

Abstract
The vast majority of human plague cases currently occur in sub-Saharan Africa. The primary route of transmission of Yersinia pestis, the causative agent of plague, is via flea bites. Non-pathogenic flea-associated bacteria may interact with Y. pestis within fleas and it is important to understand what factors govern flea-associated bacterial assemblages. Six species of fleas were collected from nine rodent species from ten Ugandan villages between October 2010 and March 2011. A total of 660,345 16S rRNA gene DNA sequences were used to characterize bacterial communities of 332 individual fleas. The DNA sequences were binned into 421 Operational Taxonomic Units (OTUs) based on 97% sequence similarity. We used beta diversity metrics to assess the effects of flea species, flea sex, rodent host species, site (i.e. village), collection date, elevation, mean annual precipitation, average monthly precipitation, and average monthly temperature on bacterial community structure. Flea species had the greatest effect on bacterial community structure with each flea species harboring unique bacterial lineages. The site (i.e. village), rodent host, flea sex, elevation, precipitation, and temperature also significantly affected bacterial community composition. Some bacterial lineages were widespread among flea species (e.g. Bartonella spp. and Wolbachia spp.), but each flea species also harbored unique bacterial lineages. Some of these lineages are not closely related to known bacterial diversity and likely represent newly discovered lineages of insect symbionts. Our finding that flea species has the greatest effect on bacterial community composition may help future investigations between Yersinia pestis and non-pathogenic flea-associated bacteria. Characterizing bacterial communities of fleas during a plague epizootic event in the future would be helpful.
Introduction
Since 2000, greater than 95% of reported cases of human plague have occurred in sub-Saharan Africa [1]. Models incorporating 10-year meteorological data and human plague incidence data found that, in Uganda, plague risk increases in sites located above 1,300 meters in elevation and with increased (but not continuous) rains in February, October, and November [2]. In addition, the number of human plague cases in the West Nile region was negatively associated with dry season rainfall and positively with rainfall during the interval rainy season that immediately precedes plague transmission season [3]. Increased rainfall may increase primary production, which may, in turn, increase rodent and flea abundance [2,4,5]. Increases in flea abundance are predicted to increase the risk of a plague epizootic event [6].
In addition to flea abundance, flea-associated microbial communities may also contribute to plague transmission. Yersinia pestis, the causative agent of plague, reduces the abundance of or completely eliminates specific bacterial lineages within fleas [7], and exposing laboratory-reared fleas to diverse wild-type microbial communities increases transmission of Y. pestis [8]. Although determining the presence of particular lineages (e.g. Rickettsia spp., Bartonella spp., Yersinia pestis) in wild fleas has been routine, the characterization of entire bacterial communities of wild fleas has been limited [9,10]. The microbial compositions of two closely related fleas (Oropsylla hirsuta vs. Oropsylla tuberculata cynomuris) and of two more distantly related fleas (Orchopeas leucopus vs. Ctenophthalmus pseudagyrtes) were not found to differ [9,10]. However, flea-associated bacterial communities shifted drastically over three years in both species of flea studied [10]. Shifts in microbial communities over time are often due to concomitant shifts in environmental conditions [11–15], but environmental conditions have not previously been explored in relation to insect-associated bacterial communities.
Symbionts of disease vectors may mediate the spread of disease through negative or positive interactions with pathogens. Wolbachia-positive mosquitos have suppressed rates of infection by dengue virus, Chikungunya virus, West Nile virus, and Plasmodium spp. [16–18], and introducing Wolbachia-positive mosquitos to a natural population has proven to be an effective means to decrease the number of potential vectors of human disease [19]. The entire insect-associated microbiome can also influence pathogen persistence; dengue virus titers in sterile Aedes aegypti midguts are significantly higher than titers in A. aegypti with wildtype microbiomes [20]. These negative effects of vector-associated microbes on pathogens do not seem to occur between flea-associated bacteria and Y. pestis: exposing ‘germ-free’ fleas to wildtype microbes increases transmission of Y. pestis [8] and infecting wild fleas (with wild-type microbiomes) with Y. pestis eliminates specific bacterial lineages within fleas [7]. Flea-borne viruses have yet to be studied, but interactions between viruses and Y. pestis may also alter the ability of fleas to transmit Y. pestis and would be a novel research pursuit in the future.
In this study, we characterized the bacterial communities of six flea species collected from nine species of rodents in March 2011 from ten sites in a plague-endemic area of Uganda. For the two most abundant flea species, we analyzed additional samples collected in October and December 2010. Due to our sampling strategy, we can test for the effects of rodent host, flea species, site, environmental conditions, and time on flea-associated bacterial communities.
Materials and Methods
Flea Samples
Field collection permits are not required in Uganda, but all protocols for this work were reviewed and approved by the Uganda Virus Research Institute Science and Ethics Committee, the Uganda National Council of Science and Technology, and the Uganda President’s Office. All research protocols involving animals (e.g. trapping animals to capture fleas) was also approved by the Animal Care and Use Committee of the Division of Vector-Borne Disease at the United States Centers for Disease Control and Prevention. Fleas were collected at three times across ten sites in Uganda [21]. Briefly, Tomahawk and Sherman live traps were used, traps were set at dusk and and retrieved the next morning, animals were released after sampling, and no animals died during trapping; for full details, please see the original publication that details flea and rodent diversity across these sites [21]. The three collection times correspond to the peak of the primary rainy season (October 2010), the dry season (November/December 2010), and the beginning of the secondary rainy season (March 2011). We used a subset of these fleas to investigate flea-associated bacterial communities. Xenopsylla brasiliensis and Xenopsylla cheopis were examined from the three different collection periods; Ctenophthalmus calceatus cabirus, Dinopsyllus lypusus, Stivalius torvus, and Xenopsylla nubica were examined from the March 2011 collection (Table 1). Fleas analyzed in this study were collected from nine taxa of rodents: Aethomys hindei, Arvicanthis niloticus, Crocidura spp., Lophuromys flavopunctatus, Lemniscomys striatus, Mastomys spp., Rattus rattus, Taterillus emini, and Tatera valida (Table 2).
Table 1. Number of individual fleas analyzed from each flea species across three collection periods.
| Site | Ccc | Dl | St | Xb | Xc | Xn | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mar | Mar | Mar | Oct | Nov/Dec | Mar | Oct | Nov/Dec | Ma | Mar | |
| 1 | - | - | - | - | - | - | 2 | 4 | 7 | - |
| 2 | - | - | - | - | - | - | 9 | 15 | 16 | - |
| 3 | - | - | - | - | - | - | 13 | 10 | 14 | - |
| 4 | - | - | - | - | - | - | 11 | 6 | 13 | - |
| 5 | - | - | - | - | - | - | 8 | 8 | 17 | 7 |
| 6 | - | - | - | - | - | - | 3 | 7 | 14 | 9 |
| 7 | - | - | - | - | - | 4 | - | - | 1 | - |
| 8 | - | - | - | 9 | 7 | 12 | - | - | - | - |
| 9 | 13 | 12 | 16 | 9 | 10 | 14 | - | - | - | - |
| 10 | - | 4 | - | 2 | 9 | 16 | - | - | - | - |
| Total | 13 | 16 | 16 | 20 | 26 | 46 | 46 | 50 | 82 | 16 |
Ccc: Ctenophthalmus calceatus cabirus, Dl: Dinopsyllus lypusus, St: Stivalius torvus, Xb: Xenopsylla brasiliensis, Xc: Xenopsylla cheopis, Xn: Xenopsylla nubica.
Oct: October, 2010; Nov/Dec: November/December, 2010; Mar: March, 2011.
Table 2. Distribution of fleas across mammalian hosts.
| Host | Ccc | Dl | St | Xb | Xc | Xn |
|---|---|---|---|---|---|---|
| A. hindei | 8 | |||||
| A. niloticus | 9 | 12 | 37 | 72 | ||
| Crocidura spp. | 1 | 15 | 3 | 8 | ||
| L. flavopunctatus | 1 | |||||
| L. striatus | 3 | |||||
| Mastomys spp. | 4 | 8 | 15 | |||
| R. rattus | 44 | 75 | ||||
| T. emini | 2 | |||||
| T. valida | 14 | |||||
| Total | 13 | 16 | 16 | 92 | 178 | 16 |
Fleas: Ccc: Ctenophthalmus calceatus cabirus, Dl: Dinopsyllus lypusus, St: Stivalius torvus, Xb: Xenopsylla brasiliensis, Xc: Xenopsylla cheopis, Xn: Xenopsylla nubica.
Hosts: A. hindei: Aethomys hindei; A. niloticus: Arvicanthis niloticus; Crocidura spp.: Crocidura species; L. flavopunctatus: Lophuromys flavopunctatus; L. striatus: Lemniscomys striatus; M. natalensis: Mastomys species; R. rattus: Rattus rattus; T. emini: Taterillus emini; T. valida: Tatera valida.
DNA Extractions.
Fleas were stored in 70% ethanol upon collection. Prior to DNA extraction, individual fleas were surface-sterilized by soaking in 10% bleach for 30 seconds and then washed twice with 100% ethanol. Surface-sterilized fleas were subjected to 20 minutes of mechanical lysis using a Retsch MM301 homogenizer, and then DNA was extracted using the MO BIO PowerSoil-htp 96 Well Soil DNA Isolation Kit (Carlsbad, CA) with the standard protocol.
DNA Sequencing
We amplified the V1 and V2 hypervariable regions of the 16S rRNA gene using previously described primers: the forward primer (5’-GCCTTGCCAGCCCGCTCAGTCAGAGTTTGATCCTGGCTCAG-3’) contains the 16S rRNA gene 27f primer, the 454 Life Sciences primer B sequence, and a two-base ‘TC’ linker; the reverse primer (5′-GCCTCCCTCGCGCCATCAGNNNNNNNNNNNNCATGC TGCCTCCCGTAGGAGT-3′) contains a 12 bp error-correcting barcode, the 16S rRNA gene 338r primer, the Life Sciences primer A sequence, and a two-base ‘CA’ linker [22]. We amplified the DNA samples using the following conditions: Initial denaturation at 94°C for 5 min; then 35 cycles of 94°C for 45s, 50°C for 30s, 72°C for 90s; with a final extension at 72°C for 10 min. Each amplification was performed in triplicate and PCR products from the three independent reactions were combined and cleaned using the MO BIO UltraClean-htp 96 Well PCR Clean-Up Kit (Carlsbad, CA). The concentration of each sample was estimated using the Quant-iT PicoGreen dsDNA Assay Kit (Life Technologies, Carlsbad, CA). Normalized and cleaned bar-tagged PCR products were combined into a single sample and sent to EnGenCore (Columbia, SC) for DNA sequencing on a Roche Genome Sequencer FLX using Titanium reagents.
Sequence Analysis
We analyzed DNA sequence data using QIIME v1.8 [23]. Sequences were assigned to their flea sample based on unique barcodes and were filtered using QIIME’s default quality settings. Sequences were truncated to 280 basepairs and Operational Taxonomic Units (OTUs) were selected using the uclust algorithm and a 97% sequence similarity threshold [24]. The most abundant sequence within an OTU was chosen as its representative sequence, and representative sequences were aligned using PyNAST [25]. Aligned sequences were filtered against the greengenes core set alignment and screened for chimeras using ChimeraSlayer and chimeric sequences were removed from the dataset. DNA sequences representing less than 0.005% of all sequences were removed from the dataset. Flea samples with less than 300 DNA sequences were removed from the dataset. The final dataset included 660,345 DNA sequences from 332 fleas (range: 305–4279 DNA sequences per flea). These DNA sequences were binned into 421 OTUs (Accession #’s: KT589425 –KT589833). We assigned taxonomic classifications to the OTUs based on the RDP database, as implemented within QIIME. We estimated a phylogeny of the OTUs using FastTree [26].
Alpha Diversity
Alpha diversity is a measure of diversity at a local scale [27]; here it refers to the amount of bacterial diversity found within an individual flea. We used flea samples represented by at least 1,000 DNA sequences (n = 282) to estimate alpha diversity. We rarefied the dataset to 1,000 (i.e. 1000 DNA sequences were randomly chosen from each flea sample). From this rarefied dataset, alpha diversity was estimated in two ways using QIIME v1.8: observed species and phylodiversity. Observed species is simply the number of unique OTUs represented in each sample. Phylodiversity is a phylogenetic alpha diversity metric and represents the sum of branch lengths represented by a single community given a phylogenetic tree constructed using all potential community members [28].
Beta Diversity
For beta diversity measurements, we rarefied the dataset to 300 (i.e. 300 DNA sequences were randomly chosen from each flea sample). Pairwise dissimilarity matrices were created in three ways using QIIME v1.8: Bray-Curtis, UniFrac, and Weighted UniFrac [29–31]. These metrics differ in how they assess community membership: the Bray-Curtis distance uses bacterial OTU presence and abundance to compare communities, but does not account for phylogenetic relatedness of the OTUs; UniFrac is a measure of shared phylogenetic diversity, as assessed by shared branch lengths between the communities; Weighted UniFrac is similar to UniFrac but also accounts for the relative abundance of OTUs. Each of these metrics provides dissimilarity values between individual communities with the value ranging from 0 (exact same communities in both samples) to 1 (no overlap in community membership). The effects of flea species, site, host, and elevation on bacterial community composition across all samples were tested using an Analysis of Similarity as implemented in QIIME v1.8. Likewise, we used an Analysis of Similarity to test the effects of host, site, collection date, and flea sex on bacterial community composition within flea species. We also tested the effects of environmental conditions on bacterial community composition. We used Euclidean distances to create pairwise dissimilarity matrices of elevation, mean annual precipitation, average monthly precipitation, and average monthly temperature for each sample. Each pairwise dissimilarity matrix for each environmental variable was compared individually to the bacterial community dissimilarity matrices using a Mantel test implemented in QIIME v1.8. Environmental conditions were previously described [21]. Finally, principal coordinates of the bacterial community dissimilarity matrices were created and used to generate 2-dimensional plots based on flea species.
Results
A total of 660,345 DNA sequences were grouped into 421 OTUs based on 97% sequence similarity. The vast majority of bacteria within the six flea species belonged to four bacterial phyla: Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria (Fig 1). Within C. calceatus cabirus, a Spiroplasma species was also common (Table 3).
Fig 1. Average relative abundances of bacterial phyla within flea species.

Proteobacteria were further divided based on Class: Alphaproteobacteria, Betaproteobacteria, and Gammaproteobacteria. Ccc: Ctenophthalmus calceatus cabirus, Dl: Dinopsyllus lypusus, St: Stivalius torvus, Xb: Xenopsylla brasiliensis, Xc: Xenopsylla cheopis, Xn: Xenopsylla nubica. Xenopsylla nubica were not sexed.
Table 3. Average relative abundances of most common bacterial OTUs detected in Ugandan fleas.
| Taxonomic Classification | BLAST | Ccc (f) | Ccc (m) | Dl (f) | Dl (m) | St (f) | St (m) | Xb (f) | Xb (m) | Xc (f) | Xc (m) | Xn |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (# individuals) | % | (5) | (8) | (8) | (8) | (7) | (9) | (40) | (35) | (89) | (84) | (16) |
| Bartonella sp. | 99 | 7.4% | 23.8% | 35.6% | 19.7% | 8.3% | 58.7% | 1.2% | 1.0% | 3.0% | 3.6% | 49.5% |
| Wolbachia sp. | 98 | 1.7% | 1.1% | 23.7% | 13.0% | 9.3% | 1.1% | 35.8% | 5.9% | 69.4% | 16.1% | 4.0% |
| Wolbachia sp. | 98 | 24.8% | 10.5% | 0.0% | 0.4% | 71.4% | 0.3% | 0.0% | 0.5% | 0.0% | 0.0% | 0.0% |
| Lariskella sp.* | 100 | 3.6% | 4.2% | 35.9% | 54.0% | 0.0% | 1.4% | 0.0% | 0.0% | 0.8% | 0.0% | 0.0% |
| Cardinium sp. | 99 | 0.1% | 0.2% | 0.0% | 0.0% | 0.0% | 0.4% | 34.8% | 36.8% | 1.0% | 0.1% | 0.0% |
| Spiroplasma sp.* | 99 | 26.3% | 29.7% | 0.0% | 0.1% | 0.0% | 0.2% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
| Bartonella sp.* | 100 | 0.5% | 7.3% | 0.0% | 0.0% | 3.4% | 22.1% | 0.0% | 0.0% | 0.8% | 0.0% | 0.0% |
| Pasteurellaceae (family) | 90 | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 10.1% | 21.1% | 0.0% | 0.0% | 0.0% |
| Pasteurellaceae (family) | 96 | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.8% | 0.5% | 4.9% | 21.1% | 1.7% |
| Betaproteobacteria (class) | 93 | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 1.7% | 0.3% | 1.0% | 21.3% | 4.5% |
| Propionibacterium acnes | 100 | 3.8% | 0.8% | 0.1% | 0.5% | 0.1% | 0.4% | 3.3% | 5.0% | 1.8% | 3.1% | 1.7% |
| Pasteurellaceae (family) | 95 | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.1% | 0.2% | 15.4% |
Ccc: Ctenophthalmus calceatus cabirus, Dl: Dinopsyllus lypusus, St: Stivalius torvus, Xb: Xenopsylla brasiliensis, Xc: Xenopsylla cheopis, Xn: Xenopsylla nubica. Taxonomy of bacterial DNA sequences was determined using the Ribosomal Database Project classification scheme.
*: The taxonomy of DNA sequence was further classified using BLAST against GenBank’s nucleotide database.
Of the 421 OTUs detected, only 12 represented at least 1% of bacteria detected across all flea species, on average (Table 3). The most common and widespread OTU was a lineage within the Bartonella genus, and the next four most common lineages were those related to known endosymbionts (e.g. Wolbachia, Cardinium). Besides the most common Bartonella lineage and the most common Wolbachia lineage, most of the common lineages tended to be dominant community members within one flea species but rare community members in other flea species. Three lineages within the Pasteurellaceae were abundant in one species, but rare or absent in other species (Table 3).
The observed number of OTUs within fleas ranged from 8.1, on average, in S. torvus females to 36.9, on average, in X. cheopis males (Fig 2A). Females had significantly less observed OTUs than males in D. lypusus, S. torvus, and X. cheopis. The phylodiversity ranged from 1.36%, on average, in female S. torvus to 4.65%, on average, in male X. cheopis (Fig 2B). Females had significantly less phylodiversity than males in D. lypusus, S. torvus, X. brasiliensis, and X. cheopis.
Fig 2. Estimates of alpha diversity for flea-associated bacterial communities.
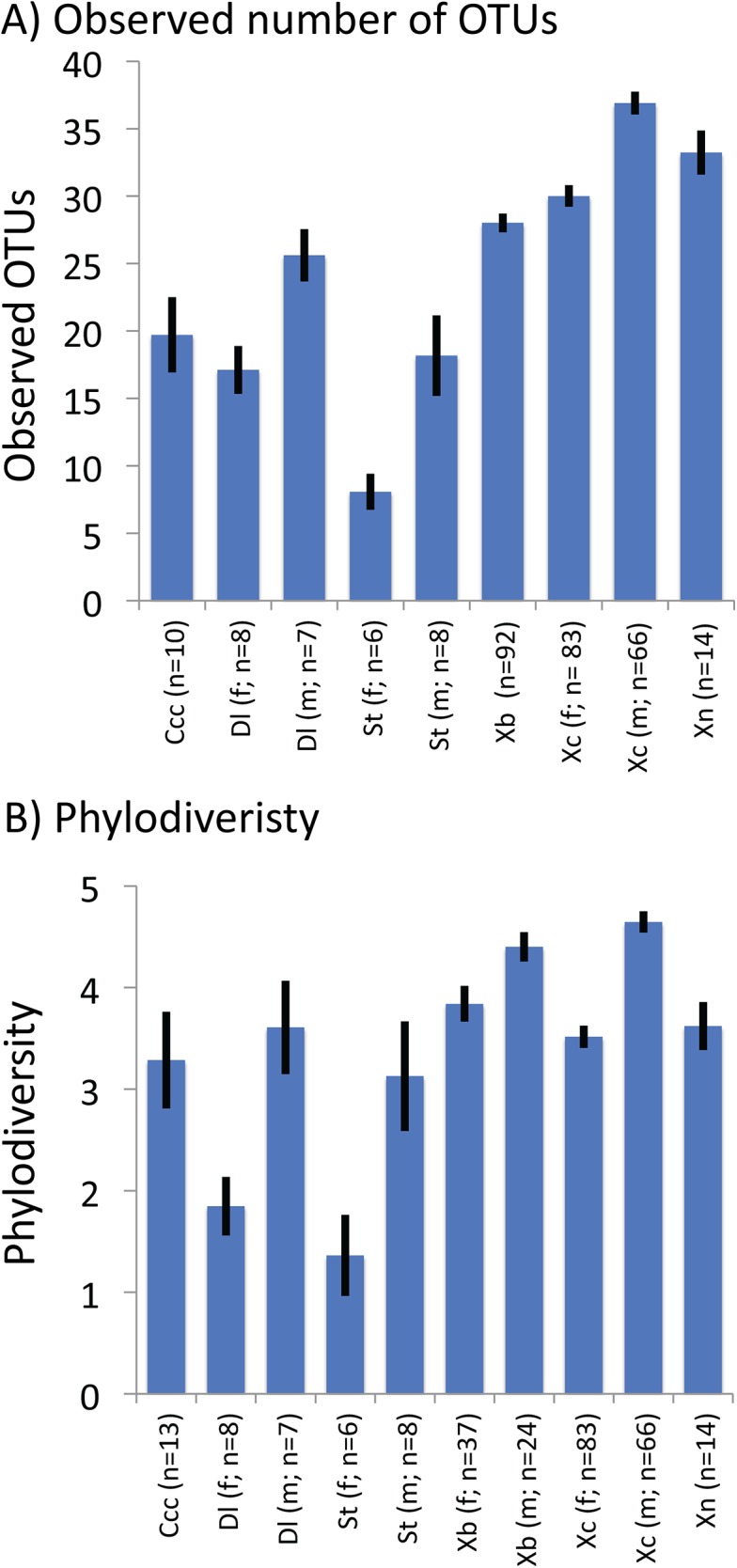
Alpha diversity was measured as the total number of observed OTUs detected in a subset of 1000 randomly chosen sequences from a sample (A) and as phylodiversity of bacteria within a sample (B). Diversity did not significantly differ between males and females of C. c. cabirus and sex was not determined for X. nubica. The number of observed species did not significantly differ in X. brasiliensis. In all other comparisons, male fleas harbored significantly more diversity based on student’s t-tests. Ccc: Ctenophthalmus calceatus cabirus, Dl: Dinopsyllus lypusus, St: Stivalius torvus, Xb: Xenopsylla brasiliensis, Xc: Xenopsylla cheopis, Xn: Xenopsylla nubica.
The flea species had a large and significant effect on the bacterial community (Fig 3; Table 4); site, rodent host, and elevation (above 1,300m vs. below 1,300m) also varied significantly with community composition when all samples were analyzed simultaneously (Table 4). The effects of host, site, collection date, and sex on bacterial communities within flea species varied depending on the flea species, the factor analyzed, and the metric used to compare communities (Table 5). The effect of sex was widespread across different flea species with males and females having different bacterial communities in D. lypusus (UniFrac only), S. torvus, X. brasiliensis, and X. cheopis. Host, site, and collection date also significantly affected bacterial communities in Xenopsylla species, depending on the metric used to compare communities (Table 5).
Fig 3. Principal coordinate analysis (PCoA) of flea-associated bacterial communities based on flea species.

PCoA was performed based on Bray-Curtis dissimilarities (A) and on weighted UniFrac distances (B). The percentage of variation explained by axes one and two are presented in parentheses. Green: Ctenophthalmus calceatus cabirus, Purple: Dinopsyllus lypusus, Yellow: Stivalius torvus, Blue: Xenopsylla brasiliensis, Red: Xenopsylla cheopis, Orange: Xenopsylla nubica.
Table 4. Analysis of Similarity of bacterial communities (All Samples).
| Species | Site | Host | Elevation* | |||||
|---|---|---|---|---|---|---|---|---|
| R | p-value | R | p-value | R | p-value | R | p-value | |
| Bray-Curtis | 0.601 | 0.001 | 0.237 | 0.001 | 0.110 | 0.001 | 0.420 | 0.001 |
| UniFrac | 0.343 | 0.001 | 0.151 | 0.001 | 0.106 | 0.001 | 0.245 | 0.001 |
| Weighted UniFrac | 0.458 | 0.001 | 0.105 | 0.001 | 0.110 | 0.001 | 0.278 | 0.001 |
*: Elevation was categorized as above 1300 meters (plague positive) and below 1300 meters (plague negative).
Table 5. Analysis of Similarity of bacterial communities (Within Flea Species).
| Host | Site | Date | Sex | |||||
|---|---|---|---|---|---|---|---|---|
| R | p-value | R | p-value | R | p-value | R | p-value | |
| Bray-Curtis | ||||||||
| C. cabirus | -0.25 | 0.985 | - | - | - | - | -0.17 | 0.918 |
| D. lypusus | -0.12 | 0.799 | -0.06 | 0.632 | - | - | 0.04 | 0.253 |
| S. torvus | - | - | - | - | - | - | 0.75 | 0.001 |
| X. brasiliensis | 0.07 | 0.039 | 0.10 | 0.002 | 0.19 | 0.001 | 0.15 | 0.001 |
| X. cheopis | 0.05 | 0.021 | 0.12 | 0.001 | 0.00 | 0.388 | 0.44 | 0.001 |
| X. nubica | 0.28 | 0.182 | 0.09 | 0.232 | - | - | - | - |
| UniFrac | ||||||||
| C. cabirus | -0.09 | 0.742 | - | - | - | - | 0.02 | 0.309 |
| D. lypusus | -0.21 | 0.911 | -0.06 | 0.618 | - | - | 0.16 | 0.038 |
| S. torvus | - | - | - | - | - | - | 0.39 | 0.007 |
| X. brasiliensis | 0.17 | 0.001 | 0.05 | 0.23 | 0.09 | 0.017 | 0.08 | 0.003 |
| X. cheopis | 0.08 | 0.004 | 0.06 | 0.001 | 0.05 | 0.004 | 0.21 | 0.001 |
| X. nubica | 0.30 | 0.086 | 0.28 | 0.019 | - | - | - | - |
| Weighted UniFrac | ||||||||
| C. cabirus | -0.11 | 0.805 | - | - | - | - | -0.10 | 0.818 |
| D. lypusus | -0.06 | 0.612 | -0.09 | 0.774 | - | - | 0.03 | 0.263 |
| S. torvus | - | - | - | - | - | - | 0.74 | 0.001 |
| X. brasiliensis | 0.11 | 0.002 | 0.11 | 0.002 | 0.20 | 0.001 | 0.06 | 0.012 |
| X. cheopis | 0.08 | 0.009 | 0.10 | 0.001 | -0.02 | 0.821 | 0.28 | 0.001 |
| X. nubica | 0.10 | 0.209 | 0.00 | 0.428 | - | - | - | - |
Elevation, mean annual precipitation, and average monthly temperature significantly co-varied with bacterial community composition across all samples (Table 6). Within X. cheopis, bacterial communities co-varied with elevation, mean annual precipitation, and average monthly temperature; within X. brasiliensis, bacterial communities co-varied with average monthly precipitation (Table 6).
Table 6. Mantel Tests comparing bacterial community structure to environmental parameters.
| Elevation | MAP | Precipitation | Temp (avg) | |||||
|---|---|---|---|---|---|---|---|---|
| R | p-value | R | p-value | R | p-value | R | p-value | |
| Bray-Curtis | ||||||||
| All samples | 0.28 | 0.001 | 0.20 | 0.001 | -0.02 | 0.337 | 0.16 | 0.001 |
| X. brasiliensis | 0.01 | 0.842 | 0.05 | 0.087 | 0.14 | 0.001 | 0.08 | 0.021 |
| X. cheopis | 0.12 | 0.001 | 0.15 | 0.001 | 0.00 | 0.978 | 0.08 | 0.001 |
| UniFrac | ||||||||
| All samples | 0.19 | 0.001 | 0.12 | 0.001 | -0.01 | 0.712 | 0.07 | 0.001 |
| X. brasiliensis | -0.01 | 0.750 | 0.04 | 0.273 | 0.06 | 0.191 | 0.10 | 0.003 |
| X. cheopis | 0.00 | 0.965 | -0.04 | 0.104 | 0.00 | 0.968 | -0.02 | 0.503 |
| Weighted UniFrac | ||||||||
| All samples | 0.17 | 0.001 | 0.11 | 0.001 | -0.04 | 0.123 | 0.11 | 0.001 |
| X. brasiliensis | 0.01 | 0.759 | 0.05 | 0.096 | 0.15 | 0.003 | 0.10 | 0.009 |
| X. cheopis | 0.12 | 0.002 | 0.15 | 0.001 | -0.01 | 0.784 | 0.07 | 0.036 |
MAP: Mean Annual Precipitation, Precipitation: Average monthly precipitation.
Discussion
Bacterial lineages within Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria dominated community membership of the six flea species examined in this study (Fig 1). These same bacterial phyla have repeatedly been shown to dominate bacterial communities in previous studies of fleas [9,10], other disease vectors [9,32–35], a wide diversity of insects [36–38], and animals in general [39–41]. It is worth nothing that no primer pair will detect all bacteria and that primer choice will always affect detection of bacterial lineages, but the region analyzed here has low non-coverage rates for most phyla (exceptions include Aquificae, Armatimonadetes, Chlamydiae, Planctomycetes, and Verrucomicrobia; these lineages are not common members of insect-associated bacterial communities) [42].
Many of the most common lineages detected are related to known symbionts previously detected in fleas (e.g. Bartonella spp. [43–45], Cardinium sp. [46], Wolbachia spp. [44,47–49], Spiroplasma spp. [46,50,51], Lariskella sp. [52]). However, some lineages commonly detected in this study are not related to known insect symbionts. Three of the most common lineages were within the Pasteurellaceae family and were abundant within a single flea species but rare or non-existent in other flea species (Table 3). This pattern suggests species-specific symbiosis between the flea species and its corresponding Pasteurellaceae lineage. While this is common with certain groups of bacteria (e.g. Rickettsiales, Bacteroidetes), this species-specific relationship has not been seen previously within the Pasteurellaceae. The Pasteurellaceae lineages discovered here share only 90–95% sequence similarity with other previously sequenced bacteria, suggesting that the lineages discovered here represent new lineages of insect-associated bacterial symbionts.
Flea species had the greatest effect on bacterial community composition (Table 4; Fig 3). This supports previous work that demonstrated a substantial effect of insect host taxonomy on bacterial community composition [36,37]. However, our results differ from previous research on flea-associated bacterial communities that found no differences in bacterial communities between flea species [9,10]. Here, Wolbachia spp. and Bartonella spp. were commonly found in different flea species, but each flea species also harbored a unique bacterial lineage (Table 3). These lineages unique to specific flea species are likely responsible for the effect of flea species on community composition (Table 4).
To our knowledge, this is the first study to assess environmental effects on insect-associated bacterial communities. We were able to compare flea-associated bacterial communities to environmental variables such as temperature, precipitation, and elevation (Table 6). Elevation, mean annual precipitation, and mean monthly temperature all co-varied significantly with bacterial community composition when using all flea samples (Table 6). This is result is somewhat driven by non-random distribution of flea species across sites (Table 1) and the strong effect of flea species on bacterial community composition (Table 4). Nevertheless, significant effects of elevation, mean annual precipitation, and temperature are also found within X. cheopis, suggesting that environmental effects may contribute to bacterial community composition. The environmental effects are rather weak, however, and this is somewhat surprising because outbreaks of Y. pestis are often attributed to environmental change [3,5,53–55].
Bacterial communities of X. brasiliensis and X. cheopis changed slightly across the collection periods. A previous study of flea-associated bacteria found communities to vary substantially across time [10], but that study compared bacterial communities collected three years apart whereas this study spans only six months. It is becoming increasingly clear that although insect species harbor unique symbionts within an insect population, the dominant symbionts within a population shift across time and among populations. For example, here a common symbiont of D. lypusus was a Lariskella sp. (Table 3), which has previously been detected in X. cheopis and a variety of stinkbugs [52]; here it was rarely detected in X. cheopis, demonstrating both its ability to colonize different insect hosts and its variability of prevalence across populations. This pattern is seen across many insect-associated bacteria and is likely due to a combination of stochastic effects and fitness benefits for the insect of particular insect-bacteria associations. If these symbionts interact with pathogens such as Y. pestis, their presence or absence may alter the likelihood of successful plague transmission and their variable prevalence across flea-subpopulations may contribute to the patchy distribution in both time and space of plague epizootics.
Supporting Information
(TXT)
(TXT)
(TXT)
Acknowledgments
Many thanks to the field crews in Uganda for collecting flea specimens and to the Ugandan citizens who allowed flea collections to take place in their homes and villages. Thanks to the Fierer lab at the University of Colorado for access to laboratory equipment that was necessary to generate the data.
Data Availability
DNA sequence data has been deposited in GenBank, and accession numbers are included within the paper. Mapping files, OTU table, and Fasta files are uploaded as Supporting Information.
Funding Statement
Funding was provided by the Centers for Disease Control and Prevention.
References
- 1. World Health Organization. Human plague: review of regional morbidity and mortality, 2004–2009. Weekly Epidemiological Record. 2010; 85: 40–45. [Google Scholar]
- 2. MacMillan K, Enscore RE, Ogen-Odoi A, Borchert JN, Babi N, Amartre G, et al. Landscape and residential variables associated with plague-endemic villages in the West Nile region of Uganda. Am J Trop Med Hyg. 2011; 84: 435–442. 10.4269/ajtmh.2011.10-0571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moore SM, Monaghan A, Griffith KS, Apangu T, Mead PS, Eisen RJ. Improvement of disease prediction and modeling through the use of meteorological ensembles: human plague in Uganda. PLoS One. 2012; 7(9): e44431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Collinge SK, Johnson WC, Ray C, Matchett R, Grensten J, Cully JF, et al. Testing the generality of a trophic-cascade model for plague. Ecohealth. 2005; 2: 102–112. [Google Scholar]
- 5. Enscore RE, Biggerstaff BJ, Brown TL, Fulgham RF, Reynolds PJ, Engelthaler DM, et al. Modeling relationships between climate and the frequency of human plague cases in the southwestern United States, 1960–1997. Am J Trop Med Hyg. 2002; 66: 186–196. [DOI] [PubMed] [Google Scholar]
- 6. Eisen RJ, Gage KL. Adaptive strategies of Yersinia pestis to persist during inter-epizootic and epizootic periods. Vet Res. 2009; 40(2): 01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jones RT, Vetter SM, Montenieiri J, Holmes J, Bernhardt SA, Gage KL. Yersinia pestis infection and laboratory conditions alter flea-associated bacterial communities. ISME J. 2013; 7: 224–228. 10.1038/ismej.2012.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jones RT, Vetter SM, Gage KL. Short report: Exposing laboratory-reared fleas to soil and wild flea feces increases transmission of Yersinia pestis . Am J Trop Med Hyg. 2013; 89: 784–787. 10.4269/ajtmh.13-0138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hawlena H, Rynkiewicz E, Toh E, Alfred A, Durden LA, Hastriter MW, et al. The arthropod, but not the vertebrate host or its environment, dictates bacterial community composition of fleas and ticks. ISME J. 2013; 7: 221–223. 10.1038/ismej.2012.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jones RT, Knight R, Martin AP. Bacterial communities of disease vectors sampled across time, space, and species. ISME J. 2010; 4: 223–231. 10.1038/ismej.2009.111 [DOI] [PubMed] [Google Scholar]
- 11. Bell CW, Acosta-Martinez V, McIntyre NE, Cox S, Tissue DT, Zak JC. Linking microbial community structure and function to seasonal differences in soil moisture and temperature in a Chihuahuan desert grassland. Microb Ecol. 2009; 58: 827–842. 10.1007/s00248-009-9529-5 [DOI] [PubMed] [Google Scholar]
- 12. Ghiglione JF, Larcher M, Lebaron P. Spatial and temporal scales of variation in bacterioplankton community structure in the NW Mediterranean Sea. Aquat Microb Ecol. 2005; 40: 229–240. [Google Scholar]
- 13. Gonzalez A, King A, Robeson MS II, Song S, Shade A, Metcalf JL, et al. Characterizing microbial communities through space and time. Curr Opin Biotechnol. 2012; 23: 431–436. 10.1016/j.copbio.2011.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haack SK, Fogarty LR, West TG, Alm EW, McGuire JT, Long DT, et al. Spatial and temporal changes in microbial community structure associated with recharge-influenced chemical gradients in a contaminated aquifer. Environ Microbiol. 2004; 6: 438–448. [DOI] [PubMed] [Google Scholar]
- 15. Schmidt SK, Costello EK, Nemergut DR, Cleveland CC, Reed SC, Weintraub MN, et al. Biogeochemical consequences of rapid microbial turnover and seasonal succession in soil. Ecology. 2007; 88: 1379–1385. [DOI] [PubMed] [Google Scholar]
- 16. Glaser RL, Meola MA. The Native Wolbachia endosymbionts of Drosophila melanogaster and Culex quinquefasciatus increase host resistance to West Nile Virus infection. PLoS One; 2010; 5(8): e11977 10.1371/journal.pone.0011977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hughes GL, Koga R, Xue P, Fukatsu T, Rasgon JL. Wolbachia infections are virulent and inhibit the human malaria parasite Plasmodium falciparum in Anopheles gambiae . PLoS Pathog; 2011; 7(5): e1002043 10.1371/journal.ppat.1002043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moreira LA, Iturbe-Ormaetxe I, Jeffery JA, Lu G, Pyke AT, Hedges LM, et al. A Wolbachia symbiont in Aedes aegypti limits infection with dengue, chikungunya, and Plasmodium . Cell. 2009; 139: 1268–1278. 10.1016/j.cell.2009.11.042 [DOI] [PubMed] [Google Scholar]
- 19. Hoffmann AA, Montgomery BL, Popovici J, Iturbe-Ormaetxe I, Johnson PH, Muzzi F et al. Successful establishment of Wolbachia in Aedes populations to suppress dengue transmission. Nature. 2011; 476(7361): 454–457. 10.1038/nature10356 [DOI] [PubMed] [Google Scholar]
- 20. Xi Z, Ramirez JL, Dimopoulos G. The Aedes aegypti toll pathway controls dengue virus infection. PLoS Pathog. 2008; 4(7): e1000098 10.1371/journal.ppat.1000098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eisen RJ, Borchert JN, Mpanga JT, Atiku LA, MacMillan K, Boegler KA, et al. Flea diversity as an element for persistence of plague bacteria in an East African plague focus. PLoS One. 2012; 7(4): e35598 10.1371/journal.pone.0035598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fierer N, Hamady M, Lauber CL, Knight R. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Pro Natl Acad Sci U S A. 2008; 105: 17994–17999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010; 7(5): 335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010; 26(19): 2460–2461. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 25. Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010; 26(2): 266–267. 10.1093/bioinformatics/btp636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Price MN, Dehal PS, Arkin AP. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol and Evol. 2009; 26(7): 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Whittaker RH. Evolution and measurement of species diversity. Taxon. 1972; 21: 213–251. [Google Scholar]
- 28. Faith DP. Conservation evaluation and phylogenetic diversity. Biol Conserv. 1992; 61(1): 1–10. [Google Scholar]
- 29. Bray JR, Curtis JT. An ordination of the upland forest communities of southern Wisconsin. Ecol Monogr. 1957; 27: 325–349. [Google Scholar]
- 30. Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005; 71(12): 8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol. 2007; 73(5): 1576–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Andreotti R, de Leon AAP, Dowd SE, Guerrero FD, Bendele KG, Scoles GA. Assessment of bacterial diversity in the cattle tick Rhipicephalus (Boophilus) microplus through tag-encoded pyrosequencing. BMC Microbiol. 2011; 11(1):6 10.1186/1471-2180-11-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Minard G, Mavingui P, Moro CV. Diversity and function of bacterial microbiota in the mosquito holobiont. Parasit Vectors. 2013; 6:146 10.1186/1756-3305-6-146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schabereiter-Gurtner C, Lubitz W, Rolleke S. Application of broad-range 16S rRNA PCR amplification and DGGE fingerprinting for detection of tick-infecting bacteria. J Microbiol Methods. 2003; 52(2): 251–260. [DOI] [PubMed] [Google Scholar]
- 35. Wang Y, Gilbreath TM III, Kukutla P, Yan G, Xu J. Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS One. 2011; 6(9): e24767 10.1371/journal.pone.0024767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Colman DR, Toolson EC, Takacs-Vesbach CD. Do diet and taxonomy influence insect gut bacterial communities? Mol Ecol. 2012; 21(20): 5124–5137. 10.1111/j.1365-294X.2012.05752.x [DOI] [PubMed] [Google Scholar]
- 37. Jones RT, Sanchez LG, Fierer N. A cross-taxon analysis of insect-associated bacterial diversity. PLoS One. 2013; 8(4): e61218 10.1371/journal.pone.0061218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yun JH, Roh SW, Whon TW, Jung MJ, Kim MS, Park DS, et al. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl Environ Microb. 2014; 80(17): 5254–5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, et al. Evolution of mammals and their gut microbes. Science. 2008; 320(5883): 1647–1651. 10.1126/science.1155725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sullam KE, Essinger SD, Lozupone CA, O'Connor MP, Rosen GL, Knight R, et al. Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Mol Ecol. 2012; 21(13): 3363–3378. 10.1111/j.1365-294X.2012.05552.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wei S, Morrison M, Yu Z. Bacterial census of poultry intestinal microbiome. Poult Sci. 2013; 92(3): 671–683. 10.3382/ps.2012-02822 [DOI] [PubMed] [Google Scholar]
- 42. Ghyselinck J, Pfeiffer S, Heylen K, Sessitsch A, De Vos P. The effect of primer choice and short read sequences on the outcome of 16S rRNA gene based diversity studies. PLoS One. 2013; 8(8): e71360 10.1371/journal.pone.0071360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chomel BB, Kasten RW, FloydHawkins K, Chi BH, Yamamoto K, Roberts-Wilson J, et al. Experimental transmission of Bartonella henselae by the cat flea. J Clin Microbiol. 1996; 34(8): 1952–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rolain JM, Franc M, Davoust B, Raoult D. Molecular detection of Bartonella quintana, B. koehlerae, B. henselae, B. clarridgeiae, Rickettsia felis, and Wolbachia pipientis in cat fleas, France. Emerg Infect Dis. 2003; 9(3): 338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sackal C, Laudisoit A, Kosoy M, Massung R, Ererneeva ME, Karpathy SE, et al. (2008) Bartonella spp. and Rickettsia felis in Fleas, Democratic Republic of Congo. Emerg Infect Dis. 2008; 14(12): 1972–1974. 10.3201/eid1412.080610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jones RT, Bernhardt SA, Martin AP, Gage KL. Interactions among symbionts of Oropsylla spp. (Siphonoptera: Ceratophyllidae). J Med Entomol. 2012; 49(3): 492–496. [DOI] [PubMed] [Google Scholar]
- 47. Casiraghi M, Bordenstein SR, Baldo L, Lo N, Beninati T, Wernegreen JJ, et al. Phylogeny of Wolbachia pipientis based on gltA, groEL and ftsZ gene sequences: clustering of arthropod and nematode symbionts in the F supergroup, and evidence for further diversity in the Wolbachia tree. Microbiology. 2005; 151(Pt 12): 4015–4022. [DOI] [PubMed] [Google Scholar]
- 48. Dittmar K, Whiting MF. New Wolbachia endosymbionts from nearctic and neotropical fleas (Siphonaptera). J Parasitol. 2004; 90(5): 953–957. [DOI] [PubMed] [Google Scholar]
- 49. Gorham CH, Fang QQ, Durden LA. Wolbachia endosymbionts in fleas (Siphonaptera). J Parasitol. 2003; 89(2): 283–289. [DOI] [PubMed] [Google Scholar]
- 50. Hornok S, Meli ML, Perreten A, Farkas R, Willi B, Beugnet F, et al. Molecular investigation of hard ticks (Acari: Ixodidae) and fleas (Siphonaptera: Pulicidae) as potential vectors of rickettsial and mycoplasmal agents. Vet Microbiol. 2010; 140(1–2): 98–104. 10.1016/j.vetmic.2009.07.013 [DOI] [PubMed] [Google Scholar]
- 51. Pornwiroon W, Kearney MT, Husseneder C, Foil LD, Macaluso KR. Comparative microbiota of Rickettsia felis-uninfected and infected colonized cat fleas, Ctenocephalides felis . ISME J. 2007; 1(5): 394–402. [DOI] [PubMed] [Google Scholar]
- 52. Matsuura Y, Kikuchi Y, Meng XY, Koga R, Fukatsu T. Novel clade of alphaproteobacterial endosymbionts associated with stinkbugs and other arthropods. Appl Environ Microbiol. 2012; 78(12): 4149–4156. 10.1128/AEM.00673-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ari TB, Gershunov A, Tristan R, Cazelles B, Gage K, Stenseth NC. Interannual variability of human plague occurrence in the Western United States explained by tropical and North Pacific Ocean climate variability. Am J Trop Med Hyg. 2010; 83(3): 624–632. 10.4269/ajtmh.2010.09-0775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Brown HE, Ettestad P, Reynolds PJ, Brown TL, Hatton ES, Holmes J, et al. Climatic predictors of the intra- and inter-annual distributions of plague cases in New Mexico based on 29 Years of animal-based surveillance data. Am J Trop Med Hyg. 2010; 82(1): 95–102. 10.4269/ajtmh.2010.09-0247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Parmenter RR, Yadav EP, Parmenter CA, Ettestad P, Gage KL. Incidence of plague associated with increased winter-spring precipitation in New Mexico. Am J Trop Med Hyg. 1999; 61(5): 814–821. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TXT)
(TXT)
(TXT)
Data Availability Statement
DNA sequence data has been deposited in GenBank, and accession numbers are included within the paper. Mapping files, OTU table, and Fasta files are uploaded as Supporting Information.