

Abstract
We found earlier that apoA-I variants that induced hypertriglyceridemia (HTG) in mice had increased affinity to TG-rich lipoproteins and thereby impaired their catabolism. Here, we tested whether a naturally occurring human apoA-I mutation, Lys107del, associated with HTG also promotes apoA-I binding to TG-rich particles. We expressed apoA-I[Lys107del] variant in Escherichia coli, studied its binding to TG-rich emulsion particles, and performed a physicochemical characterization of the protein. Compared with WT apoA-I, apoA-I[Lys107del] showed enhanced binding to TG-rich particles, lower stability, and greater exposure of hydrophobic surfaces. The crystal structure of truncated, Δ(185-243), apoA-I suggests that deletion of Lys107 disrupts helix registration and disturbs a stabilizing salt bridge network in the N-terminal helical bundle. To elucidate the structural changes responsible for the altered function of apoA-I[Lys107del], we studied another mutant, apoA-I [Lys107Ala]. Our findings suggest that the registry shift and ensuing disruption of the inter-helical salt bridges in apoA-I[Lys107del] result in destabilization of the helical bundle structure and greater exposure of hydrophobic surfaces. We conclude that the structural changes in the apoA-I[Lys107del] variant facilitate its binding to TG-rich lipoproteins and thus, may reduce their lipolysis and contribute to the development of HTG in carriers of the mutation.
Keywords: apolipoprotein A-I, natural mutation, high plasma triglyceride levels, triglyceride, very low density lipoprotein
Hypertriglyceridemia (HTG) is a lipid disorder characterized by elevated plasma levels of TGs. About one-third of the US population has elevated plasma TG levels (1), and the increase in plasma TG levels in recent years has been linked to the increasing prevalence of obesity (2). Conversely, low levels of TGs have been linked to longevity (3). HTG is identified as an independent risk factor of cardiovascular disease (1, 4, 5). Yet, the etiology of HTG in humans is poorly understood. A number of common metabolic conditions, including type 2 diabetes, metabolic syndrome, uremia, nephrotic syndrome and chronic alcohol consumption, are typically associated with HTG, mainly due to overproduction of TG-rich lipoproteins, primarily VLDLs, or due to reduced lipolysis of these lipoproteins (6). Single gene mutations that lead to deficiency of enzymes or other proteins involved in catabolism of TG-rich lipoproteins may result in severe HTG. Deficiency of LPL or its cofactor, apoC-II, is an example of such mutations (7, 8). However, in most cases of HTG, the causes of the disorder are unknown (8).
Several engineered variants of apoA-I have been found to induce HTG when expressed in animal models (9–11). A naturally occurring apoA-I mutation, a single lysine deletion, K107del (apoA-IHelsinki), is associated with HTG in humans (12, 13). Another natural human apoA-I mutation associated with HTG, apoA-INashua, was identified when this manuscript was in preparation (14). apoA-I is a major protein component of HDLs that provides the structural integrity of the lipoprotein particles and is critical for their function (15, 16). Plasma levels of apoA-I and HDL correlate inversely with the incidence of cardiovascular disease and atherosclerosis, and the mechanisms of these relationships relate to the key role of apoA-I and HDLs in the pathways of reverse cholesterol transport [(15–17) and references cited therein]. Accordingly, most human naturally occurring apoA-I mutations and engineered apoA-I mutations studied in animal models are associated with deficiency of apoA-I, HDLs, and HDL cholesterol [(9–16) and references cited therein].
The mechanisms by which certain apoA-I mutations may lead to increased plasma TG levels in humans or animals are not well understood. We showed earlier that the engineered apoA-I variants that induced HTG in animal models, apoA-I[E110A/E111A], apoA-I[del(61-78)], and apoA-I[D89A/E91A/E92A], had enhanced ability to bind to TG-rich emulsion particles in vitro (11, 18). In vivo studies of mice expressing these apoA-I variants showed increased apoA-I concentrations in plasma TG-rich lipoproteins and reduced lipolysis of these lipoproteins (9–11). Compared with the WT apoA-I, all the apoA-I mutants associated with HTG in animal models have destabilized conformation with greater exposure of hydrophobic surfaces in solution, which apparently facilitates the protein binding to TG-rich lipoproteins (18). We hypothesized that structural mutations in apoA-I that increase the protein’s ability to bind to TG-rich lipoproteins may, in some cases, contribute to HTG in humans. In the current study, we tested this hypothesis by studying the natural apoA-I mutation, K107del, that is associated with HTG in humans. We set out to investigate whether, similar to the engineered apoA-I mutations associated with HTG in animal models, the human K107del mutation also might lead to enhanced binding of apoA-I to TG-rich lipoproteins, thus contributing to the development of HTG in carriers.
The apoA-I[K107del] variant is found in Caucasian and Asian populations and is the most frequent apoA-I variant in the population of Germany (1:5,000) (13, 19). It is associated with HTG, hypoalphacholesterolemia, and amyloidosis (12, 13, 19, 20). Several groups have investigated the properties of the apoA-I[K107del] in order to clarify the molecular mechanisms underlying the development of the hypoalphacholesterolemia in carriers of this variant. It was found that the apoA-I[K107del] had increased catabolic rate (21) and reduced binding to HDLs (22), that likely leads to faster clearance of this variant from the circulation and ensuing apoA-I deficiency. It does not seem clear whether the K107del mutation affects the LCAT activation ability of apoA-I. The LCAT activation ability of the apoA-I[K107del] variant isolated from heterozygous individuals was found to be either lower than (19, 23) or similar to (24) that of WT apoA-I isolated from the same individuals. Also, studies of recombinant pro-apoA-I did not find any effect of the K107del mutation on the LCAT activation ability of apoA-I (22). Recent in vitro studies of recombinant apoA-I[K107del] revealed molecular features of this protein that may underlie its amyloidogenic propensity (25). However, regardless of the published studies on the apoA-I[Lys107del] variant, the mechanisms underlying the development of HTG in carriers of this mutation have not been addressed.
In the current study, we show that the K107del mutation results in enhanced binding of apoA-I to TG-rich emulsion particles. We also investigate the physicochemical properties of apoA-I[K107del] and show that this variant has a destabilized conformation with increased exposure of hydrophobic surfaces that may underlie its enhanced ability to bind to TG-rich particles. According to our proposed model for monomeric apoA-I based on the high resolution crystal structure of the C-terminally truncated human apoA-I, apoA-I[Δ(185-243)] (Figs. 1, 2) (26), the K107del mutation is expected to disrupt one of the major salt bridge networks stabilizing the protein structure and also result in a registry shift in the apolar face of the amphipathic helical conformation responsible for lipid interaction. In order to test whether the stabilizing salt bridge network involving K107 or the registry shift are responsible for the altered conformation and function of the apoA-I[K107del], we generated and investigated another apoA-I mutation, K107A, that was expected to disrupt the stabilizing salt bridge network involving the charged residue K107 without disturbing the registration.
Fig. 1.

Location of K107 in the crystal structure of the C-terminally truncated apoA-I. A: X-ray crystal structure of apoA-I[Δ(185-243)] showing a crystallographic dimer (Protein Data Bank entry number 3R2P). The crystallographic 2-fold axis that passes through the middle of the central helix 5 (residues 121-142) is shown. The red dashed rectangle outlines the part of the structure shown in (B). B: A major salt bridge network involving Lys107. The network consists of inter- and intra-molecular salt bridges. The distances between residues involved in the salt bridge network are indicated. The figure shows the region of the crystal structure outlined in (A) by the red dashed rectangle; the view is perpendicular to the red dashed rectangle plane.
Fig. 2.

Structure of the lipid-free C-terminally truncated apoA-I monomer. Location of K107 is shown. A: Overall structure of one molecule of apoA-I[Δ(185-243)] from the crystallographic dimer shown in Fig. 1A. The circular arrow around the dimer 2-fold axis (in the middle of Helix 5) shows the direction of folding back the helical repeats 6 and 7 upon the conversion of the apoA-I dimer conformation to the monomer conformation [shown in (B)]. B: Structure presumed for the C-terminally truncated apoA-I monomer in solution. This monomer structure of apoA-I[Δ(185-243)] was obtained from the structure of one molecule in the crystallographic dimer [shown in (A)] that is folded back in the middle of the central helix 5. The two conformational states [(A) and (B)] are proposed to interconvert dependent on protein concentration [(A) at high concentrations, when dimers are formed, and (B) at low concentrations, when protein is monomeric]. The red dashed rectangle outlines a part of the proposed monomer structure that corresponds to the part of the crystallographic dimer outlined similarly in Fig. 1.
EXPERIMENTAL PROCEDURES
Protein expression and purification
WT apoA-I, apoA-I[K107A], and apoA-I[K107del] were expressed in Escherichia coli BL21(DE3) CodonPlus-RIL cells (Stratagene, LaJolla, CA) using a His6-MBP-TEV expression system as described (26, 27). The mutations were made using the QuickChange mutagenesis kit (Stratagene) and confirmed by DNA sequencing. Protein purification was performed by subsequent chromatographies using Histrap columns (GE Healthcare) with a fast-protein liquid chromatography (FPLC) system (Pharmacia Biotech) as previously described (26). The purified proteins generated by this system contain a single glycine at the N-terminus derived from the TEV cleavage site (26, 27). Protein purity was analyzed by 12% SDS-PAGE followed by staining with Coomassie blue R250 and by Western blotting using monoclonal antibodies against human apoA-I. Protein identity and purity were verified by MALDI-TOF mass spectrometry (performed at the Molecular Biology Core Facilities at the Dana-Farber Cancer Institute and Tufts University Core Facility, Boston, MA). The determined molecular masses were 28,136 ± 7, 28,080 ± 6, and 28,005 ± 8 (mean ± SD of at least triple measurements) for WT apoA-I, apoA-I[K107A], and apoA-I[K107del], correspondingly, in agreement with the calculated masses of the designed protein sequences. Freshly purified proteins, with purity higher than 95%, were rapidly frozen in small aliquots using liquid nitrogen and stored at −80°C. Before each experiment, a protein aliquot was thawed and freshly refolded by dialysis against 4 M guanidine hydrochloride (GdnHCl) followed by extensive dialysis against Tris buffer, pH 7.6. The protein samples prepared for the circular dichroism (CD) experiments were refolded and then extensively dialyzed against 10 mM sodium phosphate, pH 7.6. The dialyzed proteins were stored at 4°C and used within 2 weeks. The proteins were periodically checked for proteolytic degradation by SDS-PAGE to make sure they remained intact.
Preparation of TG-rich emulsion particles
Binding of apoA-I to the emultion particles.
TG-rich emulsion particles were prepared by sonication of a triolein/egg yolk phosphatidylcholine (PC) mixture (4.5:1, w:w) in TBS and isolated by ultracentrifugation as previously described (18, 28). Isolated emulsions were visualized by negative staining electron microscopy to analyze morphology and to estimate the size of the particles. TG and PC contents of the emulsions were determined by the InfinityTM TG kit (Thermo Electron) and by the Bartlett phosphorus assay (29), respectively. Binding assays were performed within 2–3 days of isolation of the emulsion; no changes in morphology or particle size were detected during this period.
The binding of apoA-I to emulsion particles was assayed according to the protocol used in our previous emulsion-binding studies of the engineered apoA-I variants associated with HTG (11, 18). Briefly, a fixed amount (120 μg) of each apoA-I form was incubated with increasing amounts of emulsion in 1.8 ml of Tris buffer to give a PC:protein molar ratio ranging from 200 to 720. After incubation for 1 h at 27°C with gentle shaking, emulsion-bound and free apoA-I were separated by ultracentrifugation. Four 1 ml fractions were recovered after ultracentrifugation of each sample; all the fractions were assayed for their protein, phospholipid, and TG content, and in most cases, analyzed by electron microscopy. Protein concentrations were determined by the modified Lowry assay (30) in the presence of SDS or chloroform; TG and PC concentrations were determined as indicated above.
Electron microscopy
Negative staining electron microscopy analysis was performed to visualize isolated emulsions and fractions recovered after centrifugation of emulsion/protein mixtures as described (18, 28). The samples were applied to carbon-coated grids, stained with sodium phosphotungstate, and visualized in the Philips CM-12 electron microscope (Philips Electron Optics, Eindhoven, The Netherlands) and photographed. The major diameter of particles was determined from electron micrographs as an average of 100 to 120 particles.
DMPC turbidity clearance studies
The solubilization of dimyristoyl phosphatidylcholine (DMPC) multilamellar vesicles by apoA-I was monitored by the decrease in absorbance at 325 nm following the administration of apoA-I to a suspension of DMPC in Tris buffer as described previously (28), except for the final concentrations of DMPC and the protein in the mixture that were adjusted to 40 μg/ml and 16 μg/ml, respectively. DMPC clearance took place in the cuvettes within the spectrophotometer holders maintaining the controlled temperature of 24°C.
CD spectroscopy
Thermal and denaturant-induced unfolding.
Far-UV spectra of apoA-I in 10 mM sodium phosphate buffer (pH 7.4) were recorded at 25°C on AVIV 62DS or AVIV 215 spectropolarimeters (AVIV Associates, Inc.) at the protein concentration 25–60 μg/ml, as previously described (31, 32). Spectra were recorded at several protein concentrations, normalized, and expressed as mean residue ellipticity, [Θ]. The α-helical content was determined from the mean residue ellipticity at 222 nm, [Θ222] (33). Thermal and GdnHCl-induced unfolding of lipid-free apoA-I was monitored by changes in ellipticity at 222 nm. Thermal unfolding was carried out by heating the sample in the cuvette of the spectropolarimeter from 2 to 98°C with 1° increments. GdnHCl-induced unfolding experiments were performed at 25°C with a titration unit (Hamilton Company, USA) interfaced with the spectropolarimeter. Concentrations of GdnHCl increased from 0 to 2.7 M in 0.1 M increments. The melting (midpoint) temperature, Tm, and van’t Hoff enthalpy, ΔHv, were determined from van’t Hoff analysis of the melting curves, and the conformational stability, ΔGD0, m-values and the midpoint of chemical denaturation, D1/2, were determined from the GdnHCl-induced denaturation curves as described previously (31, 32).
8-Anilino-2-naphthalene-sulfonate fluorescence measurements
Fluorescence measurements were performed on a FluoroMax-2 fluorescence spectrometer (Instruments S.A., Inc.) at 25°C. Fluorescence emission spectra were recorded for 8-anilino-2-naphthalene-sulfonate (ANS) (0.25 mM) alone or in the presence of 0.05 mg/ml of lipid-free WT apoA-I, apoA-I[K107del], or apoA-I[K107A] in PBS buffer as previously described (32). The wavelength of maximum fluorescence (WMF) and fluorescence intensity at the WMF, I, were measured from the spectra after subtraction of the buffer baseline.
RESULTS
Influence of the mutations on binding of apoA-I to emulsion particles
The size of emulsion particles was determined from electron micrographs. For a typical emulsion, the average diameter was 65 ± 23 nm (mean ± SD; n = 108). The triolein:PC weight ratio in the isolated emulsions was 5.0 ± 0.4 (mean ± SD, for three isolated emulsions). These data show that, as judged by the average particle size, the broad size distribution, and the high content of triolein (about 83% of the total mass), the isolated emulsion particles resembled VLDLs, which justifies the use of the particles as a model for plasma TG-rich lipoproteins.
After incubations of emulsions with apoA-I and subsequent ultracentrifugation of the mixtures, the fractions recovered were characterized by composition (TG, PC, and protein concentrations) and visualized by electron microscopy. The analysis showed that bottom fractions contained lipid-free apoA-I and the top fractions contained emulsion-bound apoA-I. Control samples containing emulsion only or apoA-I only were incubated and spun in each experiment. The TG and PC concentrations and the particle size distribution of the fractions collected after centrifugation of the protein-emulsion mixtures were close to those collected after centrifugation of the corresponding control samples containing emulsion only. This result is consistent with binding of apoA-I to emulsion particles versus solubilization of emulsion lipids by apoA-I. The trace concentrations of free protein determined in the top fractions recovered after centrifugation of the control “protein-only” samples were used as the “background” free apoA-I concentrations to calculate the amounts of emulsion-bound apoA-I as described (28).
For each incubation mixture, the portion of bound protein was determined as the fraction of emulsion-bound apoA-I from the total amount of apoA-I in the mixture (bound and unbound). The portion of bound protein for each apoA-I form was plotted versus the PC:protein molar ratio in the incubation mixtures (Fig. 3A), in the manner described for the engineered apoA-I forms associated with HTG (18). This presentation of the binding data was chosen over plotting the concentration of emulsion-bound protein versus the concentration of unbound protein (34) because our assays were performed with a fixed amount of protein and increasing amounts of emulsion to minimize the amount of recombinant proteins required for the assays (18, 28). The plots in Fig. 3A illustrate representative binding curves obtained with one of three isolated emulsions. Binding assays performed with emulsions from different preparations might be conducted at slightly different PC:protein ratios (28); the standard deviations for the portion of bound protein were calculated for the PC:protein ratios for which at least three binding assays were performed. The values for the portion of bound protein (mean ± SD) for WT apoA-I and each apoA-I variant at three different PC:protein molar ratios in the incubation mixtures are compared in Table 1. Regardless of the initial proportion between apoA-I and emulsion during incubations, the portion of bound protein is significantly higher for apoA-I[K107del] than for WT apoA-I, indicating enhanced ability of the variant to bind to TG-rich particles. The binding curves for the apoA-I[K107A] were very close to those for WT apoA-I in all binding assays.
Fig. 3.

Binding of WT apoA-I and the apoA-I variants to TG-rich emulsion particles. apoA-I was incubated with TG-rich emulsion particles at various ratios of emulsion PC to protein, then emulsion-bound and unbound proteins were separated. A: Portion of bound protein. *, WT apoA-I; Δ, apoA-I[K107A]; ○, apoA-I[K107del]. B: Number of bound apoA-I molecules per emulsion particle. WT apoA-I (dark gray with diagonal pattern), apoA-I[K107A] (light gray), apoA-I[K107del] (black).
TABLE 1.
Binding of lipid-free WT apoA-I and the apoA-I mutant forms to TG-rich emulsion particles
| 257 ± 7 PC:Protein Molar Ratio | 351 ± 3 PC:Protein Molar Ratio | 697 ± 20 PC:Protein Molar Ratio | ||||
| Portion of the Bound Proteina | Protein on the Emulsion Particlesb | Portion of the Bound Proteina | Protein on the Emulsion Particlesb | Portion of the Bound Proteina | Protein on the Emulsion Particlesb | |
| WT apoA-I | 0.23 ± 0.02 | 0.34 ± 0.02 | 0.28 ± 0.01 | 0.22 ± 0.01 | 0.35 ± 0.02 | 0.15 ± 0.01 |
| apoA-I[K107A] | 0.22 ± 0.01 | 0.34 ± 0.01 | 0.28 ± 0.02 | 0.21 ± 0.01 | 0.35 ± 0.02 | 0.17 ± 0.01 |
| apoA-[K107del] | 0.30 ± 0.02c | 0.41 ± 0.02c | 0.36 ± 0.03c | 0.28 ± 0.02c | 0.48 ± 0.03d | 0.21 ± 0.02c |
Values are mean ± SD from at least three binding assays. Each apoA-I form was incubated with emulsion at various PC:protein molar ratios. Data are shown for three different PC:protein molar ratios in the incubation mixtures.
Portion of emulsion-bound protein of the total amount of protein added to emulsion.
Parameter derived from composition of apoA-I- emulsion complexes recovered by ultracentrifugation following the incubation of apoA-I with emulsion. Number of amino acids/molecule of PC.
P < 0.05 compared with the value for WT apoA-I.
P < 0.01 compared with the value for WT apoA-I.
The resultant protein-emulsion complexes were characterized by relative protein content, in terms of a number of amino acids of bound protein per molecule of PC (Table 1). These values were calculated from the weight ratio of emulsion-bound protein to PC in the top fractions recovered after ultracentrifugation. At each ratio of PC:protein in incubation mixtures, the resultant particles containing apoA-I[K107del] had a higher number of amino acids per PC molecule, suggesting that the larger surface area of these particles is covered with protein, as compared with the particles containing WT apoA-I. The number of amino acids per PC molecule in the emulsion-protein complexes containing apoA-I[K107A] were not significantly different from those in the complexes containing WT apoA-I. We also characterized the resultant protein-emulsion complexes by the average numbers of protein, phospholipid, and triolein molecules per particle. These numbers were calculated on the basis of the average particle diameter (estimated from the electron micrographs) and the composition of the resultant complexes, as described previously (18). The number of phospholipid and triolein molecules per particle [(19 ± 1.5) × 103 and (80 ± 4) × 103, correspondingly] did not change significantly between all the protein-emulsion complexes. However, the average numbers of apoA-I molecules per particle were higher for the particles containing apoA-I[K107del] than for the particles containing WT apoA-I at each ratio of PC:protein in the incubation mixtures (Fig. 3B). The average numbers of apoA-I molecules per particle were not significantly different for the particles containing apoA-I[K107A] or WT apo A-I. These data support the conclusion that the K107del mutation, but not the K10A mutation, increases the ability of apoA-I to bind to TG-rich particles.
DMPC turbidity clearance
To compare the ability of the apoA-I variants to bind lipids, we performed standard DMPC-binding assays that examined the kinetics of solubilization of DMPC multilamellar vesicles by the proteins. The low concentration of the protein (16 μg/ml) was used to avoid protein oligomerization. The time courses of DMPC turbidity clearance by the apoA-I forms (Fig. 4) show that, compared with WT apoA-I, apoA-I[K107del] solubilizes the lipids at a faster rate, while apoA-I[K107A] solubilizes the lipids at a slower rate. The clearance curves were fitted to an exponential decay function and half-times (t1/2) corresponding to a 50% decrease in turbidity were determined for each protein. For WT apoA-I, t1/2 was 19.5 ± 1.0 min (mean ± SD of three experiments). Compared with the value of t1/2 for WT apoA-I, t1/2 for the apoA-I[K107del] was reduced (14.4 ± 0.9 min, P < 0.01), while t1/2 for apo A-I[K107A] was increased (23.0 ± 1.2 min, P < 0.05).
Fig. 4.
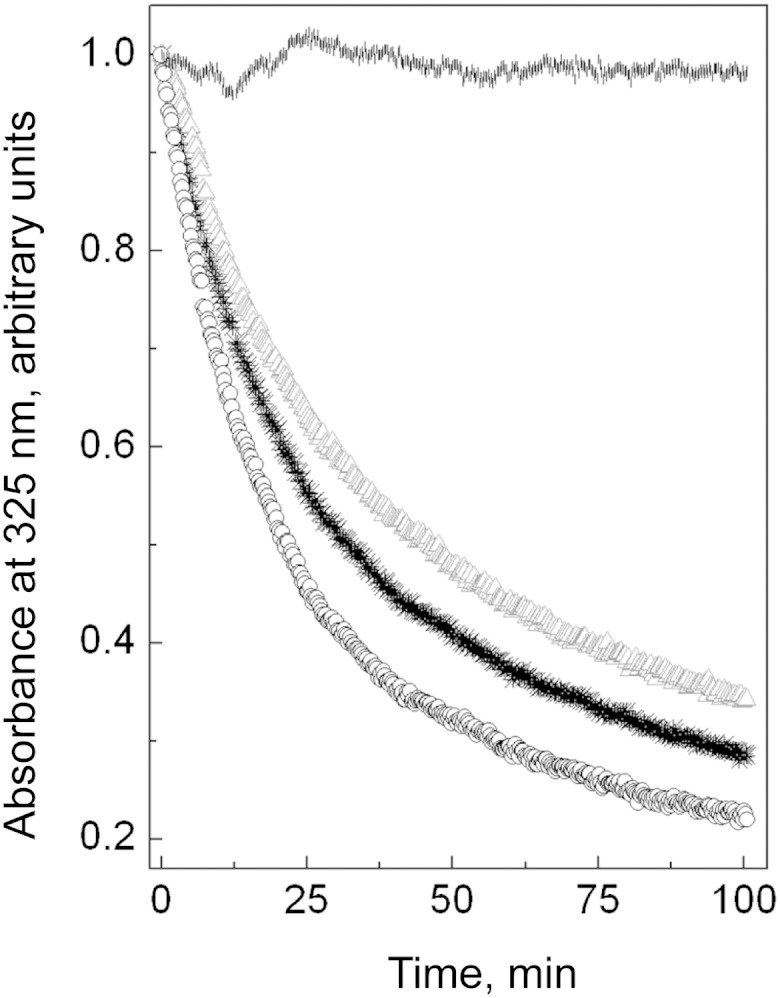
The time course of DMPC turbidity clearance by the apoA-I forms or in buffer alone. DMPC multilamellar vesicles (40 μg/ml lipids) were preincubated at 24°C in the cuvette within the spectrophotometer, and the clearance was triggered by addition of lipid-free protein (final concentration in cuvette 16 μg/ml). *, WT apoA-I; Δ, apoA-I[K107A]; ○, apoA-I[K107del]; |, buffer alone.
CD analysis of the α-helical content and stability
Normalized far-UV CD spectra of apoA-I variants (not shown) were used to determine the average α-helical content in the lipid-free proteins. The spectra were recorded at protein concentrations lower than 55 μg/ml to avoid protein aggregation. No changes in the normalized spectra were observed with variations in the protein concentrations within the range of the protein concentrations studied, which is consistent with the absence of the protein self-association. The apoA-I[K107del] variant had slightly lower α-helical content than WT apoA-I (Table 2), but the difference was not statistically significant. The K107A mutation did not affect the α-helical content.
TABLE 2.
α-Helical content, thermodynamic parameters, and ANS fluorescence for lipid-free WT apoA-I and the apoA-I mutant forms
| α-Helixa(%) | Tmb(°C) | ΔHvb(kcal/mol) | Δ Gc (kcal/mol) | mc[kcal (moles of apoA-I)−1 (moles of GdnHCl)−1] | D1/2c (M) | Id (relative units) | WMFd (nm) | |
| WT apoA-I | 58 ± 2 | 63 ± 1 | 43 ± 2 | 4.3 ± 0.1 | 4.0 ± 0.2 | 1.0 ± 0.05 | 1.0 | 474 |
| apoA-I[K107A] | 58 ± 3 | 62 ± 1 | 46 ± 2 | 4.6 ± 0.2 | 4.5 ± 0.1e | 1.0 ± 0.0 | 0.8 | 477 (+3) |
| apoA-[K107del] | 55 ± 1 | 57 ± 2e | 29 ± 1g | 3.0 ± 0.1e | 3.7 ± 0.1 | 0.8 ± 0.0f | 1.5 | 468 (−6) |
Values are mean ± SD from at least three experiments.
Estimated from the [Θ222] at 25°C.
The melting temperature, Tm, and van’t Hoff enthalpy, ΔHv, were determined from van’t Hoff analysis of thermal unfolding curves monitored by CD.
The conformational stability, ΔGD0, m-values and midpoint of chemical denaturation, D1/2, were determined by the linear extrapolation method from the curves for CD-monitored GdnHCl-induced unfolding.
Parameters of ANS fluorescence were determined in the presence of WT apoA-I, apoA-I[K107A], or apoA-[K107del]. I, fluorescence intensity in relative units compared with the fluorescence in the presence of WT apoA-I.
P < 0.05 compared with the values for WT apoA-I.
P < 0.01 compared with the values for WT apoA-I.
P < 0.005 compared with the values for WT apoA-I.
The thermal and GdnHCl-induced unfolding of the apoA-I forms was monitored by the ellipticity at 222 nm; the typical recorded unfolding curves are shown in Fig. 5A, B, correspondingly. The average parameters determined from the unfolding curves are listed in Table 2. The K107del mutation led to a substantial shift of the melting curve to lower temperatures indicating protein destabilization and made unfolding less cooperative (Fig. 5A). Accordingly, the analysis of the melting curves shows a significant (∼6°C) decrease in the melting temperature Tm, and a large (∼14 kcal/mol) decrease in the effective enthalpy ΔHv. In contrast, the K107A mutation had no significant effect on the shape of the melting curve or parameters of thermal unfolding of apoA-I, indicating that this mutation does not change the stability of apoA-I. The shift of the Gdn-induced unfolding curve for the apoA-I[K107del] variant to the lower denaturant concentrations (Fig. 5B) and the corresponding decrease (∼0.2 M) in the midpoint of denaturation, D1/2, along with a large reduction (∼1.3 kcal/mol) in the conformational stability, ΔGD0, indicate a strong destabilizing effect of the K107del mutation, in accord with the results from the thermal unfolding experiments. The K107A mutation did not affect the midpoint of chemical denaturation, and the small increase in the ΔGD0 was not statistically significant. However, the K107A mutation led to a small (∼0.5 kcal/mol apoA-I/mol GdnHCl) but statistically significant increase in the m-value, suggesting the increased cooperativity of the protein unfolding.
Fig. 5.

Unfolding of the lipid-free WT apoA-I and the apoA-I variants monitored by ellipticity at 222 nm. A: Thermal unfolding was induced by heating the protein from 2 to 98°C in the cuvette within the CD spectrometer holder. B: Chemical unfolding was induced by adding GdnHCl, in 0.1 M increments, to the protein inside the cuvette within the CD spectrometer holder. *, WT apoA-I; Δ, apoA-I[K107A]; ○, apoA-I[K107del].
ANS fluorescence
To assess the effect of the mutations on exposure of hydrophobic surfaces or cavities of apoA-I, ANS fluorescence was recorded alone in buffer and in the presence of WT apoA-I or each mutant apoA-I form (Fig. 6). Table 2 shows the values for WMF and the fluorescence intensity at the WMF, I; the latter is expressed in relative units, taking the ANS fluorescence intensity in the presence of WT apoA-I as one. A 50% increase in the fluorescence intensity and a significant (∼6 nm) “blue” shift of the ANS spectrum in the presence of apoA-I[K107del], compared with the spectrum in the presence of WT apoA-I, indicate a significantly increased exposure of hydrophobic surfaces in the apoA-I[K107del] mutant. This observation suggests that the K107del mutation results in a more loosely folded tertiary conformation of the protein. In contrast, the ANS spectrum in the presence of apoA-I[K107A] had lower intensity (∼20% decrease) and was “red” shifted (by ∼3 nm) compared with the ANS spectrum in the presence of WT apoA-I, indicating lesser exposure of hydrophobic surfaces in apoA-I[K107A] that is consistent with a more compact folding of this variant.
Fig. 6.
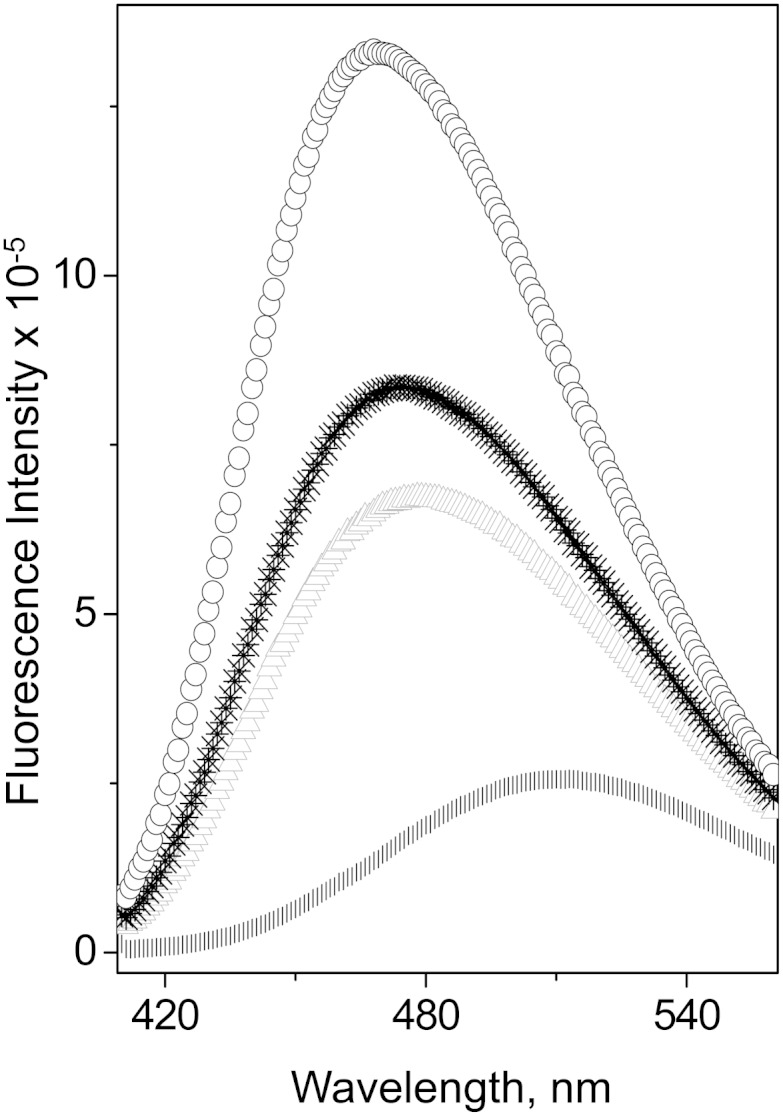
Fluorescence spectra of ANS in the presence of lipid-free apoA-I variants. Final sample concentrations are 250 μM ANS and 50 μg/ml protein. *, WT apoA-I; Δ, apoA-I[K107A]; ○, apoA-I[K107del]; |, in buffer alone.
DISCUSSION
We showed earlier that the engineered apoA-I variants that cause HTG in mice have increased ability to bind to TG-rich emulsion particles in vitro (11, 18). TG-rich lipoprotein fractions from mice expressing these apoA-I variants had significantly increased content of apoA-I, and catabolism of these lipoproteins was impaired (9–11). In this study, we wanted to determine whether the naturally occurring apoA-I mutation, K107del, that is associated with HTG in humans, also promotes binding of apoA-I to TG-rich lipoproteins and thus may affect lipolysis of these lipoproteins. We expressed and purified the apoA-I[K107del] variant and tested it in binding assays using synthetic TG-rich emulsion particles, similar to the manner by which we tested the engineered apoA-I variants associated with HTG in mice (11, 18). The assays showed that the K107del mutation promoted binding of apoA-I to TG-rich emulsion particles and led to an increased content of apoA-I on the particles. These findings infer that, similar to the engineered apoAI variants causing HTG in mice, the natural human apoA-I[K107del] variant has an increased level of association with plasma TG-rich lipoproteins. The enhanced binding of apoA-I to TG-rich lipoproteins in vivo may have several implications for catabolism of these lipoproteins and development of HTG, as discussed previously (18). First, the higher ratio of protein to phospholipid on the surface of TG-rich lipoproteins leads to reduced fluidity of the lipid monolayer (35, 36) that may impede the access of LPL to the substrate, and thus inhibit TG hydrolysis. This concept agrees with the in vitro studies demonstrating impaired lipolysis by LPL of TG-rich lipoproteins with increased apoA-I content (9, 11). Second, enhanced binding of apoA-I to TG-rich lipoproteins may lead to displacement of apoC-II, the activator of LPL, from these lipoproteins. It was shown that engineered apoA-I variants that demonstrated enhanced binding to TG-rich emulsions in vitro (18) can displace apoC-II from plasma TG-rich lipoproteins in vivo (9, 10). The displacement of apoC-II from the lipoproteins impairs their lipolysis and may also lead to faster clearance of apoC-II from the circulation, thus further contributing to HTG. Third, apoA-I bound to TG-rich lipoproteins directly inhibits hydrolysis of these lipoproteins by hepatic lipase (37). Finally, the accumulation of apoA-I on the surface of VLDLs may diminish the surface area available for apoE molecules, and thereby result in apoE conformations with the receptor binding region concealed or the N-terminal domain displaced from the lipid surface (34, 38). These conformational changes may preclude interaction of apoE with the LDL receptor and impede clearance of remnant lipoproteins (38). All these metabolic consequences of the enhanced binding of apoA-I[K107del] to TG-rich lipoproteins may contribute to the higher plasma TG levels in carriers of the mutation.
To understand the molecular basis for the enhanced ability of apoA-I[K107del] to bind to TG-rich particles, we investigated the physicochemical characteristics of the apoA-I forms. Thermal and denaturant-induced unfolding studies indicate lower conformational stability of apoA-I[K107del]. Protein unfolding was monitored by ellipticity at 222 nm and thus, reflected changes in the secondary conformation. Ramella et al. (25) also found a destabilizing effect of the K107del mutation when apoA-I unfolding was monitored by a fluorescent probe that reflected changes in the tertiary conformation. Increased exposure of the hydrophobic surfaces of apoA-I[K107del] in solution (as indicated by ANS binding studies) and low cooperativity of unfolding (Table 2) are consistent with a partially folded or molten globular-like conformation (32, 39), which provides flexibility and adaptability for substantial conformational changes that accompany protein binding to the surface of large lipoprotein particles. Low conformational stability facilitates the conformational changes, and greater exposure of hydrophobic surfaces of apoA-I[K107del] makes its binding to the lipid surface of TG-rich lipoproteins favorable. These conformational and stability characteristics of apoA-I[K107del] can also account for the faster solubilization of DMPC vesicles by this variant.
The high-resolution crystal structure of the C-terminally truncated, Δ(185-243), human apoA-I (26) helps to understand mechanistically the structural changes in apoA-I caused by the K107del mutation. In the crystal structure (Fig. 1A), apoA-I[Δ(185-243)] forms a dimer comprised of two antiparallel molecules. The structure is stabilized by two four-segment bundles, each comprised of three helical regions and an extended section, one at each end of the dimer. Figure 1B shows the region of the crystal structure surrounding K107 and the salt bridge network involving this residue. Figure 2A shows one molecule from the crystallographic dimer, and Fig. 2B shows our proposed lipid-free structure of the apoA-I[Δ(185-243)] monomer that is derived from the structure of one molecule of the crystallographic dimer (Fig. 2A) by folding back the C-terminal part at the helix 5 region through a domain swap mechanism. The secondary structure assignment for residues in the proposed conformation (Fig. 2B) is close to that found in hydrogen exchange and mass spectrometry studies (40), and the tertiary conformation is similar to the four-helical bundle core region of apoA-I inferred from cross-linking studies (41, 42). These observations, together with recent studies that explain conformational and functional features of apoA-I and its natural variants based on the crystal structure and the proposed lipid-free conformation (43, 44), support the view that the proposed conformation for the lipid-free truncated apoA-I (Fig. 2B) represents the conformation of the corresponding region (residues 1-184) in full-length lipid-free human apoA-I. The four-segment bundle in the proposed lipid-free monomeric conformation (Fig. 2B) is similar to that observed at both ends of the crystallographic dimer (Fig. 1), but with all four segments belonging to the same molecule. Deletion of K107 is expected to eliminate two intra-helical salt bridges, K107-E110 and K107-E11, and also disrupt the registration of the apolar face of the helical segment (Fig. 1B) that may result in partial helix unfolding. The registry shift leads to the movement of residue E111 away from residues H155 and R151 of the antiparallel helix, and thus is expected to disrupt the inter-helical salt bridges E111-H155 and E111-R151 and destabilize the four-segment bundle leading to exposure of hydrophobic surfaces buried inside the bundle. Fluorescence analysis of apoA-I with scanning tryptophan mutagenesis (45) is also consistent with destabilization of the four-segment bundle in the apoA-I[K107del]. According to the fluorescence data, residue W108 is the least solvent exposed of all tryptophan residues of apoA-I, substantiating that this residue is located deep inside the hydrophobic core created by the helical bundle (45). Deletion of K107 and the ensuing registry shift result in an ∼100° turn of the position of W108 in helix, so that hydrophobic W108 becomes exposed to the solvent (in place of charged K107), thus destabilizing the bundle structure. Exactly these properties (lower stability and loosely folded conformation with greater exposure of hydrophobic surfaces) were shown for the apoA-I[K107del] variant by the physicochemical analysis, supporting the concept of disruption of the four-segment bundle structure.
The integrity of the four-segment bundle of apoA-I was reported to be crucial for the protein overall structure and function (43, 45, 46). Therefore, we surmised that the stabilizing salt bridge network involving K107 might be essential for maintaining the apoA-I conformation and function. To test this assumption, we generated and studied the K107A mutation that, similar to the K107del mutation, was expected to disrupt the stabilizing salt bridge network involving K107 but, in contrast to the K107del, would not disrupt the registration. Contrary to expectations, the apoA-I[K107A] variant was not destabilized. Furthermore, it unfolded slightly more cooperatively, solubilized DMPC vesicles more slowly, and had slightly lesser exposure of hydrophobic surfaces than WT apoA-I, substantiating that this mutant is slightly more compactly folded than WT apoA-I. Based on the crystal structure (Fig. 1B), the substitution of K107 by Ala eliminates two relatively weak intra-helical salt bridges, K107-E110 and K107-E111, but may allow residue E110 to move closer to K106, while residue E111 can turn closer toward R151 and H155 of the antiparallel helix, potentially resulting in stronger salt bridges, intra-helical (E110-K106) and inter-helical (E111-R151 and E111-H155). Thus, the K107A mutation, which does not disturb the registry and apparently leads to stronger salt bridge interactions, promotes the structural integrity of the four-segment bundle. From the fluorescence data of single tryptophan mutants of apoA-I (45), the substitution of Ala for charged Lys on the outside of the four-segment bundle would not be expected to disturb the hydrophobic core created by the bundle. In accordance, the apoA-I[K107A] mutant does not show enhanced binding to TG-rich particles. These studies support the notion that disruption of the four-segment bundle structure of apoA-I is required for enhanced binding of the protein to TG-rich particles.
Remarkably, the engineered mutations of apoA-I associated with HTG in mice (9–11, 18), i.e., E110A/E111A, D89A/E91A/E92A, and del(61-78), are expected to disrupt the four-segment bundle structure, according to the proposed lipid-free conformation of the truncated apoA-I based on the crystal structure (Figs. 1, 2B). The naturally occurring human mutation apoA-INashua [insertion of six amino acids, GARAHL, in positions 157 through 162 (14)] is also expected to eliminate the salt bridge interactions between antiparallel helixes (Figs. 1, 2B) leading to destabilization of the four-segment bundle structure that may enhance binding of apoA-INashua to TG-rich lipoproteins, and thus contribute to HTG.
Human K107del and apoA-INashua mutations are associated with HTG that is combined with low plasma HDL cholesterol (13, 14). The engineered apoA-I variants that induce HTG in mice also lead to low plasma levels of HDL cholesterol and apoA-I (9–11). Combined HTG and low plasma HDL cholesterol are also common in insulin resistance states, including metabolic syndrome and type 2 diabetes (47, 48). However, the mechanisms underlying combined HTG and low plasma HDL caused by apoA-I mutations may be different from those caused by insulin resistance. In insulin resistance states, high plasma TG levels lead to enrichment of HDL with TG, thus triggering rapid catabolism of HDL by hepatic lipase, dissociation of apoA-I from HDL and ensuing faster clearance of apoA-I from the circulation, and low plasma HDL levels (47–49). In combined HTG and low plasma HDL caused by apoA-I mutations, the altered structure of apoA-I may directly enhance its clearance from the circulation (14, 21, 22) and also affect various reverse cholesterol transport pathways that directly modulate plasma HDL cholesterol and apoA-I levels, such as LCAT activation, ABCA-1 mediated efflux of cholesterol, or the interaction with scavenger receptor class B type I (9–11, 15, 16, 50, 51), while enhanced binding of the mutated apoA-I to TG-rich lipoproteins may be one of the mechanisms contributing to HTG.
apoA-I mutations may cause various metabolic changes [(9–11, 15, 16, 23, 46, 50, 51) and references cited therein], and some of the changes may potentially be involved in the development of HTG. Further studies may unravel additional mechanisms contributing to the development of HTG associated with apoA-I mutations. It is possible, for example, that apoA-I mutations associated with HTG may impair the ability of apoA-I to liberate hepatic lipase from heparan sulfate proteoglycans (37, 52) and thus lead to a reduced amount of the active enzyme and contribute to HTG. Our study shows that enhanced binding of apoA-I to TG-rich lipoproteins may be one of the factors contributing to the development of HTG associated with human apoA-I mutation.
Acknowledgments
The authors thank Donald L. Ganz for help with electron microscopy analysis, Cheryl England and Michael Gigliotti for help with biochemical assays, and the Molecular Biology Core Facilities at the Dana-Farber Cancer Institute and Tufts University Core Facility for mass spectrometry analysis of the proteins.
Footnotes
Abbreviations:
- ANS
- 8-anilino-2-naphthalene-sulfonate
- CD
- circular dichroism
- DMPC
- dimyristoyl phosphatidylcholine
- GdnHCl
- guanidine hydrochloride
- HTG
- hypertriglyceridemia
- PC
- phosphatidylcholine
- WMF
- wavelength of maximum fluorescence
This work was supported by National Institutes of Health Grant NHLBI P0HL 26335.
REFERENCES
- 1.Miller M., Stone N. J., Ballantyne C., Bittner V., Criqui M. H., Ginsberg H. N. 2011. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 123: 2292–2333. [DOI] [PubMed] [Google Scholar]
- 2.Eckel R. H. 2011. The complex metabolic mechanisms relating obesity to hypertriglyceridemia. Arterioscler. Thromb. Vasc. Biol. 31: 1946–1948. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez-Covarrubias V., Beekman M., Uh H. W., Dane A., Troost J., Paliukhovich I., van der Kloet F. M., Houwing-Duistermaat J., Vreeken R. J., Hankemeier T., et al. 2013. Lipidomics of familial longevity. Aging Cell. 12: 426–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nordestgaard B. G., Benn M., Schnohr P., Tybjaerg-Hansen A. 2007. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 298: 299–308. [DOI] [PubMed] [Google Scholar]
- 5.Kannel W. B., Vasan R. S. 2009. Triglycerides as vascular risk factors: new epidemiologic insights. Curr. Opin. Cardiol. 24: 345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hassing H. C., Surendran R. P., Mooij H. L., Stroes E. S., Nieuwdorp M., Dallinga-Thie G. M. 2012. Pathophysiology of hypertriglyceridemia. Biochim. Biophys. Acta. 1821: 826–832. [DOI] [PubMed] [Google Scholar]
- 7.Johansen C. T., Kathiresan S., Hegele R. A. 2011. Genetic determinants of plasma triglycerides. J. Lipid Res. 52: 189–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johansen C. T., Hegele R. A. 2012. The complex genetic basis of plasma triglycerides. Curr. Atheroscler. Rep. 14: 227–234. [DOI] [PubMed] [Google Scholar]
- 9.Chroni A., Kan H-Y., Kypreos K. E., Gorshkova I. N., Shkodrani A., Zannis V. I. 2004. Substitutions of Glu110 and Glu111 in the middle helix 4 of human apoA-I by alanine affect the structure and in vitro functions of apoA-I and induce severe hypertriglyceridemia in apoA-I-deficient mice. Biochemistry. 43: 10442–10457. [DOI] [PubMed] [Google Scholar]
- 10.Chroni A., Kan H-Y., Shkodrani A., Liu T., Zannis V. I. 2005. Deletions of helices 2 and 3 of human apoA-I are associated with severe dyslipidemia following adenovirus-mediated gene transfer in apoA-I-deficient mice. Biochemistry. 44: 4108–4117. [DOI] [PubMed] [Google Scholar]
- 11.Kateifides A. K., Gorshkova I. N., Duka A., Chroni A., Kardassis D., Zannis V. I. 2011. Alteration of negatively charged residues in the 89 and 96 domain of apoA-I affects lipid homeostasis and maturation of HDL. J. Lipid Res. 52: 1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.von Eckardstein A., Funke H., Walter M., Altland K., Benninghoven A., Assmann G. 1990. Structural analysis of human apolipoprotein A-I variants. Amino acid substitutions are nonrandomly distributed throughout the apolipoprotein A-I primary structure. J. Biol. Chem. 265: 8610–8617. [PubMed] [Google Scholar]
- 13.Nofer J. R., von Eckardstein A., Wiebusch H., Weng W., Funke H., Schulte H., Köhler E., Assmann G. 1995. Screening for naturally occurring apolipoprotein A-I variants: apo A-I(delta K107) is associated with low HDL-cholesterol levels in men but not in women. Hum. Genet. 96: 177–182. [DOI] [PubMed] [Google Scholar]
- 14.Lee E. Y., Klementowicz P. T., Hegele R. A., Asztalos B. F., Schaefer E. J. 2013. HDL deficiency due to a new insertion mutation (ApoA-I Nashua) and review of the literature. J. Clin. Lipidol. 7: 169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sorci-Thomas M. G., Thomas M. J. 2002. The effects of altered apolipoprotein A-I structure on plasma HDL concentration. Trends Cardiovasc. Med. 12: 121–128. [DOI] [PubMed] [Google Scholar]
- 16.Zannis V. I., Chroni A., Krieger M. 2006. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J. Mol. Med. 84: 276–294. [DOI] [PubMed] [Google Scholar]
- 17.Rye K. A., Barter P. J. 2014. Cardioprotective functions of HDLs. J. Lipid Res. 55: 168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorshkova I. N., Atkinson D. 2011. Enhanced binding of apolipoprotein A-I variants associated with hypertriglyceridemia to triglyceride-rich particles. Biochemistry. 50: 2040–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rall S. C., Jr, Weisgraber K. H., Mahley R. W., Ogawa Y., Fielding C. J., Utermann G., Haas J., Steinmetz A., Menzel H. J., Assmann G. 1984. Abnormal lecithin:cholesterol acyltransferase activation by a human apolipoprotein A-I variant in which a single lysine residue is deleted. J. Biol. Chem. 259: 10063–10070. [PubMed] [Google Scholar]
- 20.Amarzguioui M., Mucchiano G., Häggqvist B., Westermark P., Kavlie A., Sletten K., Prydz H. 1998. Extensive intimal apolipoprotein A1-derived amyloid deposits in a patient with an apolipoprotein A1 mutation. Biochem. Biophys. Res. Commun. 242: 534–539. [DOI] [PubMed] [Google Scholar]
- 21.Tilly-Kiesi M., Lichtenstein A. H., Ordovas J. M., Dolnikowski G., Malmström R., Taskinen M. R., Schaefer E. J. 1997. Subjects with ApoA-I(Lys107→0) exhibit enhanced fractional catabolic rate of ApoA-I in Lp(AI) and ApoA-II in Lp(AI with AII). Arterioscler. Thromb. Vasc. Biol. 17: 873–880. [DOI] [PubMed] [Google Scholar]
- 22.Huang W., Matsunaga A., Li W., Han H., Hoang A., Kugi M., Koga T., Sviridov D., Fidge N., Sasaki J. 2001. Recombinant proapoA-I(Lys107del) shows impaired lipid binding associated with reduced binding to plasma high density lipoprotein. Atherosclerosis. 159: 85–91. [DOI] [PubMed] [Google Scholar]
- 23.Jonas A., von Eckardstein A., Churgay L., Mantulin W. W., Assmann G. 1993. Structural and functional properties of natural and chemical variants of apolipoprotein A-I. Biochim. Biophys. Acta. 1166: 202–210. [DOI] [PubMed] [Google Scholar]
- 24.Tilly-Kiesi M., Zhang Q., Ehnholm S., Kahri J., Lahdenperä S., Ehnholm C., Taskinen M. R. 1995. ApoA-IHelsinki (Lys107→0) associated with reduced HDL cholesterol and LpA-I:A-II deficiency. Arterioscler. Thromb. Vasc. Biol. 15: 1294–1306. [DOI] [PubMed] [Google Scholar]
- 25.Ramella N. A., Schinella G. R., Ferreira S. T., Prieto E. D., Vela M. E., Ríos J. L., Tricerri M. A., Rimoldi O. J. 2012. Human apolipoprotein A-I natural variants: molecular mechanisms underlying amyloidogenic propensity. PLoS ONE. 7: e43755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mei X., Atkinson D. 2011. Crystal structure of C-terminal truncated apolipoprotein A-I reveals the assembly of high density lipoprotein (HDL) by dimerization. J. Biol. Chem. 286: 38570–38582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nallamsetty S., Waugh D. S. 2007. A generic protocol for the expression and purification of recombinant proteins in Escherichia coli using a combinatorial His6-maltose binding protein fusion tag. Nat. Protoc. 2: 383–391. [DOI] [PubMed] [Google Scholar]
- 28.Gorshkova I. N., Kypreos K. E., Gantz D. L., Zannis V. I., Atkinson D. 2008. Biophysical properties of apolipoprotein E4 variants: implications in molecular mechanisms of correction of HTG. Biochemistry. 47: 12644–12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bartlett G. R. 1959. Phosphorus assay in column chromatography. J. Biol. Chem. 234: 466–468. [PubMed] [Google Scholar]
- 30.Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193: 265–275. [PubMed] [Google Scholar]
- 31.Gorshkova I. N., Liu T., Zannis V. I., Atkinson D. 2002. Lipid-free structure and stability of apolipoprotein A-I: probing the central region by mutation. Biochemistry. 41: 10529–10539. [DOI] [PubMed] [Google Scholar]
- 32.Gorshkova I. N., Liu T., Kan H. Y., Chroni A., Zannis V. I., Atkinson D. 2006. Structure and stability of apolipoprotein A-I in solution and in discoidal high-density lipoprotein probed by double charge ablation and deletion mutation. Biochemistry. 45: 1242–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Y. H., Yang J. T., Martinez H. M. 1972. Determination of the secondary structures of proteins by circular dichroism and optical rotatory dispersion. Biochemistry. 11: 4120–4131. [DOI] [PubMed] [Google Scholar]
- 34.Saito H., Dhanasekaran P., Baldwin F., Weisgraber K. H., Lund-Katz S., Phillips M. C. 2001. Lipid binding-induced conformational change in human apolipoprotein E. Evidence for two lipid-bound states on spherical particles. J. Biol. Chem. 276: 40949–40954. [DOI] [PubMed] [Google Scholar]
- 35.Gorshkova I. N., Menschikowski M., Jaross W. 1996. Alterations in the physicochemical characteristics of low and high density lipoproteins after lipolysis with phospholipase A2. A spin-label study. Biochim. Biophys. Acta. 1300: 103–113. [DOI] [PubMed] [Google Scholar]
- 36.Tetali S. D., Budamagunta M. S., Simion C., den Hartigh L. J., Kálai T., Hideg K., Hatters D. M., Weisgraber K. H., Voss J. C., Rutledge J. C. 2010. VLDL lipolysis products increase VLDL fluidity and convert apolipoprotein E4 into a more expanded conformation. J. Lipid Res. 51: 1273–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramsamy T. A., Neville T. A., Chauhan B. M., Aggarwal D., Sparks D. L. 2000. Apolipoprotein A-I regulates lipid hydrolysis by hepatic lipase. J. Biol. Chem. 275: 33480–33486. [DOI] [PubMed] [Google Scholar]
- 38.Narayanaswami V., Ryan R. O. 2000. Molecular basis of exchangeable apolipoprotein function. Biochim. Biophys. Acta. 1483: 15–36. [DOI] [PubMed] [Google Scholar]
- 39.Semisotnov G. V., Rodionova N. A., Razgulyaev O. I., Uversky V. N., Gripas A. F., Gilmanshin R. I. 1991. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers. 31: 119–128. [DOI] [PubMed] [Google Scholar]
- 40.Chetty P. S., Mayne L., Lund-Katz S., Stranz D., Englander S. W., Phillips M. C. 2009. Helical structure and stability in human apolipoprotein A-I by hydrogen exchange and mass spectrometry. Proc. Natl. Acad. Sci. USA. 106: 19005–19010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silva R. A., Hilliard G. M., Fang J., Macha S., Davidson W. S. 2005. A three-dimensional molecular model of lipid-free apolipoprotein A-I determined by cross-linking/mass spectrometry and sequence threading. Biochemistry. 44: 2759–2769. [DOI] [PubMed] [Google Scholar]
- 42.Pollard R. D., Fulp B., Samuel M. P., Sorci-Thomas M. G., Thomas M. J. 2013. The conformation of lipid-free human apolipoprotein A-I in solution. Biochemistry. 52: 9470–9481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gursky O., Mei X., Atkinson D. 2012. The crystal structure of the C-terminal truncated apolipoprotein A-I sheds new light on amyloid formation by the N-terminal fragment. Biochemistry. 51: 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gursky O. 2013. Crystal structure of Δ(185-243)ApoA-I suggests a mechanistic framework for the protein adaptation to the changing lipid load in good cholesterol: from flatland to sphereland via double belt, belt buckle, double hairpin and trefoil/tetrafoil. J. Mol. Biol. 425: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davidson W. S., Arnvig-McGuire K., Kennedy A., Kosman J., Hazlett T. L., Jonas A. 1999. Structural organization of the N-terminal domain of apolipoprotein A-I: studies of tryptophan mutants. Biochemistry. 38: 14387–14395. [DOI] [PubMed] [Google Scholar]
- 46.Tanaka M., Dhanasekaran P., Nguyen D., Nickel M., Takechi Y., Lund-Katz S., Phillips M. C., Saito H. 2011. Influence of N-terminal helix bundle stability on the lipid-binding properties of human apolipoprotein A-I. Biochim. Biophys. Acta. 1811: 25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rashid S., Watanabe T., Sakaue T., Lewis G. F. 2003. Mechanisms of HDL lowering in insulin resistant, hypertriglyceridemic states: the combined effect of HDL triglyceride enrichment and elevated hepatic lipase activity. Clin. Biochem. 36: 421–429. [DOI] [PubMed] [Google Scholar]
- 48.Ji J., Watts G. F., Johnson A. G., Chan D. C., Ooi E. M., Rye K. A., Serone A. P., Barrett P. H. 2006. High-density lipoprotein (HDL) transport in the metabolic syndrome: application of a new model for HDL particle kinetics. J. Clin. Endocrinol. Metab. 91: 973–979. [DOI] [PubMed] [Google Scholar]
- 49.Lamarche B., Rashid S., Lewis G. F. 1999. HDL metabolism in hypertriglyceridemic states: an overview. Clin. Chim. Acta. 286: 145–161. [DOI] [PubMed] [Google Scholar]
- 50.Sorci-Thomas M. G., Bhat S., Thomas M. J. 2009. Activation of lecithin: cholesterol acyltransferase by HDL ApoA-I central helices. Clin. Lipidol. 4: 113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith L. E., Davidson W. S. 2010. The role of hydrophobic and negatively charged surface patches of lipid-free apolipoprotein A-I in lipid binding and ABCA1-mediated cholesterol efflux. Biochim. Biophys. Acta. 1801: 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chatterjee C., Sparks D. L. 2011. Hepatic lipase, high density lipoproteins, and hypertriglyceridemia. Am. J. Pathol. 178: 1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]