

Abstract
Traumatic brain injury (TBI) can lead to secondary neuropsychiatric problems that develop and persist years after injury. Mounting evidence indicates that neuroinflammatory processes progress after the initial head injury and worsen with time. Microglia contribute to this inflammation by maintaining a primed profile long after the acute effects of the injury have dissipated. This may set the stage for glial dysfunction and hyperactivity to challenges including subsequent head injury, stress, or induction of a peripheral immune response. The purpose of this review is to discuss the evidence that microglia become primed following TBI and how this corresponds with vulnerability to a “second hit” and subsequent neuropsychiatric and neurodegenerative complications.
Keywords: Traumatic Brain Injury, Priming, Microglia, Immune Challenge, Neuroinflammation
Introduction to Traumatic Brain Injury
Traumatic brain injury (TBI) is a leading cause of neurological disability in the United States and is associated with increased risk for development of neuropsychiatric illness. TBI occurs when brain physiology and structure are either focally or diffusely disrupted following high-impact contact between the skull and another object. TBIs most commonly occur in persons aged 0–4, 15–19, and above 65 years [1]. The high incidence in pediatric and young adult populations is significant as these individuals may face a lifetime of compromised physical and mental health after TBI.
TBIs are heterogeneous, with many etiologies and clinical presentations. Diffuse injury results from movement of the brain within the skull. In contrast, focal injury results from penetrating trauma, when either a foreign body or fractured portion of the skull enters the brain. Blast injuries in military veterans are most commonly classified as concussive; however shrapnel or skull fractures can cause penetrating injury [2]. Etiology and experimental models for diffuse and focal injuries are outlined in Figure 1.
FIGURE 1. Penetrating and diffuse traumatic brain injuries have varied etiologies and experimental models.

Human traumatic brain injuries can either be penetrating/focal or diffuse in nature, with multiple etiologies falling into each of these categories. The animal models used to produce TBIs cause pathology that is in line with these two categories of injury.
Experimental paradigms in rodents, pigs, and non-human primates can model the complex pathophysiology of human brain injury. Some models produce pathology that is more consistent with a penetrating head injury and result in lesion formation, focal cell death, and infiltration of leukocytes. For example, controlled cortical impact (CCI; see Glossary) [3, 4, 5], ballistic penetrating injury [6], and lateral fluid percussion injury [7, 8] produce unilateral focal lesions. Other techniques model diffuse head injury, characterized by diffuse axonal injury that occurs in the absence of cell death or focal lesion. These models include midline fluid percussion injury (mFPI) [9] and closed head impact acceleration models [10, 11]. Modifications of these models are used to induce repeated head injuries and mild TBI [12, 13]. Animal models are integral in developing our understanding of the pathophysiology of TBI.
While it is clear that TBI causes acute inflammation that can be life threating, the majority of patients recover without significant CNS pathology or functional impairment. Nonetheless, these individuals experience higher incidence of neuropsychiatric illnesses that develop or persist after the initial injury. These neuropsychiatric sequelae include depression [14], cognitive impairment/deterioration [15], and increased risk for the development of neurodegenerative pathologies [16]. Moreover, evidence shows that inflammation persists in the brain well after injury and may progress with time. This is, in part, mediated by microglia, which maintain a heightened inflammatory status, referred to as “primed”. Primed microglia are significant because they have a lower threshold for response to secondary challenge and become hyper-activated [17]. We will review the evidence that microglia are chronically activated after TBI, and show that microglia-mediated inflammatory responses following a “second hit” may underlie psychiatric and neurodegenerative complications.
Acute Activation of Microglia after TBI
Microglia are the innate immune cells of the brain and respond rapidly following immune challenge or injury. They derive from myeloid progenitors in the yolk-sac and populate the brain and spinal cord during early embryonic development [18, 19]. Microglia represent 10–12% of the cells in the CNS and have numerous ramified processes, which rapidly respond to local chemotactic signals [20] and constantly survey the local environment [21]. These surveying processes interact with synapses in an activity-dependent manner and are capable of eliminating synapses and altering neuronal plasticity [22].
This indicates a role for microglia in learning and memory [23]. Microglia are long-lived cells with minimal turnover throughout the lifespan [18, 24], making them particularly susceptible to CNS injury. The myeloid origin of microglia and their functional similarities to tissue macrophages (e.g. TLR-mediated activation, scavenging, antigen presentation, clearance of misfolded proteins), indicate a major role in immune surveillance of the brain [25, 26, 27, 28]. Penetrating and diffuse TBI in rodents and humans induces significant inflammation, which is mediated by microglia. Microglial activation serves an important role as an immune alert system, with the production of cytokines, chemokines, and the release of secondary mediators (Figure 2) [29]. Microglial responses after CNS injury include phagocytosis, scavenging of debris, angiogenesis, and wound healing. For instance, in spinal cord injury, microglia phagocytize damaged myelin, activate other immune cells, and remove synapses from damaged axons [30]. Characteristics of classic (M1) and alternative activation (M2) of microglia and macrophages are observed after traumatic CNS injury [31, 32] and have implications in tissue destruction and repair. In vivo activation status is likely variable and heterogeneous [33].
FIGURE 2. Microglia are acutely activated following TBI and take on a variety of functional and morphological states.

Microglia in their “resting” state are constantly surveying the environment and maintaining homeostasis. Following TBI, microglia are activated by cell debris, neuronal injury, and blood components. Microglia produce the chemokine CCL2, which plays a role in attracting peripheral monocytes to the CNS. Microglia upregulate both M1 and M2 cytokines and surface markers after injury. M1 activation results in production of ROS and RNS and up regulation of pro-inflammatory cytokines and surface markers including IL-1β, TNFα, and CD14. These activated cells can become phagocytic to clear debris. M2 activation is neuroprotective and indicated by upregulation of chitinase and arginase. Rod microglia align with neuronal structures after diffuse injury and are neuroprotective in vitro.
Microglia are tightly regulated in the CNS and are activated by numerous signals from neurons, astrocytes, and other glia [17]. In the context of injury, microglia respond to damaged cells, other activated glia, and peripherally derived stimuli following breakdown of the blood brain barrier (BBB). For instance, TBI causes down-regulation of gap-junction proteins as early as 6 hours after injury [34] that persists for up to 4 days [35], allowing blood components to enter the brain. This is relevant because fibrinogen [36], a component of the clotting cascade, and ferritin [37], an iron storage molecule, cause microglial activation and chemotaxis. Nonspecific activation signals help to initiate rapid changes in microglial function, structural morphology, and protein expression in the acutely injured CNS.
Microglia rapidly increase expression and release of cytokines and chemokines after injury. This is associated with alterations in morphology and re-distribution of cell-surface markers. For example, surface expression of Iba1 and CD68 rapidly increase in cortical microglia after mFPI [38]. These cells cluster together and exhibit a deramified and hypertrophic morphology. Morphological changes following diffuse injury in mice (Mus musculus) are dependent on intact p38α MAPK signaling, a pathway that also prompts microglial pro-inflammatory cytokine production [39]. Diffuse brain injury results in transiently elevated IL-1β, TNFα, and CD14 in the cortex and hippocampus of mice as early as 4 hours after injury, which returns to baseline by 72 hours [40]. Moreover, Percoll-enriched microglia and macrophages show increased IL-1β, CD14, iNOS, and arginase expression 24 hours after diffuse injury compared to sham-injured controls [41]. Therefore, diffuse injury results in up regulation of both M1 and M2 activation markers. Evidence of mixed M1/M2 activation also occurs after focal TBI. Peak expression of iNOS, CD86, CD68, CD11b, IL-10, chitinase, TGF-β, and arginase occurs 3–5 days after CCI [5, 42]. Interventions that limit microglial activation, such as minocycline, reduce inflammation and improve functional recovery in animal models of TBI [43, 44]. In summary, both M1 and M2-associated cytokines are upregulated and indicate that both microglia-mediated inflammation and repair occur after injury.
Activation is also associated with morphological restructuring of microglia, facilitating movement to areas of damage. Roth et al. identified “jellyfish” microglia by two-photon microscopy [29]. After TBI induced by pressure on a thin skull preparation, these microglia become highly activated, move to the area of damage, and phagocytize particulate [29]. Penetrating injury causes microglia to associate with the axon initial segment within 3 hours [45] and diffuse brain injury induces microglia to form tight clusters and long rod-like structures in the cortex within 7 days [46]. Rod microglia are neuroprotective in vitro [47]; however their role in vivo is unclear. Together these findings suggest that microglia assume a variety of structural morphologies after injury that have multiple functional roles.
Along with resident microglia, there are other myeloid cells within the injured CNS (Figure 3). Bone marrow derived monocytes are recruited into the brain, differentiate into macrophages, and have neuroinflammatory and neuroprotective functions [48]. Activation of microglia following TBI results in production of chemokines, including CCL2, which attract monocytes and granulocytes to the site of injury [49]. Notably, antagonism of the CCL2 receptor, CCR2, reduces inflammation and improves cognitive function 1 month after TBI induced by CCI [50]. Differentiating the contribution of resident microglia from infiltrating macrophages can be difficult, particularly in the context of penetrating brain injury. Once monocytes differentiate into brain macrophages they are nearly indistinguishable from microglia by histology due to similarities in surface marker expression (Figure 3). Both of these cells are rapidly activated and have central roles in mediating neuroinflammatory responses after injury.
FIGURE 3. Monocytes extravasate and differentiate into brain macrophages.

Following injury, circulating monocytes extravasate through activated endothelium and can traffic into the brain parenchyma. These monocytes can differentiate into macrophages. Activated microglia and ramified brain macrophages are nearly indistinguishable by both morphology and cell-surface markers.
Clinical evidence suggests that microglial and macrophage activation occurs rapidly following TBI. Activated microglial morphologies are detected in patients 2–10 days after severe TBI on autopsy [51]. Increased CSF levels of ferritin and soluble CD163, markers of macrophage and microglial activation, were elevated in pediatric patients within 3 days of severe TBI [52]. Additionally, plasma levels of microglial activation products MMP-9 and galectin-3 were elevated within 8 hours of mild TBI in adults [53]. Inflammatory proteins in cerebrospinal fluid (CSF) or blood may be useful biomarkers of microglia/macrophage-mediated inflammation.
It is important to highlight that there are benefits of inflammation after CNS trauma that may aid in intrinsic repair processes [54]. For example, a recent study induced hippocampal lesion using the CaM/Tet-DTA model and subsequently eliminated microglia using CSF1R antagonism [55, 56]. Loss of microglia increased hippocampal neuron death compared to controls, suggesting a protective role for microglia acutely. The same study found that functional recovery and inflammatory cytokine expression was reduced with microglial elimination. Therefore, acute activation of microglia after injury is beneficial but protracted microglial activation is detrimental to recovery. Moreover, another recent study shows that repeated injection of lipopolysaccharide (LPS) moves microglia towards a novel profile, in which they migrate to the synapses of inhibitory neurons and displace them from cortical neurons [57]. This synaptic regulation by microglia was associated with neuroprotection and reduced lesion size after cryogenic brain injury. Together these findings suggest that microglia play an active role in repair and recovery despite the harmful effects of excessive or prolonged inflammation.
In summary, microglia are rapidly activated and have both pro-inflammatory and neuroprotective roles immediately after TBI. A major concern is the degree to which inflammation fails to resolve and persists after injury. Evidence suggests that a prolonged neuroinflammatory response mediated by microglia and macrophages may be detrimental after CNS injury.
Chronic Microglial Activation after Injury
A pro-inflammatory profile of microglia persists long after injury and is evident in diffuse, penetrating, and repeated head injuries. While this profile is consistent with microglial dysfunction in other settings, it is unclear if this represents a functional inflammatory response after TBI. A pro-inflammatory profile of microglia and macrophages, characterized by elevated expression of MHCII, CD68, and NADPH oxidase (NOX2), persists in the cortex and thalamus up to 12 months after CCI injury [58]. Prolonged inflammation is associated with continued lesion volume expansion and hippocampal cell death. In addition, cortical and thalamic staining of chitinase, an alternative activation marker (M2), is transiently upregulated 1 week after CCI, but undetectable at 3 and 12 months [58]. Therefore, neuroprotective repair processes (M2) may decrease over time while pro-inflammatory processes (M1) increase and persist after focal TBI.
Evidence suggests that a primed or pro-inflammatory profile of microglia persists after diffuse head injury. Increased expression of MHCII and CD68 on microglia persists up to 1 week after mFPI in rats (Rattus norvegicus) [46]. These microglia have a unique rod-like morphology along neuronal structures in the cortex [46]. One month after injury, diffuse TBI in mice increases mRNA and protein expression of MHC II on microglia. This is associated with increased cell size and Iba1 immunoreactivity of microglia in the parietal cortex and hippocampus [40]. These data provide evidence that a pro-inflammatory microglial profile persists after diffuse brain injury.
Chronic CNS and peripheral inflammation is also evident in humans after moderate or severe TBI. Increased metabolic activity and white matter abnormalities detected on imaging of TBI patients suggest persistence of inflammation [59, 60]. Elevated markers of inflammation in CSF and plasma are associated with negative clinical outcomes. For instance, elevated IL-6 in CSF is associated with impaired functional recovery in patients after severe TBI [61]. TNFα serum levels are elevated 6–12 months after moderate to severe TBI and are associated with depressive symptoms [62]. Imaging and biomarker findings do not directly indicate microglial activation, but do suggest that inflammation has a negative effect on recovery and is associated with neuropsychiatric sequelae.
In humans, microglial activation also contributes to generalized inflammation after TBI. Densely packed reactive CR3/43 (MHCII) and/or CD68 labeled microglia were detected post mortem in 28% of cases 1–18 years after moderate to severe TBI [63]. Increased labeling of amyloid precursor protein (APP), a marker of axonal injury, was associated with increased MHCII expression. Johnson et al. [16] also found that CR3/43 labeling increased by 2 weeks post-injury and was associated with increased APP labeling and reduced corpus callosum thickness. Some CR3/43+ cell bodies also labeled for myelin basic protein (MBP) in samples collected 2–8 years after injury. These results show that continued inflammation and phagocytosis of damaged myelin occur long after initial injury. It is unclear if microglia and macrophages perpetuate inflammation that causes secondary axonal damage, or if they are continuing to clear debris from the primary injury.
Parallel to post-mortem studies, in vivo positron emission tomography (PET) provides evidence of persistent microglial inflammatory profiles. PET-monitored binding of the benzodiazepine receptor ligand PK11195 to its target allows selective localization of activated microglia in vivo. Ramlackhansingh et al. [15] found that internal structures distant from the initial injury site had persistent PK11195 binding long after injury. Higher thalamic PK11195 binding was associated with poorer cognitive function, regardless of time since injury [15]. Microglial activity can also be assessed by PET using a radioactive ligand for translocator protein (TSPO), which is located on the outer mitochondrial membrane and upregulated after TBI. Coughlin et al. [64] found that TSPO binding was increased in a cohort of NFL players in the right and left supramarginal gyri and right amygdala compared to elderly healthy controls. Some players exhibited altered cognitive performance and the authors interpreted these findings to indicate that altered metabolic and inflammatory conditions after TBI may play a role in cognitive decline. Widespread activation of microglia at distant sites, visualized by PET imaging and post-mortem, may be a result of disruption of neuronal tracts and connectivity [65]. Taken together, these findings indicate that TBI causes long-lasting alterations of microglial activation states in humans.
Overall, experimental and clinical findings indicate inflammation and microglial activation persist long after functional recovery from initial injury. While pro-inflammatory cytokine production subsides, microgliado not return to their homeostatic surveying state, but instead take on a primed and pro-inflammatory phenotype.
Evidence for Microglial Priming in CNS Injury
Microglia that remain in a primed state after TBI are characterized by activated morphology and increased expression of inflammatory markers such as MHCII and CD68 (Figure 4). It is important to emphasize that primed microglia are not acutely activated. Models of focal TBI, which result in prolonged cell death and lesion expansion, may cause microglia adjacent to the lesion to be activated even at chronic time points. Despite this, there is evidence that primed microglia exist in models of diffuse injury and in regions distant from focal injury. These primed microglia have not returned to homeostasis and mount a hyper-active inflammatory response when triggered by a subsequent activating stimulus (Figure 5). Significant overlap exists between chronically reactive microglial profiles observed after TBI and primed microglial populations described in aging and preclinical neurodegenerative disease. In aging, microglial priming is characterized by increased expression of MHCII, complement receptor 3 (CD11b) and morphologic changes [66, 67, 68]. Enhanced expression of inflammatory markers corresponds with increased sensitivity and immune-reactivity of microglia.
FIGURE 4. TBI induces microglial priming and reactivity to immune challenge.
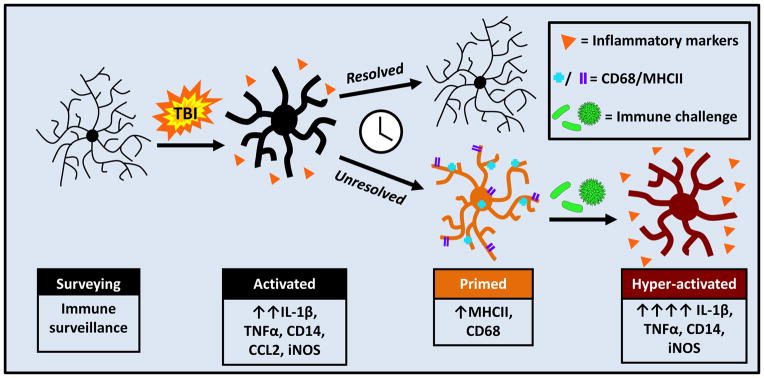
Microglia are activated by TBI and respond by producing myriad cytokines and chemokines. This immediate response resolves and some microglia return to homeostasis; however a population of microglia develops a primed or pro-inflammatory profile. This profile is characterized by increased expression of MHCII and CD68. Following secondary insult, illustrated here as a peripheral immune challenge, these primed microglia become hyper-reactive and produce an amplified and prolonged pro-inflammatory response.
FIGURE 5. TBI-induced microglial priming and reactivity to secondary insults has varied consequences.

Primed microglia become hyper-reactive following a secondary insult, which can include subsequent TBI, immune challenge, or pre-clinical neurodegenerative disease.
A central component of priming is microglial hyper reactivity following immune stimulation. Peripheral lipopolysaccharide (LPS) injection produces a transient innate immune response. LPS challenge increases microglial expression of pro-inflammatory cytokines, such as IL-1β, in aged mice compared to adult mice [68]. Moreover, MHCII+ microglia of aged mice produce more IL-1β compared to MHCII− microglia from adult mice [68], suggesting a pro-inflammatory phenotype. Consequences of amplified microglia-mediated inflammation in the aged mouse brain include exaggerated and prolonged sickness behaviors [69] and depressive-like behaviors [70]. Immune challenge also increases IL-1β, IL-6, and TNFα expression in the hippocampus and impairs spatial memory recall [71, 72]. Overall, a primed microglial phenotype develops with age and promotes exaggerated inflammation, prolonged sickness behaviors, and impaired cognition.
Reactivity of microglia to secondary immune challenge is also evident after CNS injury. In an optic nerve (ON) crush model, increased expression of CD68 in resident microglia is maintained after injury. Induction of an immune response with peripheral LPS administration 28 days after ON crush significantly increased expression of IL-1β, TNF-α, and IL-6 [73]. This heightened inflammatory response after LPS suggests that ON crush prompts development of primed CD68+ microglia. Moreover, injury induced by mFPI results in transient inflammation that resolves by 72h. Consistent with microglial priming, MHCII mRNA and protein were elevated in microglia 1 month after injury [40]. Secondary LPS challenge induced exaggerated expression of IL-1β and TNFα specifically in microglia [40]. Thus, CNS injury is a “priming event”, leaving microglia in an activated state, capable of hyper-responsiveness upon subsequent immune activation (Figure 4).
Microglial priming may also be a factor in repeated TBI, where the initial injury is the priming event and subsequent injuries result in exaggerated inflammatory responses and progressive pathology. Experimental and clinical data point to an association between multiple head injuries and negative behavioral outcomes [74, 75, 76]. Experimentally, when repeated hits are provided in the presence of inflammation, they exacerbate the neuroinflammatory response. For example, following repeated closed-head TBI, motor and cognitive performance on the Morris Water Maze were acutely impaired when compared to single injury and sham-injury controls [77]. These cognitive deficits were associated with microglial morphological changes, increased number of microglia, and axonal pathology [77]. In addition, six months following single and repeated closed-head injuries, morphologically reactive microglia, identified by thick processes and hypertrophied cell somas, were evident in the cortex and hippocampus of mice [75]. Inflammation persisted at 12 and 18 months post-injury and was associated with spatial learning deficits that were worse following repeated compared to single TBI [75]. The cognitive changes 12 and 18 months after injury were associated with white matter damage and increased Iba1 labeling, but were not associated with neurodegenerative pathology [75]. The interval between injuries may also be important. Two closed head injuries occurring 3 days apart induced an enhanced proinflammatory response, more robust axonal degeneration, and poorer performance on a spatial learning and memory task 1 month post-injury, compared to single injury or 20 day interval between TBI [76]. Therefore, multiple injuries can augment neuroinflammation and functional decline.
Head Trauma, Cognitive Decline, and Neurodegenerative Disease and Persistent Microglial Activation
The degree to which TBI increases risk of neurodegenerative diseases is of considerable interest. [78]. Meta-analyses found that head trauma is a risk factor for Alzheimer’s disease (AD) in men irrespective of family history of dementia [79] [80]. In military veterans, severe head injury increased the odds ratio of AD or dementia in a severity-dependent manner; however mild TBI did not increase risk [81]. Whether or not risk of dementia and neuropathology is increased following mild TBI or repeated mild TBI is more controversial due to confounding factors such as lack of stringent clinical criteria [82]. Nonetheless, there is considerable clinical evidence linking history of TBI to future risk of neurodegeneration.
TBI patients with neuropathology on autopsy are frequently diagnosed with chronic traumatic encephalopathy (CTE), a progressive neurodegenerative disease that is frequently associated with repeated injury. Pathology of CTE is characterized by neurofibrillary tangles (NFT), multifocal axonal varicosities, and cognitive deficits [78, 83]. Brains of younger patients (<60 years old) who died more than a year after single moderate to severe TBI had higher density and wider distribution of NFTs compared to uninjured age-matched controls [84]. Additionally, a trend of more extensive and fibrillary amyloid pathology was evident. A CTE-like pathology has also been described in rodents after repeated head injuries. For example, wild-type mice given repeated TBIs had increased immunolabeling of phosphorylated tau protein in the cortex, amygdala, and hippocampus up to 6 months after injury when compared to both sham-injured and singly-injured mice [85]. This was also associated with increased labeling of CD68+ microglia/macrophages. These findings support the idea that repeated injury increases risk for development of neuropathology in otherwise healthy mice [85]. Notably, recent studies show that TBI also speeds development of neuropathology in transgenic AD mice. For example, CCI-injured APP/PS1 mice had significantly more Aβ deposition 6 weeks after injury in the cortex and hippocampus compared to sham-injured APP/PS1 mice. Wild type (WT) TBI mice also had more Aβ labeling than sham-injured WT mice. APP/PS1 mice had cognitive deficits after TBI, which persisted up to 6 weeks [86]. Overall, these findings indicate that exposure to TBI, especially when repeated, is a risk factor for either the development or augmentation of CTE-like neuropathologies.
Improved understanding of the association between TBI and neuropathology and cognitive decline may have both prognostic and diagnostic value for patients. Currently, CTE is diagnosed on autopsy; however it is possible that PET analysis of microglial activation may provide an in vivo indication of cognitive status after TBI. TBI patients exhibit significantly lower performance on tests for processing speed than uninjured controls [15] and PK11169 binding to activated microglia inversely correlated with speed of processing complex information. Case studies of patients with history of repeated head trauma link dementia with post-mortem evidence of tau and amyloid pathologies and increased microglial immune-reactivity [87, 88]. Furthermore, meta-analyses of CTE cases verified by post-mortem pathology confirm that neuropathology is associated with progressive onset of memory impairments and emotional lability [89, 90]. While it is clear that TBI is associated with neuropathology, the mechanistic connections between head trauma and tau deposition, dementia, psychiatric symptoms, and microglia-mediated inflammation is unclear [91].
Alterations in microglial activity and corresponding changes in cognitive function after TBI may be an indication of microglial priming. Recent studies from our lab indicate that hippocampus-dependent learning and memory is intact 7 days after injury; however injured mice learn more slowly than sham-injured mice 30 days after injury (Muccigrosso et al., unpublished). This time point is associated with increased microglial expression of MHCII and IL-1β mRNA. Furthermore, LPS challenge significantly impaired memory recall in TBI mice, both compared to sham-injured mice injected with LPS and TBI mice injected with vehicle. This impairment was associated with amplified TNFα and CCL2 mRNA expression (Muccigrosso et al., unpublished). This suggests a role for primed, immune-reactive microglia in cognitive impairment after TBI, which is particularly relevant because humans face immune challenges almost daily in the form of viral and bacterial exposures. These usually innocuous pathogens may have more severe behavioral and cognitive consequences in patients with a history of brain trauma.
The Role of Primed Microglia in TBI-related Depression
Depression is also a prevalent complication after TBI. A recent study found that the rate of depression in TBI patients (53.1%) is 7.9 times higher than that of the general population (6.7%) [14]. Furthermore, an assessment of Medicare patients found that annual incidence of depression increased following TBI [92]. Therefore, these individuals are at risk for a lifetime of depressive complications that could negatively affect quality of life.
Evidence linking depression and inflammation [93] may reflect an alternate etiology for depression in TBI patients. Repeated closed head injuries in rodents are associated with development of depressive-like behaviors [94]. If these behaviors are a result of microglial priming, it is plausible that the second insult does not have to be an additional head injury. A recent study showed that immune challenge 30 days after diffuse TBI in mice was associated with prolonged social withdrawal and the development of increased resignation behavior and reduced sucrose preference [40]. Thus, a challenge to the immune system that is unrelated to the initial head injury is sufficient to promote the onset of depressive behaviors. These data can be interpreted to indicate that increased inflammation may underlie depression observed after TBI.
Summary and Conclusion
Mounting evidence indicates that microglia become chronically reactive or primed when regulation processes go awry. The stability and longevity of the microglial population makes them particularly sensitive to inflammatory exposure over time. Microglial priming is relevant because it is characterized by a lower threshold for activation to a pro-inflammatory state [95]. Therefore, inflammatory challenges, secondary injuries, and stressors, can further augment the level of microglial activation and inflammation. Here we highlight evidence that a chronically reactive, primed population of microglia develops and persists following TBI and may set the stage for a lifetime of risk of neuropsychiatric complications.
Trends Box.
Microglia are rapidly activated following TBI and produce cytokines and chemokines in addition to exhibiting morphological alterations such as hypertrophy and de-ramification of processes.
Experimental and clinical evidence indicate that microglia do not return to homeostasis after injury, but instead develop a primed and potentially hyper-reactive phenotype.
Primed microglia are characterized by exaggerated responses to secondary insults such as repeated TBI, immune challenge, or stress. This results in amplified and prolonged neuroinflammation that negatively influences cognitive and behavioral processes.
TBI patients are at increased risk for development of depression and neuropathologies (tau, Aβ) after injury.
Microglia-mediated inflammatory processes may represent a useful clinical target in the treatment of neurological and psychiatric complications associated with TBI.
Outstanding Questions.
To what extent does microglial priming after TBI worsen with age? Microglial priming develops in the aged brain, independent of insult or injury. It is important to understand how TBI, whether early or late in life, affects this process in order to prevent neuropsychiatric complications that might arise.
Are there clinical interventions that can be provided immediately after TBI that prevent microglial priming or other long-term changes in glial profiles?
Are there clinical interventions that can be provided well after injury that are effective in reversing microglial priming and chronic inflammatory states?
At what time points are interventions most beneficial after TBI to functional recovery and prevention of negative clinical outcomes? Pro-inflammatory activation may be a necessary component of the initial response to injury but likely is harmful when excessive, prolonged or unresolved.
Acknowledgments
This research was supported by an NIA grant (R01-AG-033028 to J.P.G.), College of Medicine Dean’s Discovery Grant (to J.P.G.), Funding from the Center for Brain and Spinal Cord Repair (to J.P.G.) and an American Surgical Association Fellowship (to D.S.E).
GLOSSARY
- CCI
Controlled cortical impact, method which induces focal TBI in rodents
- CD68
Found in lysosomal and surface membranes of myeloid cells, indicative of phagocytic capability
- Iba1
Calcium channel isotype used to identify microglia
- LPS
Lipopolysaccharide, component of bacterial cell wall that prompts innate immune response
- mFPI
Midline fluid percussion injury, method which induces diffuse TBI in rodents
- MHCII
Major histocompatibility complex II, surface protein used by innate immune cells to present antigens to T-cells, also referred to as OX6 (rodents) and CR3/43 (humans)
- Myeloid
Describes cells derived from myeloid progenitors in bone marrow, includes monocytes, macrophages, microglia
- PK11195
ligand which binds to benzodiazepine surface receptor on microglia, radioactive form is used to identify microglial activation by PET scan
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Faul M, et al. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2010. [Google Scholar]
- 2.Galarneau MR, et al. Traumatic brain injury during Operation Iraqi Freedom: findings from the United States Navy-Marine Corps Combat Trauma Registry. J Neurosurg. 2008;108:950–957. doi: 10.3171/JNS/2008/108/5/0950. [DOI] [PubMed] [Google Scholar]
- 3.Dixon CE, et al. A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods. 1991;39:253–262. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- 4.Chen S, et al. Time course of cellular pathology after controlled cortical impact injury. Exp Neurol. 2003;182:87–102. doi: 10.1016/s0014-4886(03)00002-5. [DOI] [PubMed] [Google Scholar]
- 5.Turtzo LC, et al. Macrophagic and microglial responses after focal traumatic brain injury in the female rat. J Neuroinflammation. 2014;11:82. doi: 10.1186/1742-2094-11-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cernak I, et al. A novel mouse model of penetrating brain injury. Front Neurol. 2014;5:209. doi: 10.3389/fneur.2014.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conti AC, et al. Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J Neurosci. 1998;18:5663–5672. doi: 10.1523/JNEUROSCI.18-15-05663.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cortez SC, et al. Experimental fluid percussion brain injury: vascular disruption and neuronal and glial alterations. Brain Res. 1989;482:271–282. doi: 10.1016/0006-8993(89)91190-6. [DOI] [PubMed] [Google Scholar]
- 9.Kelley BJ, et al. Neuroinflammatory responses after experimental diffuse traumatic brain injury. J Neuropathol Exp Neurol. 2007;66:989–1001. doi: 10.1097/NEN.0b013e3181588245. [DOI] [PubMed] [Google Scholar]
- 10.Foda MA, Marmarou A. A new model of diffuse brain injury in rats. Part II: Morphological characterization. J Neurosurg. 1994;80:301–313. doi: 10.3171/jns.1994.80.2.0301. [DOI] [PubMed] [Google Scholar]
- 11.Marmarou A, et al. A new model of diffuse brain injury in rats. Part I: Pathophysiology and biomechanics. J Neurosurg. 1994;80:291–300. doi: 10.3171/jns.1994.80.2.0291. [DOI] [PubMed] [Google Scholar]
- 12.Chen Y, et al. A modified controlled cortical impact technique to model mild traumatic brain injury mechanics in mice. Front Neurol. 2014;5:100. doi: 10.3389/fneur.2014.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Z, et al. Temporal MRI characterization, neurobiochemical and neurobehavioral changes in a mouse repetitive concussive head injury model. Sci Rep. 2015;5:11178. doi: 10.1038/srep11178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bombardier CH, et al. Rates of major depressive disorder and clinical outcomes following traumatic brain injury. JAMA. 2010;303:1938–1945. doi: 10.1001/jama.2010.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramlackhansingh AF, et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 2011;70:374–383. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- 16.Johnson VE, et al. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 2013;136:28–42. doi: 10.1093/brain/aws322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Norden DM, Godbout JP. Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol. 2013;39:19–34. doi: 10.1111/j.1365-2990.2012.01306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ginhoux F, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15:300–312. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- 20.Davalos D, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 21.Nimmerjahn A, et al. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 22.Wake H, et al. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci. 2009;29:3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parkhurst CN, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155:1596–1609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ajami B, et al. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- 25.Sterka D, Jr, Marriott I. Characterization of nucleotide-binding oligomerization domain (NOD) protein expression in primary murine microglia. J Neuroimmunol. 2006;179:65–75. doi: 10.1016/j.jneuroim.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 26.Bsibsi M, et al. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- 27.Tanaka T, et al. Engulfment of axon debris by microglia requires p38 MAPK activity. J Biol Chem. 2009;284:21626–21636. doi: 10.1074/jbc.M109.005603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meda L, et al. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 29.Roth TL, et al. Transcranial amelioration of inflammation and cell death after brain injury. Nature. 2014;505:223–228. doi: 10.1038/nature12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu R, et al. Phagocytosis of microglia in the central nervous system diseases. Mol Neurobiol. 2014;49:1422–1434. doi: 10.1007/s12035-013-8620-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kigerl KA, et al. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar A, et al. Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol Aging. 2013;34:1397–1411. doi: 10.1016/j.neurobiolaging.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abdul-Muneer PM, et al. Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumatic brain injury induced by primary blast. Free Radic Biol Med. 2013;60:282–291. doi: 10.1016/j.freeradbiomed.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blixt J, et al. Aquaporins and blood-brain barrier permeability in early edema development after traumatic brain injury. Brain Res. 2015;1611:18–28. doi: 10.1016/j.brainres.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 36.Davalos D, et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun. 2012;3:1227. doi: 10.1038/ncomms2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schonberg DL, et al. Ferritin stimulates oligodendrocyte genesis in the adult spinal cord and can be transferred from macrophages to NG2 cells in vivo. J Neurosci. 2012;32:5374–5384. doi: 10.1523/JNEUROSCI.3517-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bachstetter AD, et al. The p38alpha MAPK regulates microglial responsiveness to diffuse traumatic brain injury. J Neurosci. 2013;33:6143–6153. doi: 10.1523/JNEUROSCI.5399-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bachstetter AD, et al. Microglial p38alpha MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Abeta) J Neuroinflammation. 2011;8:79. doi: 10.1186/1742-2094-8-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fenn AM, et al. Immune activation promotes depression 1 month after diffuse brain injury: a role for primed microglia. Biol Psychiatry. 2014;76:575–584. doi: 10.1016/j.biopsych.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fenn AM, et al. Methylene blue attenuates traumatic brain injury-associated neuroinflammation and acute depressive-like behavior in mice. J Neurotrauma. 2015;32:127–138. doi: 10.1089/neu.2014.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang G, et al. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J Cereb Blood Flow Metab. 2013;33:1864–1874. doi: 10.1038/jcbfm.2013.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kovesdi E, et al. Acute minocycline treatment mitigates the symptoms of mild blast-induced traumatic brain injury. Front Neurol. 2012;3:111. doi: 10.3389/fneur.2012.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Homsi S, et al. Blockade of acute microglial activation by minocycline promotes neuroprotection and reduces locomotor hyperactivity after closed head injury in mice: a twelve-week follow-up study. J Neurotrauma. 2010;27:911–921. doi: 10.1089/neu.2009.1223. [DOI] [PubMed] [Google Scholar]
- 45.Baalman K, et al. Axon initial segment-associated microglia. J Neurosci. 2015;35:2283–2292. doi: 10.1523/JNEUROSCI.3751-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ziebell JM, et al. Rod microglia: elongation, alignment, and coupling to form trains across the somatosensory cortex after experimental diffuse brain injury. J Neuroinflammation. 2012;9:247. doi: 10.1186/1742-2094-9-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tam WY, Ma CH. Bipolar/rod-shaped microglia are proliferating microglia with distinct M1/M2 phenotypes. Sci Rep. 2014;4:7279. doi: 10.1038/srep07279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Katsumoto A, et al. Ontogeny and functions of central nervous system macrophages. J Immunol. 2014;193:2615–2621. doi: 10.4049/jimmunol.1400716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Semple BD, et al. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2−/− mice. J Cereb Blood Flow Metab. 2010;30:769–782. doi: 10.1038/jcbfm.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morganti JM, et al. CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J Neurosci. 2015;35:748–760. doi: 10.1523/JNEUROSCI.2405-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Velazquez A, et al. Widespread microglial activation in patients deceased from traumatic brain injury. Brain Inj. 2015:1–8. doi: 10.3109/02699052.2015.1018325. [DOI] [PubMed] [Google Scholar]
- 52.Newell E, et al. Cerebrospinal Fluid Markers of Macrophage and Lymphocyte Activation After Traumatic Brain Injury in Children. Pediatr Crit Care Med. 2015 doi: 10.1097/PCC.0000000000000400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shan R, et al. A New Panel of Blood Biomarkers for the Diagnosis of Mild Traumatic Brain Injury/Concussion in Adults. J Neurotrauma. 2015 doi: 10.1089/neu.2014.3811. [DOI] [PubMed] [Google Scholar]
- 54.Chen Z, Trapp BD. Microglia and neuroprotection. J Neurochem. 2015 doi: 10.1111/jnc.13062. [DOI] [PubMed] [Google Scholar]
- 55.Rice RA, et al. Elimination of Microglia Improves Functional Outcomes Following Extensive Neuronal Loss in the Hippocampus. J Neurosci. 2015;35:9977–9989. doi: 10.1523/JNEUROSCI.0336-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elmore MR, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82:380–397. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen Z, et al. Microglial displacement of inhibitory synapses provides neuroprotection in the adult brain. Nature communications. 2014;5:4486. doi: 10.1038/ncomms5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Loane DJ, et al. Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol. 2014;73:14–29. doi: 10.1097/NEN.0000000000000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brooks WM, et al. Metabolic and cognitive response to human traumatic brain injury: a quantitative proton magnetic resonance study. Journal of neurotrauma. 2000;17:629–640. doi: 10.1089/089771500415382. [DOI] [PubMed] [Google Scholar]
- 60.Kirov II, et al. Proton MR spectroscopy correlates diffuse axonal abnormalities with post-concussive symptoms in mild traumatic brain injury. Journal of neurotrauma. 2013;30:1200–1204. doi: 10.1089/neu.2012.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kumar RG, et al. Acute CSF interleukin-6 trajectories after TBI: Associations with neuroinflammation, polytrauma, and outcome. Brain, behavior, and immunity. 2015;45:253–262. doi: 10.1016/j.bbi.2014.12.021. [DOI] [PubMed] [Google Scholar]
- 62.Juengst SB, et al. Exploratory associations with Tumor Necrosis Factor-alpha, disinhibition and suicidal endorsement after traumatic brain injury. Brain, behavior, and immunity. 2014;41:134–143. doi: 10.1016/j.bbi.2014.05.020. [DOI] [PubMed] [Google Scholar]
- 63.Smith C, et al. The neuroinflammatory response in humans after traumatic brain injury. Neuropathol Appl Neurobiol. 2013;39:654–666. doi: 10.1111/nan.12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Coughlin JM, et al. Neuroinflammation and brain atrophy in former NFL players: An in vivo multimodal imaging pilot study. Neurobiol Dis. 2015;74:58–65. doi: 10.1016/j.nbd.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Banati RB. Visualising microglial activation in vivo. Glia. 2002;40:206–217. doi: 10.1002/glia.10144. [DOI] [PubMed] [Google Scholar]
- 66.Frank MG, et al. mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol Aging. 2006;27:717–722. doi: 10.1016/j.neurobiolaging.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 67.VanGuilder HD, et al. Concurrent hippocampal induction of MHC II pathway components and glial activation with advanced aging is not correlated with cognitive impairment. J Neuroinflammation. 2011;8:138. doi: 10.1186/1742-2094-8-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Henry CJ, et al. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23:309–317. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Godbout JP, et al. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 2005;19:1329–1331. doi: 10.1096/fj.05-3776fje. [DOI] [PubMed] [Google Scholar]
- 70.Godbout JP, et al. Aging exacerbates depressive-like behavior in mice in response to activation of the peripheral innate immune system. Neuropsychopharmacology. 2008;33:2341–2351. doi: 10.1038/sj.npp.1301649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barrientos RM, et al. Time course of hippocampal IL-1 beta and memory consolidation impairments in aging rats following peripheral infection. Brain Behav Immun. 2009;23:46–54. doi: 10.1016/j.bbi.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen J, et al. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav Immun. 2008;22:301–311. doi: 10.1016/j.bbi.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Palin K, et al. Systemic inflammation switches the inflammatory cytokine profile in CNS Wallerian degeneration. Neurobiol Dis. 2008;30:19–29. doi: 10.1016/j.nbd.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 74.Aungst SL, et al. Repeated mild traumatic brain injury causes chronic neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. J Cereb Blood Flow Metab. 2014;34:1223–1232. doi: 10.1038/jcbfm.2014.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mouzon BC, et al. Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann Neurol. 2014;75:241–254. doi: 10.1002/ana.24064. [DOI] [PubMed] [Google Scholar]
- 76.Weil ZM, et al. Injury timing alters metabolic, inflammatory and functional outcomes following repeated mild traumatic brain injury. Neurobiol Dis. 2014;70:108–116. doi: 10.1016/j.nbd.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 77.Shitaka Y, et al. Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal injury and microglial reactivity. J Neuropathol Exp Neurol. 2011;70:551–567. doi: 10.1097/NEN.0b013e31821f891f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McKee AC, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136:43–64. doi: 10.1093/brain/aws307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mortimer JA, et al. Head trauma as a risk factor for Alzheimer’s disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol. 1991;20(Suppl 2):S28–35. doi: 10.1093/ije/20.supplement_2.s28. [DOI] [PubMed] [Google Scholar]
- 80.Fleminger S, et al. Head injury as a risk factor for Alzheimer’s disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry. 2003;74:857–862. doi: 10.1136/jnnp.74.7.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Plassman BL, et al. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology. 2000;55:1158–1166. doi: 10.1212/wnl.55.8.1158. [DOI] [PubMed] [Google Scholar]
- 82.Gardner RC, Yaffe K. Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol Cell Neurosci. 2015 doi: 10.1016/j.mcn.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Daneshvar DH, et al. Post-traumatic neurodegeneration and chronic traumatic encephalopathy. Mol Cell Neurosci. 2015;66:81–90. doi: 10.1016/j.mcn.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 84.Johnson VE, et al. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain pathology. 2012;22:142–149. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Petraglia AL, et al. The pathophysiology underlying repetitive mild traumatic brain injury in a novel mouse model of chronic traumatic encephalopathy. Surg Neurol Int. 2014;5:184. doi: 10.4103/2152-7806.147566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tajiri N, et al. Traumatic brain injury precipitates cognitive impairment and extracellular Abeta aggregation in Alzheimer’s disease transgenic mice. PLoS One. 2013;8:e78851. doi: 10.1371/journal.pone.0078851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Saing T, et al. Frontal cortex neuropathology in dementia pugilistica. J Neurotrauma. 2012;29:1054–1070. doi: 10.1089/neu.2011.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Goldstein LE, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. 2012;4:134ra160. doi: 10.1126/scitranslmed.3003716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McKee AC, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709–735. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stern RA, et al. Long-term consequences of repetitive brain trauma: chronic traumatic encephalopathy. PM R. 2011;3:S460–467. doi: 10.1016/j.pmrj.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 91.Iverson GL, et al. A critical review of chronic traumatic encephalopathy. Neurosci Biobehav Rev. 2015;56:276–293. doi: 10.1016/j.neubiorev.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 92.Albrecht JS, et al. Patterns of Depression Treatment in Medicare Beneficiaries with Depression after Traumatic Brain Injury. J Neurotrauma. 2015 doi: 10.1089/neu.2014.3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dantzer R. Depression and inflammation: an intricate relationship. Biol Psychiatry. 2012;71:4–5. doi: 10.1016/j.biopsych.2011.10.025. [DOI] [PubMed] [Google Scholar]
- 94.Petraglia AL, et al. The spectrum of neurobehavioral sequelae after repetitive mild traumatic brain injury: a novel mouse model of chronic traumatic encephalopathy. Journal of neurotrauma. 2014;31:1211–1224. doi: 10.1089/neu.2013.3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics: the journal of the American Society for Experimental Neuro Therapeutics. 2010;7:354–365. doi: 10.1016/j.nurt.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]