

Abstract
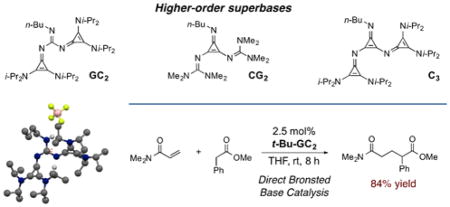
The synthesis and characterization of six new classes of higher-order superbases, including five that incorporate cyclopropenimine functionality, has been achieved. We propose a nomenclature that designates these as the CG2, GC2, PC3, PC1, C3, and GP2 classes of superbases. The pKBH+ values were measured to be between 29.0 and 35.6 in acetonitrile. Linear correlations of ten superbase basicities vs. that of their substituents demonstrated the insulating effect of the cyclopropenimine core. The molecular structures of several of these materials were obtained by single-crystal X-ray analysis, revealing interesting aspects of conformational bias and noncovalent organization. The types of superbasic cores and substituents were each shown reliably to affect selectivity for deprotonation over alkylation. Higher-order cyclopropenimine and guanidine superbase stability to hydrolysis was found to correlate to basicity. Finally, a GC2 base was found to catalyze conjugate additions of α-aryl ester pronucleophiles, representing the first report of a neutral Brønsted base to catalyze such reactions.
Graphical abstract
Introduction
Strong Brønsted bases occupy an important place in the organic chemist’s arsenal due to the large number of chemical reactions that involve deprotonation as a key activation step. Within the broader category of Brønsted bases, the so-called superbases are those species derived from the cooperative combination of two or more constituent bases,1 a synergism that typically results in high thermodynamic basicities.2 Many superbases including amidines, guanidines, and phosphazenes, which rely on the combined action of multiple amino substituents, have found important application in organic synthesis, including asymmetric catalysis.3,4 For obvious reasons, strength of basicity is a key parameter that affects what substrates are amenable to activation with a given Brønsted base. In this regard, the superbase concept can be further extended to species that involve the combination of multiple superbases to form “higher-order superbases”, 5–12 and here truly remarkable basicities have been registered. Although such bases have great potential utility,13 the variety of available functionalities remains limited, and problems of stability and difficulties of preparation make the identification of new higher-order superbases an important goal.
We recently introduced 2,3-diaminocyclopropenimines as a new class of organic superbase.14 These cyclopro-penimines are as basic as the P1 phosphazenes but have dramatically improved stability profiles in non-inert atmosphere. Given the growing recognition of the utility of cyclopropenimine bases, we sought to develop a number of higher-order cyclopropenimine superbases with the goal of realizing enhanced basicities and unique functional properties. In this Article, we describe the synthesis and characterization of four new classes of super-bases that incorporate cyclopropenimine functionality (Figure 1) as either the core group (blue box), the substituents (red box), or both (purple box). In addition, we report the first example of a bisphosphazenylguanidine higher-order superbase, as well as a monocyclopropeniminyl phosphazene base (not shown).
Figure 1.

Higher-order superbases, including those with a cyclopropenimine core (blue box), cyclopropenimine substituents (red box), or both (purple box).
Nomenclature
Before beginning the discussion of this work, a brief explanation of the nomenclature for the materials described herein is appropriate. The combination of different superbases offers a variety of possibilities, and so in order to aid in the description of these variants we propose a classification scheme inspired by the convention already in use for the phosphazene bases.6 Thus, each base is first assigned a letter according to the functionality that comprises its basic core: “G” for guanidinyl, “P” for phosphazenyl, and “C” for cyclopropeniminyl. This letter is then followed by the letters corresponding to the su-perbase substituents on that core (G, P, C), with a numerical subscript indicating the number of substituents. Thus a trisguanidinylphosphazene is a “PG3” base, while a biscyclopropeniminylguanidine is a “GC2” base. In the cases where the core and substituents are from the same class, the descriptor can be distilled to a single letter, e.g. “P4”, “G3”, and “C3”. Herein we describe the first synthesis of members of the CG2, GC2, PC3, PC1, C3 and GP2 classes of higher-order superbases.
Synthesis
Typically, higher-order superbases are prepared from the reaction of a nucleophilic superbase with an appropriate electrophile. However, such procedures can be complicated by (1) the use of highly moisture-sensitive electrophiles such as PCl5 or chloroformamidinium salts, and (2) the need for twice the equivalents of the nucleo-philic superbase in order to neutralize the acid that forms during substitution. Moreover, this type of strategy requires the use of a neutral superbase, which is often less convenient to handle and store than its conjugate acid.
In contrast to the aforementioned electrophiles, cyclo-propenimines are generally prepared from tetrachlorocy-clopropene (C3Cl4),15 which is reactive with nitrogen nucleophiles but relatively resistant to hydrolysis. As such, we were able to develop a convenient procedure to prepare the C3 base 2: cyclopropenimine 1•HCl (2 equiv) was treated with tetrachlorocyclopropene (C3Cl4) and KOH (4 equiv) in a biphasic mixture of CH2Cl2 and water at 0 °C (Figure 2a). After 1 h, the layers were separated, n-BuNH2 was added, and the mixture was stirred for 21 h at rt. After aqueous workup, ion exchange with NaBF4, and recrystallization, we obtained nearly 8 g of 2 as its HBF4 salt in 76% yield. Importantly, this convenient neutralization of HCl by aqueous KOH both enabled the use of the more conveniently handled HCl salt of the cyclo-propenimine starting material, and obviated the need for any excess of this reactant.
Figure 2.

Syntheses of higher-order superbases.
The CG2 base 4 was prepared by a similar route, but since we have observed that tetramethylguanidine (3) readily adds three times to tetrachlorocyclopropene at rt, some modifications were needed (Figure 2b). Thus the guanidine was added in dry CH2Cl2 at –78 °C to prevent the third substitution, and we employed a sufficient excess of the free base (4 equiv) to neutralize HCl. From this procedure, 2.51 g of 4•HBF4 was isolated in 58% yield.
We also developed a new route to higher-order guani-dine superbases since traditional approaches involving chloroformamidinium16 or thiouronium17 salts were consistently problematic in our hands. Compared to these electrophiles, carbonimidic dichloride18 5 provided a more desirable balance of reactivity with nucleophiles and ease of handling. The preparation of the GC2 base 6 was otherwise comparable to that of the C3 base. Cyclo-propenimine 1•HCl (2 equiv) and aqueous KOH (4 equiv) was added to 5 in CH2Cl2 (Figure 2c). After stirring at rt for 3 d, workup and ion exchange furnished 5.67 g of 6•HBF4 in 79% yield.
While the n-butyl head groups were selected for most higher-order superbases in this study for structural consistency and to facilitate synthetic procedures, we also sought a more hindered version of a GC2 base. We therefore reacted tert-butyl carbonimidic dichloride 7 with 1, although in this case 4 equiv of the neutral base were needed since the use of KOH as a base led to significant amounts of hydrolysis byproducts (Figure 2d). Workup similar to that used for the isolation of 6 provided 3.44 g of 8•HBF4 in 67% yield.
This strategy was also adapted to prepare the first reported example of a GP2 base 10 (Figure 2e). The reaction was conducted in toluene at a higher temperature (95 °C) to compensate for slower substitution, and was again cleaner when using the free phosphazene 9 (4 equiv) as both the nucleophile and the base. After workup and ion exchange, we isolated 2.04 g of 10•HBF4 in 89% yield.
The carbonimidic dichloride method similarly provided 11 HCl in 63% yield (Figure 2f), offering an alternative to the recently reported synthesis of peralkylated tri-guanides using the sensitive Vilsmeier salt.5
The PC3 base 12 was prepared by a process used in the synthesis of guanidinylphosphazenes7 (Figure 2g). Isolation of 12 was most convenient as the HPF6 salt, although purification of this material has thus far proven to be an intractable problem, possibly due to its relative sensitivity, and so the obtained characterization data is of impure material.
In light of our difficulties in obtaining a clean sample of a PC3 base, we also targeted the synthesis of a partially monocyclopropeniminylphosphazene (PC1) in order to fully characterize the first example of a molecule combining cyclopropenimine and phosphazene moieties. We observed that 1 was reluctant to add more than once to trichloroiminophosphorane 13,19 so reaction of these components in a 2:1 ratio at −78 °C followed by treatment with excess piperidine at 0 °C provided 2.71 g of PC1 base 14 as its HBF4 salt after the appropriate workup (Figure 2h).
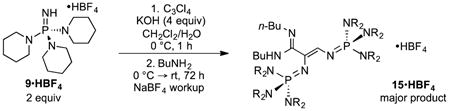
Notably, all attempts at the preparation of a CP2 base have failed due to facile ring-opening20 of the cyclopropene core (eq 1). Although we have been unable to purify the major product, spectroscopic data (NMR, MS) suggest that its structure is the amidine 15•HBF4.
 |
(1) |
All members of the new classes of superbases that we successfully isolated could be liberated on treatment with KOt-Bu (1 equiv) at rt in THF (see Supporting Information). While we isolated and characterized all of the corresponding free bases in good yields, they were typically sensitive to air and moisture, so these procedures needed to be conducted in a glove box. Therefore, our catalytic studies with these superbases (vide infra) were performed by deprotonating the base in situ immediately prior to adding the reactants.
Basicity measurements
With robust synthetic routes in hand, we sought to determine the basicities of these new higher-order super-bases. The pKBH+ values in acetonitrile are shown in Table 1. Notably, while the basicity trend of the parent su-perbases is guanidine < cyclopropenimine ≈ phos-phazene (16, 17, and 18), the trend for the higher-order superbases is cyclopropenimine < guanidine < phos-phazene (compare 11, 4, and 19 or 6, 2, and 12). These data indicate that the cyclopropene core is relatively less effective at communicating the basicity of the substituents to the head imino group. The greater insulating character of the cyclopropenimine functionality can be understood by consideration of the conjugate acid form, the core of which is a cyclopropenium ring. As a closed-shell aromatic, the cyclopropenium group gains less in stability from the donation of electron density than other cations, a trend long-established for the chemistry of the cyclopropenium unit.21
Table 1.
Basicities of higher-order superbases.a

|
Italicized numbers represent pKBH+ values. Values below 33 were measured in acetonitrile. Values above 33 were extrapolated from measurements in THF. See Supporting Information.
Literature values, see Supporting Information. (NR2) = piperidinyl.
This phenomenon as it relates to superbasicity can be quantified by plotting the relationship of the pKBH+ values of guanidine, phosphazene and cyclopropenimine su-perbases versus the basicities of their substituents (amino, guanidino, cyclopropenimino, phosphazeno) as shown in Figure 3. The slope of each line is a measure of the degree to which the core functionality transmits the electron density of the substituents to the head imino group. For comparison purposes, the slopes must be normalized per substituent; for example, the guanidine slope of 1.13 corresponds to a value of 0.57 for each individual substituent. (A “perfect” normalized value would be 1.00). From this analysis, it can be seen that the phosphazene (0.55) and guanidine (0.57) core groups have approximately the same transmissibility, while the cyclopropenimine (0.21) is less than half as effective. The upshot of these trends is that the CG2 and G3 bases have essentially the same basicity.
Figure 3.

Plot of superbase pKBH+ vs. substituent basicities for phosphazene (
 ), guanidine (
), guanidine (
 ), and cyclopropen-imine (
), and cyclopropen-imine (
 ) superbases.
) superbases.
The y-intercepts of the trend lines in Figure 3 represent the basicity of hypothetical species bearing substituents with pKBH+ values of 0. In this case, the cyclopro-penimine value (19.36) greatly exceeds that of both phosphazene (−3.22) and guanidine (2.99), which again is a reflection of the far greater stability of the aromatic cyclopropenium ion compared to either phosphonium or carbenium ions. From these trends, it can be predicted that cyclopropenimines bearing substituents that are less π-donating than amino groups (e.g. aryl) should maintain a relatively high level of basicity.
Selectivity: elimination vs. alkylation
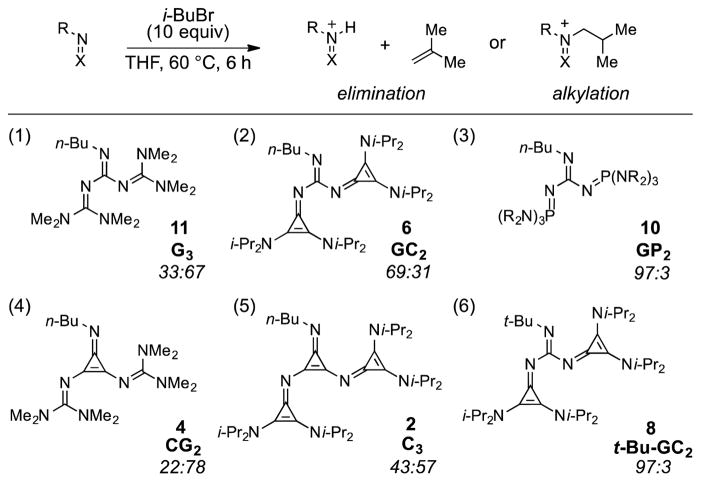
One potential complication to the use of strong Brønsted bases is their capacity to function instead as nucleophiles (i.e. Lewis bases). A strong preference for Brønsted over Lewis base reactivity is thus a crucial parameter for optimal Brønsted base catalysts. Schwe-singer has thoroughly investigated such selectivities in the context of phosphazenes6 by measuring the extent to which these bases react with alkyl halides via elimination versus alkylation.6 Using this same approach, we examined the elimination vs. alkylation selectivity of the new classes of superbases we have prepared. The super-bases were thus treated with i-butyl bromide under conditions that completely consumed the free base (10 equiv i-BuBr, 60 °C for 6 h), and the selectivity was measured by integrating the ratio of protonated and alkylated products (Table 2). As can be seen from comparison of guanidine bases with the same n-Bu head group, the selectivity for protonation increases according to substituent size in the order G3 < GC2 < GP2 (entries 1–3). The effect of the superbase core also reveals a mild size correlation, with the guanidines showing higher protonation selectivity than the cyclopropenimines (compare entries 1 and 4 or 2 and 5), due to the greater congestion of the former. However, the steric demand of the imino substituent appears to have the largest effect on selectivity, as can be seen from comparison of the n-Bu-GC2 and t-Bu-GC2 bases 6 and 8 (entries 2 and 6), with the selectivity of the latter being strongly in favor of protonation.
Table 2.
Elimination vs. alkylation with higher-order superbases and iso-butyl bromide.a
|
Italicized numbers correspond to ratios of elimination vs. alkylation as determined by the ratios of protonated vs. alkylated superbases observed by 1H NMR, See Supporting Information.
Molecular structures
The molecular structures of several higher-order su-perbase salts are shown in Figure 4. Notably, the C3 salt 2•HBF4 (Figure 4a), the CG2 salt 6•HBF4 (Figure 4b), and the GP2 salt 10•HBF4 (Figure 4e) all display gearing of the n-butyl and cyclopropeniminyl / phosphazenyl substituents, due to rather significant steric congestion. For the C3 salt 2•HBF4 and the CG2 salt 6•HBF4, the cy-clopropeniminyl substituent that is syn to the N-H group is torqued such that it is significantly out of the plane of the core π-system, at respective angles of 66° and 45°. While these distortions may be partially explained by steric effects, they appear also to enable intramolecular CH---N bonds between the imino nitrogen atom of the torqued cyclopropeniminyl substituent and one of the isopropyl CH protons of the adjacent substituent. The CH---N distances are 2.35 Å in the C3 salt and 2.50 Å in the GC2 salt, well within the expected range for such an interaction. Notably, the type of conformational organization observed in Figures 4a and 4b raises intriguing possibilities for the design of asymmetric catalysts, since it places the H-bonded counterion in close contact with the distal amino groups of the cyclopropeniminyl substituent.
Figure 4.

Molecular structures of (a) 2•HBF4, (b) 6•HBF4, (c) 4•HBF4, (d) 14•HBF4, and (e) 10•HBF4. (c) and (e) also contain second structures that are qualitatively similar to those shown here (see the SI for complete structures).
In contrast, the CG2 salt 4•HBF4 (Figure 4c) does not exhibit such a gearing effect, and indeed the guanidinyl substituents are arranged syn to one another and appear to be engaged in a π-π interaction. However, the crystal lattice was disordered, so caution should be taken in drawing conclusions from this structure. Interestingly, the GP2 salt 10•HBF4 (Figure 4d) features P=N–C bond angles noticeably larger than the ideal 120° (see the Supporting Information), a feature not observed in the other structures shown. Such a distortion most likely arises from steric conflict of the extremely bulky tris(piperidinyl)phosphazenyl substituents.
Hydrolytic Stability
Although higher-order superbases tend to be robust species for most applications, they can be susceptible to nucleophilic attack, especially by solvolysis. To probe the limits of stability of the new superbases, we measured their decomposition rates during heating in aqueous methanol in the presence of 10 equiv of sodium methox-ide (Table 3).
Table 3.
Relative stabilities of superbases in basic aqueous methanol.a
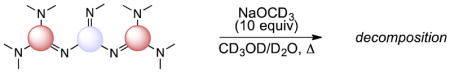
| ||||||
|---|---|---|---|---|---|---|
| entry | class | compound | temp (°C) | t1/2(h) | t1/2 rel | pKBH+ |
| 1 | GP2 | 10 | 140 | ≫24 | >104 | 34.3 |
| 2 | PC1 | 14 | 140 | ≫24 | >104 | 31.8 |
| 3 | P1 | 18 | 140 | ≫24 | >104 | 27.8 |
| 4 | PG3 | 19 | 140 | 32 | 1200 | 37.9 |
|
| ||||||
| 5 | GC2 | 6 | 140 | 2.3 | 81 | 35.6 |
| 6 | C3 | 2 | 140 | 1.0 | 36 | 31.6 |
| 7 | C3 | 2 | 80 | 9.0 | 36 | 31.6 |
| 8 | G3 | 11 | 80 | 2.0 | 8.0 | 29.5 |
| 9 | C1 | 17 | 80 | 1.0 | 4.0 | 27.6 |
| 10 | G1 | 16 | 80 | 0.67 | 2.7 | 24.8 |
| 11 | CG2 | 4 | 80 | 0.25 | 1.0 | 29.0 |
Superbase (0.075 mmol) and NaOCD3 (0.75 mmol) were heated in a mixture of CD3OD (0.45 mL) and D2O (0.25 mL) in sealed NMR tubes until 50% of the initial amount of su-perbase remained (vs. sodium 3-(trimethylsilyl)propanesulfonate internal standard).
The superbases fell roughly into 2 categories. The first encompassed highly robust molecules, which showed no measurable decomposition after 24 h at 140 °C. Among this group were the GP2, PC1, and the P1 bases (entries 1–3).6 The PG3 base is also quite robust under these conditions (entry 4), but does undergo an observable slow decomposition.
The remaining superbases had a measurable half-life (<10 h) at 140 °C (entries 5 and 6) or 80 °C (entries 7–11) in a sealed tube. For the majority of this group, there is a direct relationship between the basicity and the stability of the molecules under these conditions (entries 5–10). The exception in the CG2 base (entry 11), which despite its intermediate basicity is the least stable of all the superbases studied. We note that the CG2 base also displayed the poorest selectivity for elimination vs. alkylation (see Table 2), and we speculate that both effects result from the relatively small guanidine substituents coupled with the expanded cyclopropenimine core leaving the imino function relatively open to C-nucleophilic or N-electrophilic attack. The clear conclusion from Table 3 is that the bases in entries 6–11 should be used at temperatures well below 80 °C for maximal effectiveness.
Catalysis
The purpose of designing more strongly basic super-bases is to enable reactivity with a broader range of less-acidic substrates, and thus the new superbase platforms described here are expected to offer important new opportunities for method development. To illustrate this potential, we examined the conjugate addition of indole (23) to crotonitrile (22) under superbase catalysis (eq 2). Notably, while the C1 base 17 did not catalyze this reaction to any appreciable extent over 24 h, the C3 base 2 and GC2 base 8, which are 4 and 8 orders of magnitude more basic respectively than 17, catalyzed the formation of 24 in 89% and 95% yields after 3 h.
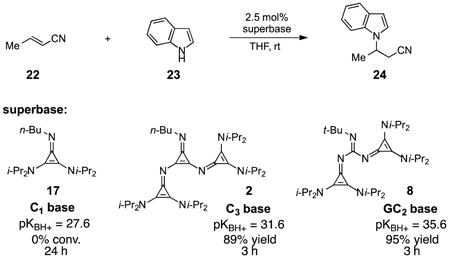 |
(2) |
In addition, we found the t-Bu-GC2 base 8 to be effective for catalyzing the conjugate addition of α-aryl esters to Michael acceptors, a reaction that has not been reported using direct neutral Brønsted base catalysis.22,23 As shown in Table 4, reaction of a range of α-aryl donors and various Michael acceptors in the presence of 2.5 mol% of 8 produced Michael adducts in excellent yields after 8–12 h. Michael acceptors including an unsaturated ester (entry 1), nitrile (entry 2a), and amide (entry 3) were found to be viable, although in the case of the amide, β-substitution was not compatible. Notably, in a direct comparison, the P4 base – the only commercially available higher-order superbase stronger than the GC2 base—resulted in only modest conversion (entry 2b) in comparison to the GC2 base (entry 2a). Reaction with a β-alkyl Michael acceptor was also productive (entry 4). In terms of scope of the aryl substituent, ortho substituents (entry 5), as well as electron withdrawing (entry 6), and electron donating (entry 7) substituents were well-tolerated. In addition, a heteroaromatic-containing product was accessible in quantitative yield (entry 8). Finally, we found that the generation of a quaternary carbon center could be accomplished in high yield with the use of an α-branched pronucleophile (entry 9). In all cases, diastereoselectivity was predictably low, given that the catalyst 8 does not incorporate any of the hydrogen-bonding or other organizational motifs that would be present in chiral analogues. Nevertheless, the ability to engage simple α-aryl esters and nitriles in direct Brønsted base chemistry represents an important advance for the area of strong neutral base catalysis.
Table 4.
Substrate scope of higher-order superbase-catalyzed conjugate additions.a
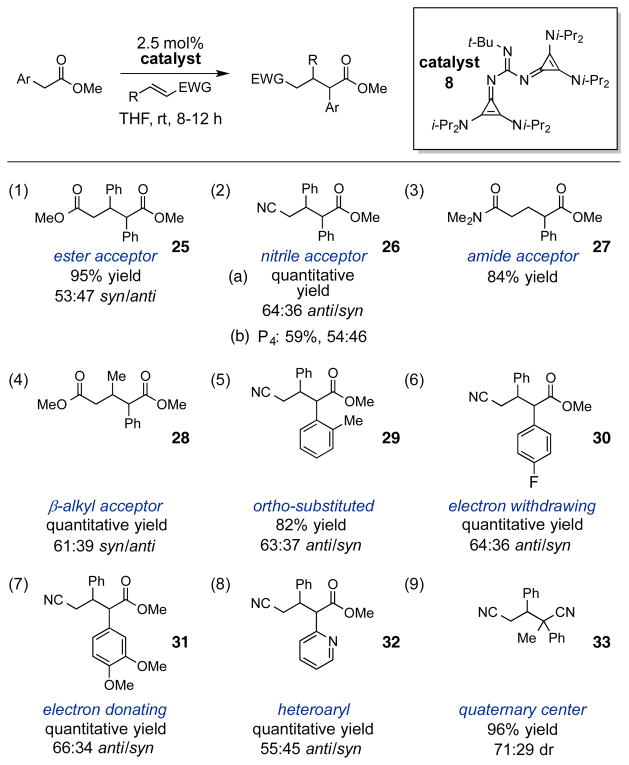
|
Pronucleophile and Michael acceptor (1.0 mmol of limiting reagent) were added to a solution of catalyst (0.025 mmol) in THF (3 mL) and stirred for 8–12 h. See Supporting Information for details.
Conclusion
Higher-order superbases offer the potential to realize very strong basicities in the context of neutral, organic-soluble reagents. The current work significantly expands the scope of available materials in this category with the introduction of higher-order superbases bearing cyclo-propenimine functionality. The enhanced basicity of these new bases coupled with their relatively straightforward synthetic accessibility offer important new opportunities for method development in the area of Brønsted base catalysis.
Experimental Section
Synthesis of C3 base 2•HBF4
3 M KOH (20.25 mL, 60.75 mmol, 4.03 equiv) was added to 1•HCl (8.71 g, 30.3 mmol, 2.01 equiv) and C3Cl415 (1.85 mL, 15.1 mmol, 1.00 equiv) in CH2Cl2 (300 mL) at 0 °C. After stirring for 1 h, the layers were separated, and the organic layer was dried over Na2SO4 and quickly filtered into a second flask at 0 °C. n-BuNH2 (1.4 mL, 14 mmol, 4.8 equiv) was added and the solution was stirred for 21 h while warming slowly to rt. The reaction was then washed with a combination of 0.5 M Na2CO3 and 0.5 M NaBF4 (2 × 100 mL), dried over Na2SO4, and concentrated. The residue was redissolved in EtOAc (300 mL), washed with sat. NH4Cl (3 × 50 mL) and a combination of 0.5 M Na2CO3 and 1 M NaBF4 (50 mL), dried over Na2SO4, and concentrated to provide a tan solid (11.75 g). This material was dissolved in hot EtOAc (125 mL), cooled to rt, and hexanes was diffused in for 3 days, followed by standing at −20 °C overnight. The product crystallized as large yellow prisms (8.00 g) The recrystallization was repeated on ~ 80% of the previous scale and deposited white prisms (7.93 g, 76%). 1H NMR (400 MHz, CDCl3) δ 5.73 (s, 1H), 3.84 (hept, J = 6.7 Hz, 8H), 3.30 (app q, 2H), 1.66 (m, 2H), 1.42 – 1.18 (overlapping signals, 50H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 128.0, 124.2, 122.3, 119.7, 50.2, 46.5, 32.7, 21.9, 19.6, 13.6. HRMS (ESI+) for C37H65N7 [MH]+ m/z calcd 608.5380, found 608.5383.
Synthesis of CG2 base 4•HBF4
Tetramethylguani-dine (3) in CH2Cl2 (5.50 mL TMG diluted to a volume of 19.6 mL, used 18.2 mL, 40.7 mmol TMG, 3.99 equiv) was added at −78 °C over 1 h to a solution of C3Cl415 (1.25 mL, 10.2 mmol, 1.0 equiv) in CH2Cl2 (50 mL). After stirring for 3 h, the reaction was warmed to rt for 30 min, cooled to 0°C, and n-BuNH2 (2.5 mL, 25 mmol, 2.5 equiv) was added. After 16 h, the reaction was concentrated, redissolved in 1:1 EtOAc/CH2Cl2,, and extracted into sat. NH4Cl (3 × 40 mL). The combined aqueous extracts were extracted with CH2Cl2 (3 × 40 mL), then these combined organic layers were washed with sat. NH4Cl (2 × 30 mL) and a combination of 0.5 M Na2CO3 and 1 M NaBF4 (2 × 20 mL), dried over Na2SO4, and concentrated. The resulting brown syrup was purified by flash chromatography (5% → 8% MeOH/CH2Cl2) on silica gel (250 mL) to provide a yellow semisolid. This material was recrystallized from ~ 4:1 EtOAc/hexanes (30 mL) while diffusing in further hexanes at −20 °C to provide the HBF4 salt of the title compound as a yellow solid (2.51 g, 58%). 1H NMR (400 MHz, CDCl3) δ 6.58 (t, J = 5.3 Hz, 1H), 3.36 (app q, 2H), 2.96 (s, 24H), 1.67 (m, 2H), 1.38 (hex, J = 7.5 Hz, 2H), 0.93 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.7, 128.5, 124.4, 123.4, 46.3, 39.8, 32.2, 19.3, 13.5. HRMS (ESI+) for C17H33N7 [MH]+ m/z calcd 336.2876, found 336.2881.
Synthesis of GC2 base 6•HBF4
3 M KOH (13.75 mL, 41.25 mmol, 4.04 equiv) was added to 1•HCl (5.88 g, 20.4 mmol, 2.00 equiv) and 516c (1.65 g, 10.7 mmol, 1.00 equiv) in CH2Cl2 (100 mL) at 0 °C. After stirring for 72 h, the CH2Cl2 was evaporated, the residue was dissolved in EtOAc (250 mL), extracted with 1 M HCl (3 × 75 mL), and extracted from the combined aqueous layers with CH2Cl2 (3 × 75 mL). The combined CH2Cl2 layers were washed with 5% Na2CO3 (75 mL) and concentrated. This residue was dissolved in EtOAc (150 mL), washed with sat. NH4Cl (50 mL), 50% sat. NH4Cl (2 × 50 mL), and a combination of 5% Na2CO3 and 1 M NaBF4 (2 × 25 mL), dried over Na2SO4, and concentrated. The resulting pale yellow solid was recrystallized from ~10:1 EtOAc/hexanes (125 mL) for 6 days at −20 °C to provide a white solid (5.67 g, 79%). 1H NMR (500 MHz, CDCl3) δ 4.52 (t, J = 5.6 Hz, 1H), 3.89 (hept, J = 6.7 Hz, 8H), 3.31 (dt, J = 7.4, 5.8 Hz, 2H), 1.59 (m, 2H), 1.39 – 1.18 (overlapping signals, 50H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 161.0, 121.9, 120.4, 50.3, 42.9, 32.1, 22.1, 20.3, 14.0. HRMS (ESI+) for C35H65N7 [MH]+ m/z calcd 584.5380, found 584.5374.
Synthesis of GC2 base 8•HBF4
716c (1.17 g, 7.60 mmol, 1.00 equiv) in CH2Cl2 (8 mL) was added to a solution of 1 (7.68 g, 30.6 mmol, 4.02 equiv) in CH2Cl2 (200 mL) at 0 °C. After stirring for 42 h, the CH2Cl2 was evaporated, the residue was suspended in EtOAc (200 mL), washed with 50% sat. NH4Cl (2 × 75 mL) and a combination of 5% Na2CO3 and 1 M NaBF4 (2 × 25 mL), dried over Na2SO4, and concentrated. This solid was recrystal-lized from ~2:1 EtOAc/hexanes (50 mL) for 2 days at −20 °C to provide a white solid (3.44 g, 67%). 1H NMR (500 MHz, CDCl3) δ 4.09 (s, 1H), 3.87 (hept, J = 6.8 Hz, 8H), 1.43 (s, 9H), 1.29 (d, J = 6.8 Hz, 48H). 13C NMR (126 MHz, CDCl3) δ 159.4, 121.5, 120.4, 51.9, 50.3, 29.3, 22.1. HRMS (ESI+) for C35H65N7 [MH]+ m/z calcd 584.5380, found 584.5374.
Synthesis of PC1 base 14•HBF4
1319 (1.05 g, 5.04 mmol, 1.00 equiv) in THF (5 mL) was added dropwise to a solution of 1 (2.82 g, 11.2 mmol, 2.23 equiv) in THF (70 mL) at −78 °C. After 4 h, the reaction was warmed to 0°C, and after 2 h, piperidine (2.5 mL, 25 mmol, 5.0 equiv) was added. After 24 h while warming to rt, the reaction was concentrated, and the residue was suspended in EtOAc (150 mL), washed with sat. NH4Cl (2 × 75 mL), 50% sat. NH4Cl (75 mL), and a combination of 0.5 M Na2CO3 and 1 M NaBF4 (2 × 20 mL), dried over Na2SO4, and concentrated. The resulting yellow solid (3.21 g) was recrystallized from ~ 2:1 EtOAc/hexanes (100 mL) while diffusing in further hexanes at −20 °C to provide the phosphazene•HBF4 as a white solid (2.71 g, 88%).1H NMR (500 MHz, CDCl3) δ 3.92 (hept, J = 6.9 Hz, 4H), 3.28 (d, J = 12.1 Hz, 1H), 3.19 (m, 4H), 3.08 (m, 4H), 1.63 – 1.52 (m, 12H), 1.33 (d, J = 7.0 Hz, 24H), 1.29 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 119.4 (d, JPC = 22.1 Hz), 118.5, 52.1, 49.7, 46.2, 31.4 (d, JPC = 3.9 Hz), 26.1 (d, JPC = 5.3 Hz), 24.4, 22.0. 31P NMR (121 MHz, CDCl3) δ 16.18. HRMS (ESI+) for C29H57N6P [MH]+ m/z calcd 521.4461, found 521.4469.
Liberation of higher-order superbases
In a glove box, KOt-Bu (1 M in THF, 1.0 equiv) was added to the superbase conjugate acid salt in THF (~0.2 M) at room temperature. After 5–10 minutes, the solution was filtered (0.2 μm PTFE), washed with THF, concentrated, redissolved in PhMe, filtered again, and concentrated.
pKBH+ measurements
Stock solutions of the HX salt of the ‘substrate’ superbase being studied (0.0667 M, 0.60 mL, 0.040 mmol) and a reference free base (0.200 M, 0.20 mL, 0.040 mmol) were mixed in NMR tubes under inert atmosphere. CD3CN was employed for all experiments, which were performed using a dual manifold, except for the GC2 (6) and GP2 (10) bases, which employed d8-THF and were performed in a glove box. The mixture was analyzed by 1H NMR spectroscopy (as well as by 13C NMR for measurements in d8-THF).
The extents of protonation of both the substrate superbase and the reference base were determined by comparison to the spectra of the HX salt and the free base of each component. These data were used to calculate the relative basicities of the substrate superbase and the reference base, and this value was compared to the known pKBH+ value of the reference base to obtain the pKBH+ value of the substrate. Measurements were performed in triplicate. Corrections were made for the observed relative NMR integrations and to convert THF data to the acetonitrile scale.
The reference bases used were DBU (pKBH+ = 24.34, MeCN)24 for G1 (16); P1-tBu(pyrr)3 (pKBH+ = 28.35, MeCN)19 for C1 (17), P1 (18), G3 (11), CG2 (4), C3 (2), and PC1 (14); and P2-Et (pKBH+ = 32.94, MeCN)6c for GC2 (6), and GP2 (10). The remaining superbases whose pKBH+ values discussed herein are literature values, except the PC3 base 12 which was estimated from Fig. 3. See the SI for complete details.
Preparation of single crystals for X-ray diffraction. In a 2 dram vial, higher-order superbase salts were dissolved in hot EtOAc, cooled to rt, then allowed to stand. In some cases, the vial was also placed in a sealed jar containing a layer of hexanes to slowly diffuse these vapors into the EtOAc solution.
C3 base 2•HBF4: salt (150 mg) in EtOAc (3 mL), standing with hexanes diffusion for 3 days.
CG2 base 4•HBF4: salt (100 mg) in EtOAc (3 mL), seeded with a few grains of the salt, then standing alone for 1 day and then with hexanes diffusion for 5 days.
GC2 base 6•HBF4: salt (150 mg) in EtOAc (3 mL), standing with hexanes diffusion for 2 days.
PC1 base 14•HBF4: salt (150 mg) in EtOAc (5 mL), standing alone for 3 days and then with hexanes diffusion for 1 day.
GP2 base 10•HBF4: salt (100 mg) in EtOAc (5 mL), standing alone for 2 days.
Conjugate additions of α-aryl esters and nitriles
KOt-Bu (1 M in t-BuOH or THF, 25 μL, 0.025 mmol, 0.025 equiv) was added to 8•HBF4 (20.3 mg, 0.0302 mmol, 0.030 equiv) in THF (3 mL), followed by the α-aryl ester or nitrile and Michael acceptor (1 mmol scale, see SI for details). After stirring for 8–12 hours, acetic acid was added (2 drops), and the reaction was concentrated. The residue was purified by chromatography (EtOAc/hexanes) on silica gel (40 mL) to afford the desired adducts. Experiments were also performed using isolated free bases 8 or P4-tBu from a glove box.
Supplementary Material
Acknowledgments
Financial support was provided by NIHGMS (R01 GM102611). EDN is grateful for the Arun Guthikonda Memorial Graduate Fellowship. We thank Serge Ruccolo, Michelle Neary, and the Parkin group for X-ray structure determination, and the National Science Foundation (CHE-0619638) is thanked for acquisition of an X-ray diffractometer. We also thank the Owen group for the use of their glove box.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information. Experimental details, characterization data, pKBH+ measurements, and crystallographic data (CIF). This material is available free of charge via the Inter-net at http://pubs.acs.org.”
References
- 1.Caubere P. Chem Rev. 1993;93:2317. [Google Scholar]
- 2.Ishikawa T, editor. Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalysts. John Wiley & Sons, Ltd; Chichester, UK: 2009. [Google Scholar]
- 3.Recent reviews: Palomo C, Oiarbide M, López R. Chem Soc Rev. 2009;38:632. doi: 10.1039/b708453f.Ishikawa T, Kumamoto T. Synthesis. 2006:737.Leow D, Tan C-H. Chem Asian J. 2009;4:488. doi: 10.1002/asia.200800361.Sohtome Y, Nagasawa K. Synlett. 2010:1.Leow D, Tan CH. Synlett. 2010:1589.Terada M. J Synth Org Chem Japan. 2010;68:1159.Uraguchi D, Ooi T. J Synth Org Chem Japan. 2010;68:1185.
- 4.Seminal publications: Chinchilla R, Nájera C, Sánchez-Agulló P. Tetrahedron: Asymmetry. 1994;5:1393.Iyer MS, Gigstad KM, Namdev ND, Lipton M. J Am Chem Soc. 1996;118:4910. doi: 10.1007/BF00807935.Corey EJ, Grogan MJ. Org Lett. 1999;1:157. doi: 10.1021/ol990623l.Ishikawa T, Araki Y, Kumamoto T, Isobe T, Seki H, Fukuda K. Chem Commun. 2001:245.Kita T, Georgieva A, Hashimoto Y, Nakata T, Nagasawa K. Angew Chem Int Ed. 2002;41:2832. doi: 10.1002/1521-3773(20020802)41:15<2832::AID-ANIE2832>3.0.CO;2-Q.Allingham MT, Howard-Jones A, Murphy PJ, Thomas DA, Caulkett PWR. Tetrahedron Lett. 2003;44:8677.Terada M, Ube H, Yaguchi Y. J Am Chem Soc. 2006;128:1454. doi: 10.1021/ja057848d.Shen J, Nguyen TT, Goh YP, Ye W, Fu X, Xu J, Tan CH. J Am Chem Soc. 2006;128:13692. doi: 10.1021/ja064636n.Terada M, Nakano M, Ube H. J Am Chem Soc. 2006;128:16044. doi: 10.1021/ja066808m.Yu Z, Liu X, Zhou L, Lin L, Feng X. Angew Chem Int Ed Engl. 2009;48:5195. doi: 10.1002/anie.200901337.Dong S, Liu X, Chen X, Mei F, Zhang Y, Gao B, Lin L, Feng X. J Am Chem Soc. 2010;132:10650. doi: 10.1021/ja1046928.Misaki T, Takimoto G, Sugimura T. J Am Chem Soc. 2010;132:6286. doi: 10.1021/ja101216x.Uraguchi D, Yoshioka K, Ueki Y, Ooi T. J Am Chem Soc. 2012;134:19370. doi: 10.1021/ja310209g.Wu L, Li G, Fu Q, Yu L, Tang Z. Org Biomol Chem. 2013;11:443. doi: 10.1039/c2ob26950c.Dai Q, Huang H, Zhao JCG. J Org Chem. 2013;78:4153. doi: 10.1021/jo4001806.Zou L, Wang B, Mu H, Zhang H, Song Y, Qu J. Org Lett. 2013;15:3106. doi: 10.1021/ol401306h.Núñez MG, Farley AJM, Dixon DJ. J Am Chem Soc. 2013;135:16348. doi: 10.1021/ja409121s.
- 5.Štrukil V, Lekšić E, Meštrović E, Eckert-Maksić M. Aust J Chem. 2014;67:1129. [Google Scholar]
- 6.(a) Schwesinger R, Schlemper H. Angew Chem Int Ed. 1987;26:1167. [Google Scholar]; (b) Schwesinger R, Hasenfratz C, Schlemper H, Walz L, Peters EM, Peters K, von Schnering HG. Angew Chem Int Ed. 1993;32:1361. [Google Scholar]; (c) Schwesinger R, Schlemper H, Hasenfratz C, Willaredt J, Dambacher T, Breuer T, Ottaway C, Fletschinger M, Boele J, Fritz H, Putzas D, Rotter HW, Bordwell FG, Satish AV, Ji G, Peters EM, Peters K, von Schnering HG, Walz L. Liebigs Ann. 1996:1055. [Google Scholar]
- 7.Kolomeitsev AA, Koppel IA, Rodima T, Barten J, Lork E, Röschenthaler GV, Kaljurand I, Kütt A, Koppel I, Mäemets V, Leito I. J Am Chem Soc. 2005;127:17656. doi: 10.1021/ja053543n. [DOI] [PubMed] [Google Scholar]
- 8.Vazdar K, Kunetskiy R, Saame J, Kaupmees K, Leito I, Jahn U. Angew Chem Int Ed. 2014;53:1435. doi: 10.1002/anie.201307212. [DOI] [PubMed] [Google Scholar]
- 9.(a) Lensink C, Xi SK, Daniels LM, Verkade JG. J Am Chem Soc. 1989;111:3478. [Google Scholar]; (b) Tang J, Dopke J, Verkade JG. J Am Chem Soc. 1993;115:5015. [Google Scholar]
- 10.Raab V, Kipke J, Gschwind RM, Sundermeyer J. Chem – Eur J. 2002;8:1682–1693. doi: 10.1002/1521-3765(20020402)8:7<1682::aid-chem1682>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 11.(a) Raab V, Gauchenova E, Merkoulov A, Harms K, Sundermeyer J, Kovacević B, Maksić ZB. J Am Chem Soc. 2005;127:15738. doi: 10.1021/ja052647v. [DOI] [PubMed] [Google Scholar]; (b) Kögel JF, Oelkers B, Kovačević B, Sundermeyer J. J Am Chem Soc. 2013;135:17768. doi: 10.1021/ja409760z. [DOI] [PubMed] [Google Scholar]
- 12.Belding L, Dudding T. Chem – Eur J. 2014;20:1032. doi: 10.1002/chem.201302959. [DOI] [PubMed] [Google Scholar]
- 13.Takeda T, Terada M. J Am Chem Soc. 2013;135:15306. doi: 10.1021/ja408296h. [DOI] [PubMed] [Google Scholar]
- 14.Bandar JS, Lambert TH. J Am Chem Soc. 2012;134:5552. doi: 10.1021/ja3015764. [DOI] [PubMed] [Google Scholar]
- 15.Tobey SW, West R. J Am Chem Soc. 1966;88:2481. [Google Scholar]
- 16.(a) Bredereck H, Bredereck K. Chem Ber. 1961;94:2278. [Google Scholar]; (b) Eilingsfeld H, Neubauer G, Seefelder M, Weidincer H. Chem Ber. 1964;97:1232. [Google Scholar]; (c) Barton DHR, Elliott JD, Géro SD. J Chem Soc Chem Commun. 1981:1136. [Google Scholar]; (d) Barton DHR, Elliott JD, Géro SD. J Chem Soc Perkin Trans. 1982;1:2085. [Google Scholar]; (d) Isobe T, Fukuda K, Ishikawa T. J Org Chem. 2000;65:7770. doi: 10.1021/jo000744v. [DOI] [PubMed] [Google Scholar]
- 17.Short JH, Biermacher U, Dunnigan DA, Leth TD. J Med Chem. 1963;6:275. doi: 10.1021/jm00339a013. [DOI] [PubMed] [Google Scholar]
- 18.(a) Kühle E, Anders B, Zumach G. Angew Chem Int Ed. 1967;6:649. [Google Scholar]; (b) Kühle E, Anders B, Klauke E, Tarnow H, Zumach G. Angew Chem Int Ed. 1969;8:20. [Google Scholar]; (c) Gober CM, Le HV, Ganem B. Tetrahedron Lett. 2012;53:4536. doi: 10.1016/j.tetlet.2012.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwesinger R, Willaredt J, Schlemper H, Keller M, Schmitt D, Fritz H. Chem Ber. 1994;127:2435. [Google Scholar]
- 20.(a) Breslow R, Yuan C. J Am Chem Soc. 1958;80:5991. [Google Scholar]; (b) Breslow R, Altman LJ, Krebs A, Mohacsi E, Murata I, Peterson RA, Posner J. J Am Chem Soc. 1965;87(48):1326. [Google Scholar]; Wu MT, Taub D, Patchett AA. Tetrahedron Lett. 1976;17:2405. [Google Scholar]
- 21.(a) Breslow R, Won Chang H. J Am Chem Soc. 1961;83:2367. [Google Scholar]; (b) Chen W, Li H, Widawsky JR, Appayee C, Venkataraman L, Breslow R. J Am Chem Soc. 2014;136:918. doi: 10.1021/ja411143s. [DOI] [PubMed] [Google Scholar]
- 22.For a report of direct, neutral base catalysis of aldol reactions using nitriles: Kisanga P, McLeod D, D’Sa B, Verkade J. J Org Chem. 1999;64:3090. doi: 10.1021/jo981858y.Tert-butoxide has also been reported as an anionic catalyst: Bun-laksananusorn T, Rodriguez AL, Knochel P. Chem Commun. 2001:745.Suto Y, Kumagai N, Matsunaga S, Kanai M, Shibasaki M. Org Lett. 2003;5:3147. doi: 10.1021/ol035206u.
- 23.Kobayashi has recently reported an enantioselective method for direct base catalysis of Michael reactions with amides and esters using an anionic base (KHMDS). Suzuki H, Sato I, Yamashita Y, Kobayashi S. J Am Chem Soc. 2015;137:4336. doi: 10.1021/jacs.5b01943.See also: Saito S, Kobayashi SJ. Am Chem Soc. 2006;128:8704. doi: 10.1021/ja061221t.
- 24.Kaljurand I, Kütt A, Sooväli L, Rodima T, Mäemets V, Leito I, Koppel IA. J Org Chem. 2005;70:1019. doi: 10.1021/jo048252w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.