known to be alive at study closure, mean clinical followup time has been 8 years, 5 months, with a median of 9 years, 8 months. Nine of the 10 patients in this cohort were able to followed to demise or to study closure date; the 10th patient (patient #8) was lost to followup two years after diagnosis.
Figure 1.
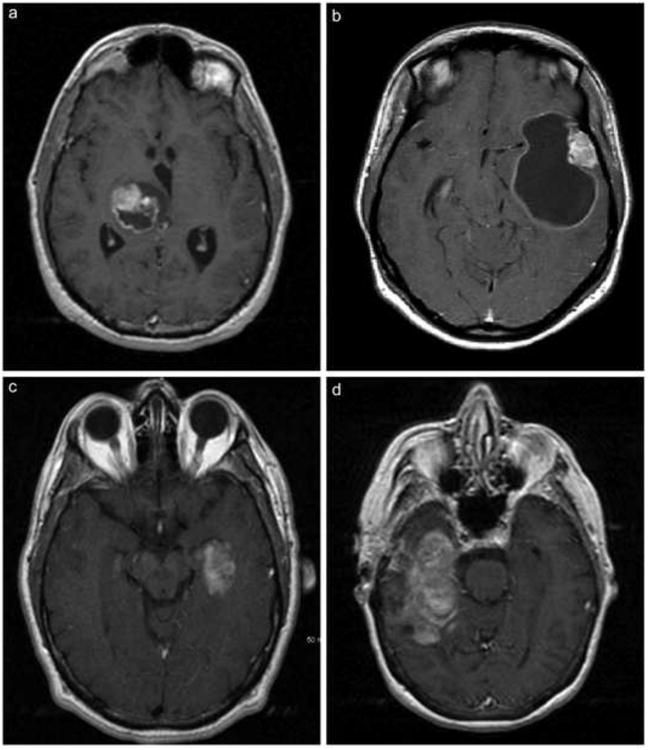
A: Axial T1 post-contrast MRI shows a heterogeneously enhancing cystic mass in the right thalamus with mass effect but minimal surrounding vasogenic edema. Patient #6 illustrated.
B. Axial T1 post-contrast MRI shows an enhancing mural nodule in the left lateral temporal cortex associated with a large rim-enhancing cyst. Patient #9 illustrated.
C. Axial T1 post-contrast MRI shows an inhomogeneously enhancing lesion in the mesial left temporal lobe. Patient #4 illustrated.
D. Axial T1 post-contrast MRI shows a large heterogeneously enhancing lesion with complex cystic component within the right temporal lobe. Note the paucity of mass effect and the relative absence of vasogenic edema. Patient #2 illustrated.
Microscopically, the majority of cases showed classic PXA areas adjacent to higher-grade glioma showing variable mitotic activity, necroses, some microvascular proliferation (see Table 2). In 7 cases the two areas were relative distinct. In 3 examples (patients #2, 4, 7), however, there was a more intimate blend between the neoplastic pleomorphic cells with xanthic or granular cell changes and smaller cells manifesting ordinary high grade glial tumor features. In some cases the higher-grade glioma manifested small cell glioblastoma features (examples: patients #1, #3, #10), a feature described years ago [25, 26]. In these instances the areas of high-grade glioma were completely indistinguishable from ordinary high-grade gliomas. This raised the obvious issue of whether sampling errors might have contributed to the diagnosis of a diffuse high grade glioma in patient #8. The areas of PXA-A contained large pleomorphic giant cells with nuclear pseudoinclusions, GFAP immunoreactivity, glassy eosinophilic pale bodies, and variable numbers of EGBs (eosinophilic granular bodies, highly refractile eosinophilic droplet-like structures) (see pictures). Several cases had rare or single EGBs (patients #2. #5, #6) and one of these (patient #5) did manifest positive BRAF V600E mutational status. Conversely, some cases (example, patient #1) had profuse numbers of EGBs, yet lacked the BRAF V600E mutation. Thus, morphological features of the PXA-As were indistinguishable based on BRAF mutational status, at least in our small case series. The histological features seen in these rare adult PXA-As are illustrated in Figures 2 and 3. The PXA features of two representative patients, one with, and one without, BRAF mutation, are shown in Figure 2. Histological features for high grade glioma areas of these tumors are shown in Figure 3.
Figure 2.
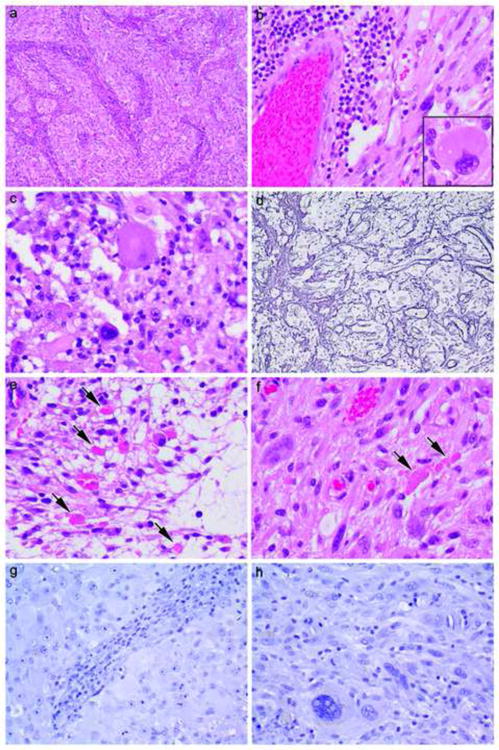
Direct comparison between PXA areas in Patient #1 (a, c, e, g) (a 27-year old female with relatively separate classic PXA and high grade glioma areas of tumor in whom no BRAF mutation was detected yet is a 10+ year survivor) versus Patient #3 (b, d, f, h) (a 68 year-old female also with relatively separate classic PXA and high grade glioma areas in whom BRAF mutation was detected yet survived less than 2 years). Note typical features of PXA including brisk non neoplastic lymphocytic collections (a, b), large pleomorphic cells (b inset, c), focally increased reticulin fibers (d), eosinophilic granular bodies (arrows, e, f) and negative immunostaining for IDH1 (g, h). Hematoxylin and eosin (a 100×, b 400×, b inset 600×, c 600×, e 600×, f 600×), reticulin stain (d 200×), and immunostaining for IDH1 with light hematoxylin counterstain (g, h, both 400×).
Figure 3.
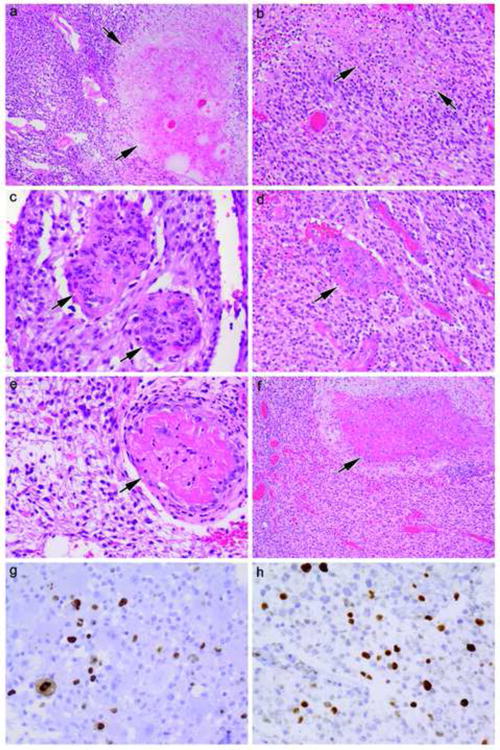
Direct comparison between high grade glioma areas in Patient #1 (a, c, e, g) (a 27-year old female with relatively separate classic PXA and high grade glioma areas of tumor in whom no BRAF mutation was detected yet is a 10+ year survivor) versus Patient #3 (b, d, f, h) (a 68 year-old female also with relatively separate classic PXA and high grade glioma areas in whom BRAF mutation was detected yet survived less than 2 years). Note hypercellularity (a, b, c, d), necrosis (arrows, a, b), microvascular proliferation (arrows, c, d), vascular thrombosis (arrows e, f), and elevated MIB-1 cell cycling rate (g, h). Hematoxylin and eosin (a 100×, b 200×, c 400×, d 200×, e 400×, f 100×,) and immunostaining for MIB-1 with light hematoxylin counterstain (g, h both 400×).
Four patients were known to have recurrences of their tumors within the study time period (see Table 1, patients #1, 4, 5, and 6). MIB-1 indices of the PXA-As at the time of the initial resections, for these patients with known recurrences were not obviously higher than for those without known recurrences. These four patients had MIB-1 rates, respectively, of 9%, 8.5%, 9% and 8.5% (Table 2). In contrast, patient #7 had an initial MIB-1 rate of 5.5% and died of his disease within 1.7 years of initial diagnosis. Small cohort size precluded any definitive conclusions about MIB-1 rate and tendency for recurrence or overall survival.
Five patients who succumbed survived less than 5 years, although 2 of these 5 died from non-tumor causes (see Table 1). Long term survivors are alive at 7-11+ years post-diagnosis. Of these long term survivors, 2 of 4 had BRAF V600E mutation in the tumor: these two patients were ages 18 and 28 years. Another young patient, 22 years old, did possess the BRAF V600E mutation, and succumbed to his disease 1.7 years after diagnosis; he received only subtotal resection and was the single patient who had refused postoperative chemo- or radiotherapy.
In our series, 3 of 4 patients over age 45 years were BRAF negative: a 68-year-old did show BRAF mutation; her survival after diagnosis was 1.9 years. A 48-year-old male without BRAF mutation survives at 9.8 years, even with thalamic location. The small sample size limits any meaningful correlation between BRAF mutational status and survival.
Discussion
We have been interested in PXA-As since 2001 when we encountered a particularly tragic case where his treatment, not tumor, led to his demise [27]. This 34-year-old man developed a radiation-induced CNS sarcoma 11 years after receiving cranial irradiation (but no chemotherapy) for a “glioblastoma” diagnosed at an outside facility [29]. On obtaining his original 1990 slides, the tumor, in retrospect, proved to be a PXA-A with copious numbers of eosinophilic granular bodies, focal lymphocytic collections, xanthic cells, Rosenthal fibers, and elongate spindle tumor cells, all features of PXA, but with superimposed necrosis, microvascular proliferation, and mitoses, features of PXA-A. He succumbed less than one year later to his radiation-induced sarcoma, despite several therapies. Clearly in this example, the risks of overly aggressive treatment were obvious. Giannini et al. also mention a patient with a high mitotic index but no necrosis at first surgery who died 5.7 years after primary surgery from a contralateral postirradiation anaplastic oligodendroglioma [3]. On the other hand, the single patient in our current study in who only a subtotal resection could be achieved and who declined any adjuvant treatment, succumbed after 1.7 years, suggesting that adjuvant radio/chemotherapy may have therapeutic benefit. At our institution, additional therapy has always been recommended, and almost always received.
Our study was confined to PXA-As in adults, which make up a smaller subset of an already-rare glioma tumor type [3], explaining the limited case numbers in our study. Most of our cases were de novo PXA-As. PXA-A may occur de novo with anaplastic features at first resection or may evolve from PXA grade II to PXA-A, but both have variable, but often poor, outcomes [3, 14-16]. One of our cases had been biopsied elsewhere and diagnosed as anaplastic astrocytoma; sampling error cannot be excluded since the high grade glioma portion of PXA-As is usually indistinguishable from ordinary diffuse high grade glioma, a point made many years ago by Kepes et al. [25] and Macaulay et al. [26].
All of our cases met criteria for PXA-A based on either mitotic rate >5/10 HPFs or presence of necrosis, and most examples had both (see Table). In addition, we conducted MIB-1 immunostaining on all cases. The likely importance of MIB-1 cell cycle labeling was anticipated by others in smaller series [10-12] and indeed, rates above 5.5% were found in all adult PXA-A cases in this series. MIB-1 may assist in distinguishing PXA-A versus PXA, WHO grade II, given the wide variation in distribution of mitotic figures in these cases, as noted previously [3, 28]. In addition, it should be noted that some patients have manifested adverse clinical outcome [10] even when fewer than the 5 mitoses/10 HPFs (recommended as a criteria for PXA-A diagnosis [2, 3]) have been identified; these did, however, have modestly elevated MIB-1 rates of 4.9% and 5.4% [10].
While our oldest patient, a 68-year-old woman, succumbed within 2 years of diagnosis, our next-oldest patient has survived for more than 9 years. Thus, we cannot reach the same conclusion as Ng et al., that advanced patient age is automatically a negative prognostic factor in adult PXA-As [9].
Similar to previous research [20-22], BRAF V600E mutation was not seen in 100% of our adult PXA-As. In our study, 50% (5/10) PXA-As demonstrated the mutation. Schindler et al. investigated a large number of PXAs (64: 38 adult/26 pediatric) and PXA-As (23: 13 adult/10 pediatric) and found the mutation in 66% of PXAs overall (63% adult, 69% pediatric) and 65% of PXA-As (38% adult, 100% pediatric) [20]. In contrast, Dias-Santagata et al. found the mutation in 60% of WHO grade II PXAs, but only in 1 of 6 PXA-As [22]. Differences may relate to the fact that only 6 PXA-As were available for analyses in the latter study [22]. Alternatively, the fact that all 6 were adults, as opposed to pediatric patients, and 5/6 were 40 years of age or older, including a 62.1-year-old female and an 86.8-year-old male, may also have influenced results [22]. Thus, there may be some differences for BRAF V600E mutation status in adult versus pediatric PXA-As. Although limited by the small number of cases, the current study contributes additional data to the issue of percentage of adult PXA-As with this mutation.
Thus, up to one-third of PXAs and PXA-As appear to be negative for BRAF V600E mutation and it is currently unclear if one, or more, as-yet-undiscovered genomic alterations might be present in this subpopulation without BRAF V600E mutation. Dougherty et al. noted that negative PXAs and PXA-As did not show KIAA1549-BRAF fusions [21]. At least in our study, we do not feel that regional heterogeneity accounts for the presence or absence of BRAF V600E mutation in our patients, since, in each case, the tissue block with the best morphological features of PXA was selected for mutational analysis and each block was confirmed to have the minimum tumor cellularity to have confidence in the sequencing assay. Whether there is heterogeneity at the single cell level cannot be discerned with this sequencing technique.
Finally, our study of PXA-As contributes to the literature on IDH1 status in these rare tumors. All of our cases were IDH1 negative by immunohistochemistry. Balss et al. assessed a total of 685 brain tumors, 7 of which were PXAs WHO grade II and all seven are negative for IDH1 mutation [23]. We now add to the literature 10 PXA-As, all in adults that are also negative for IDH1.
Acknowledgments
The authors thank Mrs. Diane Hutchinson and Ms. Diana Doyle for excellent manuscript preparation and Ms. Lisa Litzenberger for expert photographic assistance
Footnotes
Presented in abstract format at the 101st annual United States and Canadian Pathology meeting, Vancouver, B.C., March, 2012
Disclosure/Conflict of Interest: No conflicts of interest to report.
References
- 1.Kepes JJ, Rubinstein LJ, Eng LF. Pleomorphic xanthoastrocytoma: a distinctive meningocerebral glioma of young subjects with relatively favorable prognosis. A study of 12 cases. Cancer. 1979;44:1839–1852. doi: 10.1002/1097-0142(197911)44:5<1839::aid-cncr2820440543>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 2.Giannini C, Paulus W, Louis DN, Liberski P. Pleomorphic xanthoastrocytoma. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. WHO Classification of Tumours of the Central Nervous System. 3rd. IARC; Lyon: 2007. pp. 22–24. [Google Scholar]
- 3.Giannini C, Scheithauer BW, Burger PC, Brat DJ, Wollan PC, Lach B, O'Neill BP. Pleomorphic Xanthoastrocytoma. Cancer. 1999;85:2033–2045. [PubMed] [Google Scholar]
- 4.MacKenzie JM. Pleomorphic xanthoastrocytoma in a 62-year-old male. Neuropathol Appl Neurobiol. 1987;13:481–487. doi: 10.1111/j.1365-2990.1987.tb00076.x. [DOI] [PubMed] [Google Scholar]
- 5.Marton E, Feletti A, Orvieto E, Longatti P. Malignant progression in pleomorphic xanthoastrocytoma: personal experience and review of the literature. J Neurol Sci. 2007;252:144–153. doi: 10.1016/j.jns.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 6.Kros JM, Vecht CJ, Stefanko SZ. The pleomorphic xanthoastrocytoma and its differential diagnosis: A study of five cases. Hum Pathol. 1991;22:1128–1135. doi: 10.1016/0046-8177(91)90265-q. [DOI] [PubMed] [Google Scholar]
- 7.Chakrabarty A, Mitchell P, Bridges LR, Franks AJ. Malignant transformation in pleomorphic xanthoastrocytoma--a report of two cases. Br J Neurosurg. 1999;13:516–519. [PubMed] [Google Scholar]
- 8.Fu YJ, Miyahara H, Uzuka T, et al. Intraventricular pleomorphic xanthoastrocytoma with anaplastic features. Neuropathol. 2010;30:443–448. doi: 10.1111/j.1440-1789.2009.01080.x. [DOI] [PubMed] [Google Scholar]
- 9.Ng WH, Lim T, Yeo TT. Pleomorphic xanthoastrocytoma in elderly patients may portend a poor prognosis. J Clin Neurosci. 2008;15:476–478. doi: 10.1016/j.jocn.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 10.Prayson RA, Morris HH., 3rd Anaplastic pleomorphic xanthoastrocytoma. Arch Pathol Lab Med. 1998;122:1082–1086. [PubMed] [Google Scholar]
- 11.Hirose T, Ishizawa K, Sugiyama K, Kageji T, Ueki K, Kannuki S. Pleomorphic xanthoastrocytoma: a comparative pathological study between conventional and anaplastic types. Histopathology. 2008;52:183–193. doi: 10.1111/j.1365-2559.2007.02926.x. [DOI] [PubMed] [Google Scholar]
- 12.Sugita Y, Shigemori M, Okamoto K, Morimatsu M, Arakawa M, Nakayama K. Clinicopathological study of pleomorphic xanthoastrocytoma: correlation between histological features and prognosis. Pathol Int. 2000;50:703–708. doi: 10.1046/j.1440-1827.2000.01104.x. [DOI] [PubMed] [Google Scholar]
- 13.Tan TC, Ho LC, Yu CP, Cheung FC. Pleomorphic xanthoastrocytoma: report of two cases and review of the prognostic factors. J Clin Neurosci. 2004;11:203–207. doi: 10.1016/j.jocn.2003.04.003. [DOI] [PubMed] [Google Scholar]
- 14.Tekkök IH, Sav A. Anaplastic pleomorphic xanthoastrocytomas. Review of the literature with reference to malignancy potential. Pediatr Neurosurg. 2004;40:171–181. doi: 10.1159/000081935. [DOI] [PubMed] [Google Scholar]
- 15.Okazaki T, Kageji T, Matsuzaki K, et al. Primary anaplastic pleomorphic xanthoastrocytoma with widespread neuroaxis dissemination at diagnosis--a pediatric case report and review of the literature. J Neurooncol. 2009;94:431–437. doi: 10.1007/s11060-009-9876-6. [DOI] [PubMed] [Google Scholar]
- 16.Vu TM, Liubinas SV, Gonzales M, Drummond KJ. Malignant potential of pleomorphic xanthoastrocytoma. J Clin Neurosci. 2012;19:12–20. doi: 10.1016/j.jocn.2011.07.015. [DOI] [PubMed] [Google Scholar]
- 17.Korshunov A, Golanov A. Pleomorphic xanthoastrocytomas: immunohistochemistry, grading and clinico-pathologic correlations. An analysis of 34 cases from a single Institute. J Neurooncol. 2001;52:63–72. doi: 10.1023/a:1010648006319. [DOI] [PubMed] [Google Scholar]
- 18.Pahapill PA, Ramsay DA, Del Maestro R. Pleomorphic xanthoastrocytoma: case report and analysis of the literature concerning the efficacy of resection and the significance of necrosis. Neurosurgery. 1996;38:822–829. [PubMed] [Google Scholar]
- 19.Bayindir C, Balak N, Karasu A, Kasaroglu D. Anaplastic pleomorphic xanthoastrocytoma. Child's Nerv Syst. 1997;13:50–56. doi: 10.1007/s003810050040. [DOI] [PubMed] [Google Scholar]
- 20.Schindler G, Capper D, Meyer J, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011;121:397–405. doi: 10.1007/s00401-011-0802-6. [DOI] [PubMed] [Google Scholar]
- 21.Dougherty MJ, Santi M, Brose MS, et al. Activating mutations in BRAF characterize a spectrum of pediatric low-grade gliomas. Neuro-Oncology. 2010;12(7):621–630. doi: 10.1093/neuonc/noq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dias-Santagata D, Lam Q, Vernovsky K, et al. BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PLoS One. 2011;6:e17948. doi: 10.1371/journal.pone.0017948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balss J, Meyer J, Mueller W, et al. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008;116:597–602. doi: 10.1007/s00401-008-0455-2. [DOI] [PubMed] [Google Scholar]
- 24.Capper D, Weissert S, Balss J, et al. Characterization of R132H mutation-specific IDH1 antibody binding in brain tumors. Brain Pathol. 2010;20:245–254. doi: 10.1111/j.1750-3639.2009.00352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kepes JJ, Rubinstein LJ, Ansbacher L, Schreiber DJ. Histopathological features of recurrent pleomorphic xanthoastrocytomas: further corroboration of the glial nature of this neoplasm. A study of 3 cases. Acta Neuropathol. 1989;78:585–593. doi: 10.1007/BF00691285. [DOI] [PubMed] [Google Scholar]
- 26.Macaulay RJ, Jay V, Hoffman HJ, Becker LE. Increased mitotic activity as a negative prognostic indicator in pleomorphic xanthoastrocytoma. Case report. J Neurosurg. 1993;79:761–768. doi: 10.3171/jns.1993.79.5.0761. [DOI] [PubMed] [Google Scholar]
- 27.Kleinschmidt-DeMasters BK, Kang JS, Lillehei KO. The burden of radiation-induced central nervous system tumors: a single institution's experience. J Neuropathol Exp Neurol. 2006;65(3):204–216. doi: 10.1097/01.jnen.0000205146.62081.29. [DOI] [PubMed] [Google Scholar]