

Abstract
In this report, we designed a highly bright bifunctional curcumin analogue CRANAD-28. In vivo two-photon imaging suggested that CRANAD-28 could penetrate the blood brain barrier (BBB) and label plaques and cerebral amyloid angiopathy (CAAs). We also demonstrated that this imaging probe could inhibit the crosslinking of amyloid beta induced either by copper or by natural conditions.
An imaging probe and a drug could share a common structural scaffold and design concept considering that they have the same target 1, 2. They could also have similar interaction mechanisms and specificity requirements for the target. In traditional approaches, imaging probes and drugs are pursued separately, which are time-consuming and costly 3, 4. To overcome this disadvantage, developing agents that have potentials for therapy and imaging (theranostic) is highly desirable. Nanoparticle-based theranostics have been intensively explored 4; however their poor BBB penetration limits their applications for CNS diseases. Therefore, developing small molecule-based theranostics is warranted for neurodegenerative diseases such as Alzheimer’s disease 2 (AD).
Amyloid beta (Aβ), a peptide of 40 or 42 amino acids, is one of the key players in AD pathology 5. The presence of Aβ plaques is one of the hallmarks of AD 5, 6. While the degree of toxicity of amyloid plaques that contribute to the overall cognitive decline in AD remains unknown, plaques are clear sites of pathology as evidenced by the presence of dystrophic neurites and the loss of dendritic spines in their surroundings 7, 8. Furthermore, there is also some evidence that plaques may impair mitochondria function and calcium homeostasis and lead to cell death 9. Given these and other lines of evidence, reducing Aβ deposits and plaques remains an important aim for AD drug development 5, 10, 11. Imaging probes for Aβs for both clinical and preclinical research have been reported 10, 12-15. However, small molecules that can be used both for imaging and therapy of AD are still urgently needed 11.
Recently, we have developed curcumin analogues for detecting soluble and insoluble Aβs in vitro, and some of them have been successfully applied for in vivo near-infrared imaging in transgenic AD mice 16-18. Based on the limited interaction mechanism of the curcumin ligands and Aβs, we also designed imidazole-containing curcumin analogues to specifically interrupt the crosslinking of Aβs that was initialized by metal ions such as copper 19, 20. However, curcumin compounds always have low quantum yield (QY), and lack theranostic properties. In this report, we designed, synthesized and tested a bifunctional compound with high brightness for two-photon imaging and potential therapy.
To overcome the low QY limitation of curcumin analogues, we hypothesized that replacing the phenyl rings with pyrazoles could increase the brightness. Conceivably, the inductive electron-withdrawing effect of one of the nitrogens of pyrazole could lead to a low tendency of electron delocalization in the system, which will decrease tautomerization of the designed compounds. It is well known that less tautomerization can reduce non-radiative decay from the excited states, thus increase the QY 21. On the other hand, in our previous studies 17, we showed that imidazole containing curcumin analogues could specifically interfere the coordination of copper with H13 and H14 (Histidine) and thus attenuate the crosslinking of Aβ. In this report, we speculated that pyrazole could also interfere the coordination because pyrazole can coordinate with copper as well. In addition, we reasoned that phenyl substitution at the N-1 position of pyrazole could further improve the QY due to the reduction of tautomerization of pyrazole (more tautomers, more non-radiative decay) 22. Taking all the facts into consideration, CRANAD-28 was designed and synthesized (Fig.1b).
Fig.1.
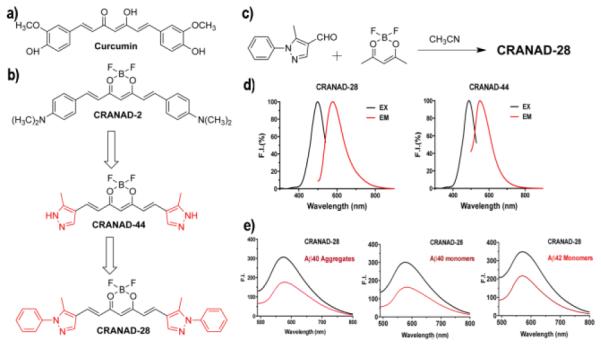
a) Structure of Curcumin. b) Design of CRANAD-28 through pyrazole replacement. c) The synthetic route for CRANAD-28. d) The excitation/emission spectra of CRANAD-28 and -44. e) Fluorescence responses of CRANAD-28 with Aβ40 aggregates, Aβ40 monomers and Aβ42 monomers. For Aβ42 dimers and oligomers, the spectra are shown in SI Fig.1.
CRANAD-28, an orange powder, was synthesized following our previously published procedures 16, 17. We first investigated its fluorescence properties such as excitation and emission, and QY. The excitation peak of CRANAD-28 is 498 nm and the emission peak is 578 nm in PBS (Fig.1d). The Stokes shift of CRANAD-28 is considerably large (80 nm), which is highly beneficial for two-photon imaging because it provides better emission penetration due to the large Stokes shift. As we expected, CRANAD-28 has a very high QY both in PBS and ethanol. The QY of CRANAD-28 is 0.32 in PBS and > 1.0 in ethanol (rhodamine B was used as a reference). Our results are consistent with several references, which showed that pyrazoles are very important moieties contributing to strong fluorescence 23-25. To confirm the N-1 phenyl substitution effect, we also synthesized CRANAD-44. As expected, the QY of CRANAD-28 was higher than that of CRANAD-44 (QY = 0.29 in PBS and 0.47 in ethanol), because the N-1 phenyl substitution can reduce the number of annular tautomers 22, and thus reduce the non-radiative emission decay (Fig.1b).
To test whether CRANAD-28 could interact with various Aβs, we first incubated the compound with soluble Aβs that included monomeric, dimeric, and oligomeric Aβs. We also tested the compound with insoluble Aβ40 aggregates. Interestingly, we observed that the fluorescence intensity of CRANAD-28 decreased upon mixing with all of the tested Aβs, indicating that CRANAD-28 could interact with these Aβs (Fig.1e and SI Fig.1). The Kd values of CRANAD-28 with Aβ40 monomers, Aβ42 monomers, Aβ42 dimers, Aβ42 oligomers, and Aβ40 aggregates were 68.8, 159.7, 162.9, 85.7 and 52.4 nM, respectively (SI Fig.1b) (see details of Kd measurement in supplemental information). The intensity decrease of CRANAD-28 is contrary to our previously reported fluorescent probes, which showed intensity increase with Aβs. No significant change was observed for CRANAD-44 with soluble Aβs (SI Fig.2b, c). It is not clear what are the quenching mechanisms for the intensity decrease of CRANAD-28. Hintersteiner et al reported that the fluorescence of imaging probe AOI987 could be slightly quenched by Aβ aggregates through an unknown mechanism 26, and Chen et al showed that Alexa fluorophores could be quenched by amino acids such as histidine, tyrosine, and methionine through static and collisional mechanisms 27. Other mechanisms that include electron-scavenging 28 and photoinduced electron transfer (PeT) 29 were also proposed for the quenching effect induced by peptides and amino acids 30. The fluorescence quenching of CRANAD-28 by Aβs could be due to one or a combination of the above mechanisms.
To further test whether the compounds could label Aβ plaques in AD brain tissue, we stained APP/PS1 mouse brain slices with CRANAD-28 and -44. Thioflavin S, a standard plaque-staining agent, was used for staining consecutive slices. We found that CRANAD-28 could provide excellent contrast for Aβ plaques (Fig.2a-c), and the distribution pattern of the highlighted plaques was similar to that with Thioflavin S staining. However, CRANAD-44 did not show obvious contrast for plaques, suggesting that the phenyl rings at the N-1 position could enhance the binding with Aβs (Fig.2 and SI Fig.3). Meanwhile, CRANAD-28 not only stained the plaques, but also labeled CAAs (Fig.2d, e), which is characterized by Aβ deposits at the exterior of the brain blood vessels that significantly contribute to pathological dementia 31.
Fig.2.
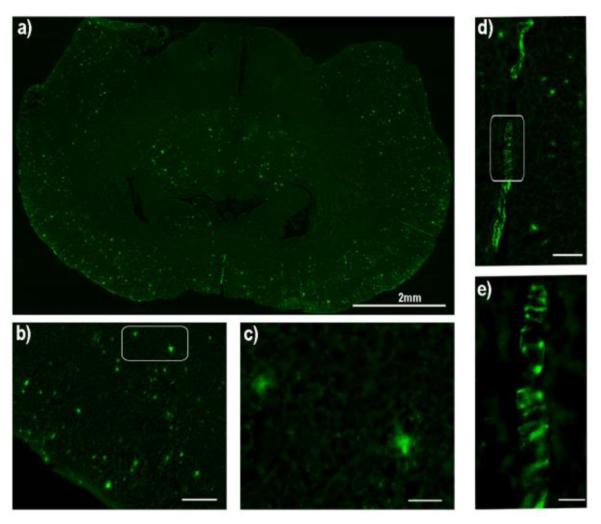
Histological staining of a brain slice of an APP/PS1 mouse with CRANAD-28. a) The image of the whole slice (2x). b) Image of the cortex area (10x) (scale bar: 200 micron), and c) Image of two plaques in the white box in b (40x) (scale bar: 100 micron). d) Image of CAAs (10x) (scale bar: 200 micron), and e) Magnified image of white box in d (40x) (scale bar: 100 micron).
To investigate whether CRANAD-28 could be used for in vivo two-photon imaging of Aβ plaques, we firstly examined its BBB penetration. To this end, CRANAD-28 was injected i.v. into a wild type mouse, which had a surgically induced thinned skull 32-34. We quantified the BBB penetration following the reported method 35, and found that CRANAD-28 reached its peak around 5 minutes after injection, followed by a slow wash out (SI Fig.4). Since CRANAD-44 was not able to label Aβ plaques in the brain tissue, we didn’t test BBB penetration of this compound.
Next, to validate the capability of CRANAD-28 for in vivo two-photon imaging of Aβ plaques, we used 9-month old APP/PS1 mice that had thinning skull surgery 32-34. This procedure is significantly less invasive compared to cranial window preparation, which requires total removal of the skull 36, 37. In addition, it causes less damage to the neuronal cells and therefore is preferable 37. After i.v. injection of CRANAD-28, CAAs were clearly highlighted around the imaged vessels (Fig.3a, white arrowhead). As expected, Aβ plaques could also be clearly labeled (Fig.3a, yellow arrowhead). In Fig.3, the blood vessels were highlighted by the injected Texas-red dextran (70,000 MW). There was a clear localization of CAAs on the wall of the vessels. These results suggested that CRANAD-28 was an effective two-photon imaging probe for both CAA and Aβ plaques. Moreover, the data also demonstrated that the longer emission of CRANAD-28 could allow for imaging to be performed with a less invasive thinning skull surgery 32-34.
Fig.3.

Two-photon in vivo images of CRANAD-28 labeling in a 9-month-old APP/PS1 mouse. (a) Images were taken through a thinned-skull window 15min after i.v. infusion of the dye. Both CAA and amyloid plaques were labeled with CRANAD-28. (b) Lower panels show zoomed-in single focal plane examples of CAA (white arrowhead, left panel) and amyloid plaque (yellow arrowhead, right panel). Blood vessels were labeled with Texas-red dextran (70,000 MW). Red punctate signals are auto-fluorescence intracellular structures. Scale bar: 25 μm.
For a specific target, a non-tagged imaging probe could serve both as a ligand and a fluorophore. Conceivably, such probe could engage with the target, and thus affect the behavior of the target. For Aβ species, CRANAD-28 could also have the capacity to change its folding/aggregating behavior, and thus to execute its anti-crosslinking effect. In our previous studies, we have demonstrated that the imidazole-containing curcumin analogue CRANAD-17 could specifically interrupt the copper induced crosslinking of Aβ, due to the imidazole moiety, which could interfere with the copper coordination with H13 and H14 of Aβ 17. In CRANAD-28, the pyrazole ring could have the capability to coordinate with copper 38, and thus may have the ability to compete with H13/H14 coordination. Therefore, it is reasonable to speculate that CRANAD-28 can also attenuate copper-induced crosslinking of Aβ. To this end, we first tested CRANAD-28 with FAM-Aβ42, a dye (FAM) conjugated Aβ42, to investigate the anti-crosslinking effect. As expected, there was significantly more (4.13-fold) monomeric Aβ remaining in CRANAD-28 treated samples than that in the control samples, indicating reduced crosslinking (Fig.4a). To further investigate whether it could inhibit the crosslinking of native Aβ42, we used western blotting to quantify the remaining Aβ momomers. Similar to the result with FAM-Aβ42, CRANAD-28 treatment resulted in considerably more monomeric Aβ free of crosslinking (1.85-fold) (Fig.4b).
Fig.4.
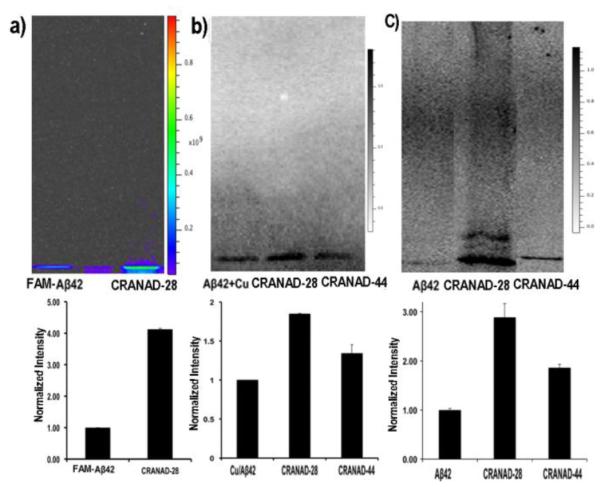
Anti-crosslinking effect of CRANAD-28. a-c) SDS-PAGE gel with FAM-Aβ42 treated with copper (a), western blot of native Aβ42 treated with copper (b) and western blotting of native Aβ42 under natural conditions (c) (up panel). Bottom panel: quantitative analysis of the monomeric bands for each condition in the upper panel (n=4). The normalization was based on FAM-Aβ42 treated with copper (a), Aβ42 treated with copper (b), and Aβ42 only (c).
Besides induction by metal ions, crosslinking of Aβ42 can occur naturally in solution, which is also an important factor contributing to its toxicity. To investigate whether CRANAD-28 could attenuate the natural crosslinking, we incubated it with Aβ42 in PBS buffer for 24 hours. Western blotting analysis showed that CRANAD-28 could significantly inhibit natural crosslinking of Aβ42 (2.89-fold higher than the control) (Fig.4c).
To investigate whether the compounds’ Aβ-specificity has any effects on anti-crosslinking induced by copper and a natural condition, we compared the monomeric bands of CRANAD-28 and CRANAD-44 on western blotting, and found that CRANAD-44 was not efficient in inhibiting the crosslinking of Aβ42 under both conditions (Fig.4b,c). This is consistent with our fluorescence studies and histological staining, which indicated that CRANAD-44 was not Aβ-specific.
In summary, we demonstrated that CRANAD-28 could be considered a theranostic agent due to its imaging and potential therapeutic functions. Through replacement of the phenyl rings with pyrazole, the resulting CRANAD-28 displayed high QY in PBS and ethanol. Previously, analogues of PiB, styrybenzene, and other compounds have been reported to be very reliable imaging probes for two-photon imaging 35, 39, 40. However, due to their short emission wavelengths, these dyes are associated with poor tissue penetrating capacities. Conceivably, CRANAD-28 has better tissue penetration because of its longer emission. Multi-color time-stamp has been considered to be a very useful tool to investigate the growth kinetics of plaques, and this technique can provide comprehensive toxicity profile of plaques 8. We believe that fluorescent dyes with longer emissions, like CRANAD-28, could be very useful for sequential multi-color labeling to avoid ex vivo section staining. In addition, CRANAD-28 is not only useful for two-photon imaging but also has the capacity to attenuate crosslinking of Aβ42 induced by metal ion and natural conditions. We believe that its bifunction could contribute to AD diagnosis and therapy development in the future.
Supplementary Material
Acknowledgement
This work was supported by K25AG036760 award to C.R and K99AG042026 award to M.A.Y. The authors would also like to thank Pamela Pantazopoulos, B.S. for proofreading this manuscript.
References
- 1.Vallabhajosula S. Molecular imaging : radiopharmaceuticals for PET and SPECT. Springer-Verlag; Berlin ; New York: 2009. [Google Scholar]
- 2.Aulic S, Bolognesi ML, Legname G. Int J Cell Biol. 2013;2013:150952. doi: 10.1155/2013/150952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de la Zerda A, Kim JW, Galanzha EI, Gambhir SS, Zharov VP. Contrast Media Mol Imaging. 2011;6:346–369. doi: 10.1002/cmmi.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie J, Lee S, Chen X. Adv Drug Deliv Rev. 2010;62:1064–1079. doi: 10.1016/j.addr.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Selkoe DJ. Nat Med. 2011;17:1060–1065. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- 6.Joachim CL, Mori H, Selkoe DJ. Nature. 1989;341:226–230. doi: 10.1038/341226a0. [DOI] [PubMed] [Google Scholar]
- 7.Tsai J, Grutzendler J, Duff K, Gan WB. Nat Neurosci. 2004;7:1181–1183. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- 8.Condello C, Schain A, Grutzendler J. Sci Rep. 2011;1:19. doi: 10.1038/srep00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie H, Guan J, Borrelli LA, Xu J, Serrano-Pozo A, Bacskai BJ. J Neurosci. 2013;33:17042–17051. doi: 10.1523/JNEUROSCI.1836-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jakob-Roetne R, Jacobsen H. Angew Chem. 2009;48:3030–3059. doi: 10.1002/anie.200802808. [DOI] [PubMed] [Google Scholar]
- 11.Yang W, Wong Y, Ng OT, Bai LP, Kwong DW, Ke Y, Jiang ZH, Li HW, Yung KK, Wong MS. Angew Chem. 2012;51:1804–1810. doi: 10.1002/anie.201104150. [DOI] [PubMed] [Google Scholar]
- 12.Ono M, Watanabe H, Kimura H, Saji H. ACS Chem. Neurosci. 2012;3:319–324. doi: 10.1021/cn3000058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui M, Ono M, Watanabe H, Kimura H, Liu B, Saji H. J Amer Chem Soc. 2014;136:3388–3394. doi: 10.1021/ja4052922. [DOI] [PubMed] [Google Scholar]
- 14.Cook NP, Ozbil M, Katsampes C, Prabhakar R, Marti AA. J Amer Chem Soc. 2013;135:10810–10816. doi: 10.1021/ja404850u. [DOI] [PubMed] [Google Scholar]
- 15.Ran C, Zhao W, Moir RD, Moore A. PLoS One. 2011;6:e19362. doi: 10.1371/journal.pone.0019362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ran C, Xu X, Raymond SB, Ferrara BJ, Neal K, Bacskai BJ, Medarova Z, Moore A. J Am Chem Soc. 2009;131:15257–15261. doi: 10.1021/ja9047043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang X, Tian Y, Li Z, Tian X, Sun H, Liu H, Moore A, Ran C. J Amer Chem Soc. 2013;135:16397–16409. doi: 10.1021/ja405239v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ran C, Moore A. Molecular Imaging Biol. 2012;14:293–300. doi: 10.1007/s11307-011-0501-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Atwood CS, Perry G, Zeng H, Kato Y, Jones WD, Ling KQ, Huang X, Moir RD, Wang D, Sayre LM, Smith MA, Chen SG, Bush AI. Biochemistry. 2004;43:560–568. doi: 10.1021/bi0358824. [DOI] [PubMed] [Google Scholar]
- 20.Thiabaud G, Pizzocaro S, Garcia-Serres R, Latour JM, Monzani E, Casella L. Angew Chem. 2013;52:8041–8044. doi: 10.1002/anie.201302989. [DOI] [PubMed] [Google Scholar]
- 21.Lakowicz J. Principles of Fluorescence Spectroscopy. 2nd Plenum Publishing Corporation; 1999. [Google Scholar]
- 22.de Paz J, Elguero J, Foces-Foces C, Llamas-Saiz A, Aguilar-Parrilla F, Klein O, Limbach H. J. Chem. Soc., Perkin Trans. 2. 1997:101–109. [Google Scholar]
- 23.Song W, Wang Y, Qu J, Madden MM, Lin Q. Angew Chem. 2008;47:2832–2835. doi: 10.1002/anie.200705805. [DOI] [PubMed] [Google Scholar]
- 24.Yu Z, Ho LY, Lin Q. J Amer Chem Soc. 2011;133:11912–11915. doi: 10.1021/ja204758c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren T, Cheng H, Zhang J, Li W, Guo J, Yang L. J. Fluoresc. 2012;22:201–212. doi: 10.1007/s10895-011-0947-7. [DOI] [PubMed] [Google Scholar]
- 26.Hintersteiner M, Enz A, Frey P, Jaton AL, Kinzy W, Kneuer R, Neumann U, Rudin M, Staufenbiel M, Stoeckli M, Wiederhold KH, Gremlich HU. Nat Biotechnol. 2005;23:577–583. doi: 10.1038/nbt1085. [DOI] [PubMed] [Google Scholar]
- 27.Chen H, Ahsan SS, Santiago-Berrios MB, Abruna HD, Webb WW. J Amer Chem Soc. 2010;132:7244–7245. doi: 10.1021/ja100500k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eftink MR, Ghiron CA. Anal Biochem. 1981;114:199–227. doi: 10.1016/0003-2697(81)90474-7. [DOI] [PubMed] [Google Scholar]
- 29.Bottari G, de la Torre G, Guldi DM, Torres T. Chem Rev. 2010;110:6768–6816. doi: 10.1021/cr900254z. [DOI] [PubMed] [Google Scholar]
- 30.Sauer M, Neuweiler H. Methods Mol Biol. 2014;1076:597–615. doi: 10.1007/978-1-62703-649-8_27. [DOI] [PubMed] [Google Scholar]
- 31.Smith EE, Greenberg SM. Stroke. 2009;40:2601–2606. doi: 10.1161/STROKEAHA.108.536839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grutzendler J, Kasthuri N, Gan WB. Nature. 2002;420:812–816. doi: 10.1038/nature01276. [DOI] [PubMed] [Google Scholar]
- 33.Grutzendler J, Yang G, Pan F, Parkhurst CN, Gan WB. Cold Spring Harb Protoc. 2011;2011 doi: 10.1101/pdb.prot065474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marker DF, Tremblay ME, Lu SM, Majewska AK, Gelbard HA. JoVE. 2010;pii:2059. doi: 10.3791/2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bacskai BJ, Hickey GA, Skoch J, Kajdasz ST, Wang Y, Huang GF, Mathis CA, Klunk WE, Hyman BT. Proc Natl Acad Sci U S A. 2003;100:12462–12467. doi: 10.1073/pnas.2034101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mostany R, Portera-Cailliau C. JoVE. 2008;pii:680. doi: 10.3791/680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu HT, Pan F, Yang G, Gan WB. Nat Neurosci. 2007;10:549–551. doi: 10.1038/nn1883. [DOI] [PubMed] [Google Scholar]
- 38.Otieno T, Blanton J, Hatfield M, Asher S, Parkin S. Acta Crystallogr C. 2002;58:m182–185. doi: 10.1107/s010827010200152x. [DOI] [PubMed] [Google Scholar]
- 39.Nesterov EE, Skoch J, Hyman BT, Klunk WE, Bacskai BJ, Swager TM. Angew Chem. 2005;44:5452–5456. doi: 10.1002/anie.200500845. [DOI] [PubMed] [Google Scholar]
- 40.Liu Z, Condello C, Schain A, Harb R, Grutzendler J. J Neurosci. 2010;30:17091–17101. doi: 10.1523/JNEUROSCI.4403-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.