

Abstract
MRI and NIR-active, multi-modal Composite NanoCarriers (CNCs) are prepared using a simple, one-step process, Flash NanoPrecipitation (FNP). The FNP process allows for the independent control of the hydrodynamic diameter, co-core excipient and NIR dye loading, and iron oxide-based nanocrystal (IONC) content of the CNCs. In the controlled precipitation process, 10 nm IONCs are encapsulated into poly(ethylene glycol) stabilized CNCs to make biocompatible T2 contrast agents. By adjusting the formulation, CNC size is tuned between 80 and 360 nm. Holding the CNC size constant at an intensity weighted average diameter of 99 ± 3 nm (PDI width 28 nm), the particle relaxivity varies linearly with encapsulated IONC content ranging from 66 to 533 mM-1s-1 for CNCs formulated with 4 to 16 wt% IONC. To demonstrate the use of CNCs as in vivo MRI contrast agents, CNCs are surface functionalized with liver targeting hydroxyl groups. The CNCs enable the detection of 0.8 mm3 non-small cell lung cancer metastases in mice livers via MRI. Incorporating the hydrophobic, NIR dye PZn3 into CNCs enables complementary visualization with long-wavelength fluorescence at 800 nm. In vivo imaging demonstrates the ability of CNCs to act both as MRI and fluorescent imaging agents.
Keywords: composite nanocarrier, multi-modal imaging, flash nanoprecipitation, MRI contrast agents, metastasis detection, theranostic
1. Introduction
While there has been significant activity in multimodal and theranostic nanoparticle science, the progress to clinical applications has been disappointingly slow. A significant obstacle has been the complexity of reported synthesis approaches. Multistep and complex nanoparticle synthesis presents a major problem in scale-up and validation of these materials for FDA approval. Schemes involving covalent coupling or repeated sequential reactions make the molecular characterization of the final materials problematic, especially when scale-up of these processes is considered. Complex, multistep assembly and separations based on ultracentrifugation or dialysis do not scale well to produce the larger samples needed for clinical studies. Covalent reactions of drugs or imaging materials makes analysis of the final purified nanomaterial challenging, since the complexity of the final composition makes tools such as NMR, HPLC and mass spectrometry difficult to apply. What is required is a simplified and scalable route for assembly of these complex nanomaterials. Advances in processing would hasten the translation of these multimodal imaging materials from the laboratory to the clinic.
We report a single-step assembly process that enables the formation of complex composite nanocarriers (CNCs), which incorporate multiple imaging agents, with control over CNC size and surface functionalization. Previously we had reported on the single step assembly of single short-wavelength fluorescent reporters in nanoparticles using a kinetically controlled, block copolymer directed rapid precipitation process called Flash NanoPrecipitation.[1] Here we extend the process to simultaneously incorporate inorganic metal oxide colloids for MRI imaging and long wavelength NIR fluorphores for whole animal fluorescence imaging. These CNCs are assembled in a single precipitation step and can be rapidly purified and concentrated by magnetic separation in a single step. An overview of previous work on processing of multimodal nanoparticles, with the number of processing steps involved, is presented in the Supplemental Information Table SI.1. Of the eight publication highlighted, all but one publication required more than three synthetic steps to form a CNC and only one demonstrated the ability to control CNC size. For example, Cho et al. present a CNC with both MRI and NIR fluorescence capabilities.[2] However, the particle synthesis involves multiple steps including a covalent coupling step.
1.1. Selection of imaging agents
1.1.1. Magnetic resonance contrast agent - iron oxide based nanocrystals (IONCs)
We are specifically interested in imaging modalities that can be applied to animal models, and eventually clinical settings. Our primary focus is on magnetic resonance imaging (MRI) [3, 4, 5] applied to the identification of metastatic tumors from non-small cell lung cancer (NSCLC), which often metastasizes to the liver.[6] Early detection of metastases in key organs, such as the liver, is vital for improved survival.[7] However, the detection of small lesions via MRI is difficult and can benefit from contrast agents.[5, 7] The CNCs we present are surface functionalized to target the liver and enhance T2 contrast between healthy and diseased tissue, which enables the detection of small metastases. As an in vivo model for the detection of small liver metastases, we used a metastatic NSCLC orthotopic mouse model. Kupffer cells are liver-specific macrophages that can be targeted by CNCs.[8] Hydroxyl groups on nanoparticles are known to initiate the complement cascade;[9] and previous work has shown that nanoparticles with terminal hydroxyl groups on the PEG stabilizing corona are avidly taken-up by macrophages and dendridic cells in contrast to methoxy terminated PEG which are not.[9-11] Therefore, hydroxyl-terminated PEG was used to stabilize the CNCs and act as the liver-targeting agent in this applied CNC example.
CNC MRI contrast is provided by 10 nm oleic acid coated super paramagnetic iron oxide nanocrystals (IONCs) to create T2- imaging agents. Unlike other processes in the literature for forming IONC-loaded CNCs,[12, 13] no complicated surface functionalization of IONCs is necessary for encapsulation via FNP. All that is required is a hydrophobic IONC surface, which is afforded by a monolayer coating of oleic acid or similar alkyl chain. Oleic acid coated IONCs are readily obtained with narrow size distributions from various methods including thermal decomposition syntheses as done in this study.[5, 13, 14] The IONC synthesis and characterization are presented in the SI. In this study we incorporated a cobalt-substituted iron oxide into the CNC core. However, we note that because the assembly of the IONCs into the CNCs through FNP is controlled primarily by their surface coating, using other iron oxide based nanocrystals should yield similar results.
1.1.2. Long wavelength fluorophore - PZn3
Whole animal optical imaging requires excitation and emission wavelengths in the “optical imaging window” (∼ 700-1100 nm) to avoid attenuation by tissue and blood at shorter wavelengths and water absorption at longer wavelengths.[15] Our approach is to encapsulate the imaging agent in the core of the NC where it does not interfere with targeting or clearance, in contrast to strategies that place the imaging agent on the corona of the NC through covalent conjugation. The NIR dyes are large, polyaromatic compounds, which, at high surface density, may initiate reticuloendothelial system interactions.[16] To ensure retention in the core, the imaging agent must be significantly hydrophobic. The dye, tris-(porphyrinato)zinc(II) dye (PZn3),[17] satisfies these criteria, having an excitation wavelength of 700 nm, an emission peak at 800 nm, and being highly hydrophobic. Previously, PZn3 has been used for NIR imaging after encapsulation in polymersomes.[18] The synthesis of the dye and the spectral characterization have been described elsewhere.[17]
1.2. Nanocarrier assembly process – Flash NanoPrecipitation (FNP)
The one-step, kinetically controlled precipitation process, Flash NanoPrecipitation (FNP), produces stable nanocarriers using amphiphilic block copolymers to direct self-assembly.[1, 19, 20] Uniform particles with tunable sizes from 40-400 nm are prepared in a scalable manner.[1, 19, 21] The key to the process is the control of micromixing, aggregation, nucleation, and growth timescales.[19] The FNP process is depicted schematically in Figure 1. In FNP, a water miscible organic stream with molecularly dissolved active pharmaceutical ingredients (API) and/or fluorophores, stabilizing block copolymers and colloidal nanostructures (ex. IONCs) are rapidly mixed against an aqueous, “anti-solvent” stream in a confined impinging jet mixing geometry. The rapid mixing, on the order of milliseconds,[22] creates homogeneous supersaturation of the dissolved species prior to nucleation.[1, 23] [24] If the timescales of the block copolymer micellization (τm), nanostructure aggregation (τagg) and API nucleation and growth (τng) are similar, the individual components self-assemble into a CNC.[19, 24] As the hydrophobic block of the copolymer adsorbs onto the particle, further growth is stopped because the hydrophilic block sterically stabilizes the CNCs, thus preventing aggregation.[1, 19] The CNC size can be tuned by adjusting the ratio of core material to stabilizing block copolymer, or by adjusting the concentration of the organic stream.[25, 26] This modular assembly method allows for the formation of a complex CNCs in a single step.
Figure 1.
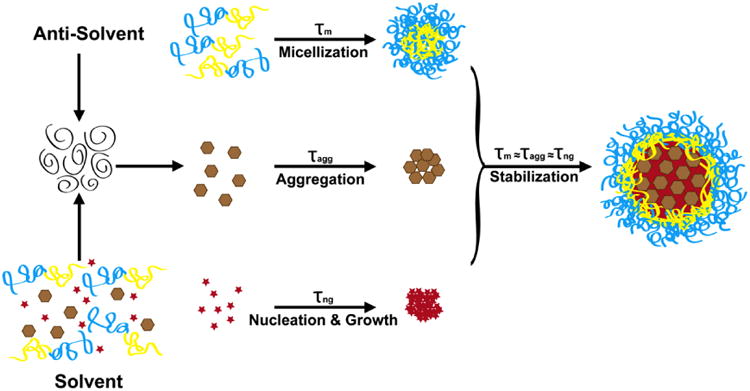
Flash NanoPrecipitation (FNP) Schematic. A water miscible organic solvent with molecularly dissolved hydrophobic active pharmaceutical ingredient (API) or fluorophore (red star), stabilizing, amphiphilic block copolymer (yellow and blue line) and colloidal nanostructures (brown hexagon) is rapidly mixed against an aqueous, anti-solvent stream. The rapid mixing, on the order of milliseconds, ensures homogenous supersaturation prior to nucleation, aggregation and micellization events. The rapid mixing allows for homogeneous supersaturation of the dissolved species prior to nucleation events and for homogeneous distribution of nanostructures prior to aggregation events. If the timescales of the block copolymer micellization (τm), nanostructure aggregation (τagg) and API nucleation and growth (τng) are similar, the individual components will self-assemble into a composite nanocarrier (CNC). As the hydrophobic block of the block copolymer (yellow block) adsorbs onto the growing particle, further growth is stopped because the hydrophilic block (blue block) sterically stabilizes the CNCs preventing further aggregation and growth.
We will demonstrate the ability of FNP to easily and controllably create CNCs with desired multimodal imaging properties as well as their imaging capabilities in vivo.
2. Results and Discussion
2.1. Formation and Characterization of Nanocrystal Loaded Composite Nanocarriers
For in vivo applications, studies have shown that CNCs smaller than 200 nm and possessing slightly negative surface charges (-10 mV to 0 mV) experience enhanced bioavailability and prolonged circulation time in vivo.[27] To act as strong T2 MRI active contrast agents, the CNCs must have high R2 relaxivities, which is a function of IONC content and size. Clustering of small IONCs results in an enhanced T2 response.[4] As demonstrated by Pöselt et al., IONC clusters between 80 and 120 nm are in the static dephasing regime (SDR),[4] where relaxivities approach the theoretical maximum values. Clusters larger or smaller display lower relaxivities based on competing physical processes. Thus control of CNC size is critical from both a safety and an imaging activity standpoint.
Using FNP, we can independently control the CNC size and IONC loading, which enables us to uniquely isolate the effect of IONC loading. To control CNC size, the core-to-stabilizing-block-copolymer ratio or the overall concentration in the organic stream can be adjusted.[25, 26] Here, we focus on the core-to-stabilizer ratio to control CNC size. When holding the stabilizing block copolymer concentration constant, and varying the 10 nm IONCs content, the CNC size can be tuned from 80 to 360 nm (Figure 2a) creating CNCs within the desired size range.
Figure 2.
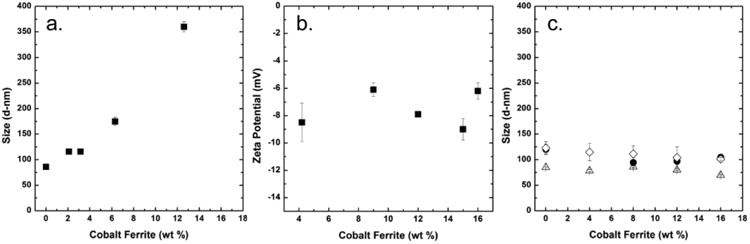
The intensity weighted diameter and zeta potential (ZP) of composite nanocarriers (CNCs) as a function of cobalt ferrite nanoparticle (IONC) loading are shown. (a.) Holding the stabilizing block copolymer concentration constant and increasing the IONC content causes an increase in particle size (■). (b.) The zeta potentials of CNCs stabilized with PLA-PEG and 11k PLA as the co-core excipient is plotted as a function of IONC content (■). The zeta potentials are not affected by the IONC content. Compositions of CNCs: 50 wt% PEG-b-PLA, X wt% IONC, and 50-X wt% co-core excipient. (c.) When the ratio of core material (IONC and co-core excipient) to stabilizing block copolymer is held constant at a 1 to 1 mass ratio, the CNC size does not vary as a function of IONC content regardless of co-core excipient. Three co-core excipients were tested, hostasol yellow dye (△), 11k PLA (◊) and 106k PLA (●).
The zeta potentials of the IONC loaded CNCs formulated with PLA as the core excipient and PLA-PEG as the stabilizing block copolymer were independent of IONC loading with an average zeta potential of -7.5 ± 1.3 mV (Figure 2c). The negative surface charge arises from the carboxylic acid end groups on PLA degradation products.[28] The screening of the surface charge by the PEG layer keeps the zeta potential within the acceptable range for long circulating particles.[28] Similar zeta potentials have been observed previously for PLA-b-PEG containing CNCs.[26]
For multimodal and theranostic CNCs it is necessary to incorporate organic fluorophores or APIs along with the IONCs in the CNC core. To more fully understand the assembly process, three hydrophobic core co-excipients were tested: hostasol yellow an organic fluorophore (HOS, Mw= 556), and poly(lactic acid) (PLA) of two molecular weights: Mw =11k and 106k). A biocompatible PLA-b-PEG (3.7k-b-5.0k) block copolymer was used as the stabilizer. The total mass ratio of the core to the stabilizing-block-copolymer was held constant at 1:1 while the IONC core content was varied from 0 to 16 wt%. Over the IONC range tested, the CNC size was essentially constant for each core component, with a mean variation in size of ∼ ±10 % (Figure 2b). All three core excipients are very hydrophobic, with LogPs above 9,[29, 30] such that high supersaturations were achieved, which enabled the control of CNC size.
However, the nanocarrier size and encapsulation efficiency did vary with the molecular weight (or size) of the co-excipient, indicating the differing roles of the co-excipients. For HOS-loaded CNCs, the average particle size was 78 ± 6 nm (PDI width (PW) 25 nm) across all formulations. For the PLA-loaded CNCs, the average particle size was 111 ± 9 nm (PW 24 nm) for the 11k PLA, and 104 ± 12 nm (PW 32 nm) for the 106k PLA To understand how the sizes of the co-excipient affected self-assembly, experiments were designed with the three co-excipients listed above.
When a polymer comes in contact with a non-solvent, as is the case with PLA and water, the polymer chain collapses into a globule over times scales much faster than particle assembly. The size can be estimated by Equation 1,
| (1) |
Where Rgl is the radius of the globule, b is the Kuhn length, N is the number of Kuhn monomers, and χ is the polymer-solvent interaction parameter.[31] The 11k and 106k PLA have globule diameters of 4 and 9 nm, respectively. (Details of the calculation can be found in the SI) Based on the molar volume, the estimated diameter of the 556 g/mol HOS dye molecule is 1 nm (Details in the SI). The collapsed chains of the polymers are on a similar size scale as the 10 nm IONCs, whereas the small molecule dye is significantly smaller.
Diffusion limited aggregation, which characterizes the rapid precipitations by FNP, depends upon three variables: the sizes, the relative number densities, and the sticking probabilities of the species.[24] The sizes, in turn, determine the diffusion coefficients of the species and their collision cross sections. For the supersaturations higher than 100 in these experiments, with these highly hydrophobic solutes, it is expected that the sticking probability should be close to unity.
Three sets of CNCs were prepared from starting compositions of 8 wt% IONC, 42 wt% co-core excipient and 50 wt% PLA-b-PEG 3.7k-b-5.0k. The CNCs were easily concentrated and purified in a single step using a magnetic column, as described in the experimental section. This fractionated the sample into a magnetically “active” fraction and a less magnetically active sample. To recover the magnetically active CNCs, the columns were demagnetized and eluted with the desired aqueous solution. The particle sizes of the two fractions were determined. For the magnetic fraction, the average particle size was ∼12 nm larger across all samples than the nominal size of the average dispersion prior to fractionation. The non-magnetic population was smaller than the original CNC size with an average size of 56 ± 7 nm (PW 25 nm) across all samples. For reference, the micelle size for PLA-b-PEG 3.7k-b-5.0k was previously determined to be 43 nm.[26]
The compositions of magnetically purified CNCs were determined via TGA and are shown in Table 1. The magnetically active CNCs are enriched in IONCs and the hydrophobic excipient above the initial starting concentrations. The concentrations of IONCs in the final CNCs were 15 wt% to 25 wt%; whereas the starting concentrations were 8 wt%. When the molecular size of the IONC (10 nm) and the collapsed 106k PLA excipient (9 nm) are similar, the mass ratio of IONC and excipient in the final CNC more closely approximates their ratios in the starting formulation: 25 wt% in the final CNC versus 19 wt% in the starting solution. When the excipient size is small (HOS ∼1 nm) the ratio of IONC to excipient is the highest: 25:46. The yield %, i.e. the fraction of the starting mass that is recovered as magnetically active CNCs, varies from 28 wt% to 42 wt%. The inverse relationship between yield (i.e. higher with the larger excipient) and IONC incorporation (i.e. lower with the larger excipient), means that the total amount of magnetic IONCs captured is about constant; it represents 75% to 100% of the IONCs charged to the system. So the non-magnetic particles, which are lost, are composed primarily of the core components and stabilizing block copolymer.
Table 1. Compositions of Magnetically Purified Composite NanoCarriers (CNCs) a.
| Co-Core Excipient | Globule Size (d-nm) | Purified CNC Composition | Relative Number Density WRT IONC | Yield (wt %) | ||
|---|---|---|---|---|---|---|
| IONC (wt %) | Co-Core Excipient (wt %) | Stabilizer (wt %) | ||||
| Original Formulation | - | 8 | 42 | 50 | - | - |
| Hostasol Yellow | 1 | 25 ± 4.8 | 45.6 ± 1.6 | 29.5 ± 3.2 | 15,000 | 28.4 ± 1.3 |
| 11k PLA | 4 | 22.0 ± 0.9 | 49.2 ± 1.5 | 31.0 ± 0.7 | 800 | 31.7 ± 0.5 |
| 106k PLA | 9 | 15.0 ± 1.1 | 61.3 ± 1.3 | 25.5 ± 2.5 | 80 | 42.1 ± 0.9 |
CNCs were prepared from initial formulations comprising 50 wt% PEG-b-PLA, 8 wt% IONC, and 42 wt% core co-excipient.
We have quantitatively modeled the aggregation of 5 nm gold particles into CNC clusters using the Smoluchowski diffusion limited aggregation model.[24] However, for this system with very different sized aggregating species and very different number densities of the components, we use the Smoluchowski analysis to guide our understanding of the differences in the fraction of magnetically active CNCs as a function of excipient size, and number density. The ratios of number densities for the excipient:IONC are 15,000:1, 800:1, and 80:1 for the HOS, 11K PLA, and 106K PLA, respectively. The Smoluchowski analysis gives the time constant (τ) for the aggregation process of a single species to be,
| (2) |
where η is the fluid viscosity, k is Boltzmann's constant, T is the temperature, and [P]0 is the initial number density of particles. The great difference in number density between the HOS and the IONC would indicate a much faster initial kinetics of aggregation for the HOS, and this initial aggregation would be expected to “scavenge” the block copolymer stabilizer from the system to create a larger polymeric micelle phase that includes few IONCs. This is consistent with the observation above that the polymer micelle nanoparticles are smaller than the magnetically active CNCs.
An example of a theranostic CNC, encapsulating both an API, the antifungal drug itraconazole, and MRI-active IONCs is described in the Supplementary Information. In this example we demonstrate 47 wt% loading of itraconazole with 3 wt% IONC. This is the highest loading of an API in a theranostic CNC that has been reported.
2.2. Composite Nanocarriers for Magnetic Resonance Imaging
The relationship between IONC content and relaxivity was investigated by varying the density of IONC clusters in CNCs of constant size, because relaxivity also varies with size.[4] The co-core excipient and stabilizer were 11k PLA and PLA-PEG. The intensity weighted size distributions of all five, magnetically purified CNCs are plotted in Figure 3a. The average particle mean size across the samples was 99 nm ± 3 nm (PW 28 nm) and all CNCs had narrow particle size distributions, similar to latex standards as discussed in the Supplementary Information. Representative TEM images of the particles are shown in Figs.3 c. to g. The relaxivity varied linearly with IONC content ranging from 66 to 533 mM-1s-1 for the samples with between 4 and 16 wt% IONC loading, (Figure 3b). The 533 mM-1s-1 relaxivity is among the highest values reported for composite nanoparticles. Using the static dephasing model for multicore particles (Equation 3), the theoretical maximum relaxivity (r2m) was calculated, in which A, N0, Z, γ and Ms are the lattice parameter, Avogadro's number, Z factor, proton gyromagnetic ratio and saturation magnetization.[12] For cobalt ferrite IONCs (CoFe2O4), the theoretical maximum relaxivity is 856 mM-1s-1. The 16 wt% IONC formulated CNC achieved 62% of the theoretical maximum. Calculation details can be found in the SI.
Figure 3.
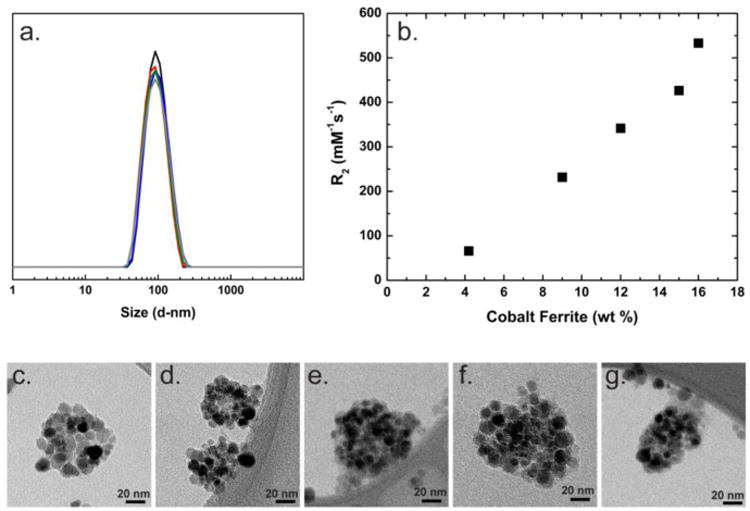
(a.) The DLS traces of the composite nanocarriers (CNCs) used to determine CNC relaxivity as a function of formulation IONC content are plotted: 4 wt% (black), 9 wt% (red), 12 wt% (blue), 15 wt% (green) and 16 wt% (grey). The particles have an intensity weighted average diameter of 100 nm. (b.) The relaxivities of 100 nm CNCs with varying IONC content are plotted. The relaxivity increases linearly with IONC content. (c.-g.) Representative TEM images of the CNCs with varying IONC content are shown: 4 wt% (c.), 9 wt% (d.), 12 wt% (e.), 15 wt% (f.) and 16 wt% (g.). The difference in IONC contrast observed is due to IONC orientation relative to the beam.
| (3) |
To demonstrate the application of CNCs as in vivo MRI contrast agents for liver metastasis detection, metastatic NSCLC orthotopic mouse models were injected with hydroxy surface functionalized CNCs and imaged via MRI. As mentioned, nanoparticles with terminal hydroxyl groups on the PEG stabilizing corona are avidly taken-up by macrophages,[9-11] including the liver-specific Kupffer cells.[8] The CNC formulation composition was 16 wt% IONCs, 34 wt% 1.6k PS and 50 wt% 1.6k-5k PS-b-PEG. The resulting CNCs had an intensity weighted average size of 93 ± 2 nm (PW 28 nm) after magnetic purification. As shown in Figure 4a, large metastases in the mouse liver were observed pre-dose, where the cancer metastases are brighter regions in the liver (i.e. regions with less MRI T2 contrast). However, 1.5 hours after a 100 mL tail vein administration of a 4.6 mg/mL CNC solution the healthy liver tissue is now significantly darker and the greater contrast now makes the metastases more visible. Several small metastases were now observable throughout the liver (Figure 4b). The smallest metastasis detectable was 0.8 mm3. Importantly, the significant contrast remained in the liver after 24 hours, indicating the potential for long-term imaging and monitoring (Figure 4c).
Figure 4.
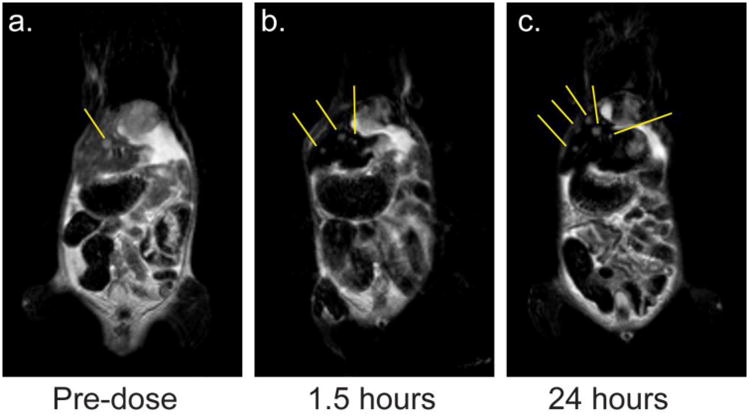
MRI scans of a mouse laden with liver metastases. (a.) In the pre-dose scan, a large metastasis in the liver is observable. (b.) 1.5 hours after CNC administration, the healthy liver tissue appears black and several small metastases are now observable. Yellow guidelines are drawn to help the reader identify the metastases. (c.) 24 hours after CNC administration, the CNC contrast persists and the healthy liver tissue continues to appear black. More metastases are now observable. Yellow guidelines are drawn to help the reader identify the metastases.
2.3. Composite Nanocarriers for Multi-Modal Imaging
To form the multi-modal CNCs, IONCs and the NIR dye, PZn3, were used as the T2-active MRI contrast agent and the NIR fluorescent agent, respectively. This imaging particle combines the sensitivity of fluorescence imaging and the anatomical detail of MRI. The imaging agents were encapsulated in a PS matrix and stabilized with hydroxy-terminated PS-PEG for liver targeting. The CNC formulation composition was 2 wt% PZn3, 16 wt% IONC, 33 wt% PS, and 49 wt% PS-PEG. The resulting CNCs had an intensity weighted average size of 121 ± 2 nm (33 nm) after magnetic purification (Figure 5a). A representative TEM image of the multi-modal CNC is shown in Figure 5b. The particles were stable in 10 mM PBS, which is in accordance to other CNCs formed via FNP.[10, 29]
Figure 5.
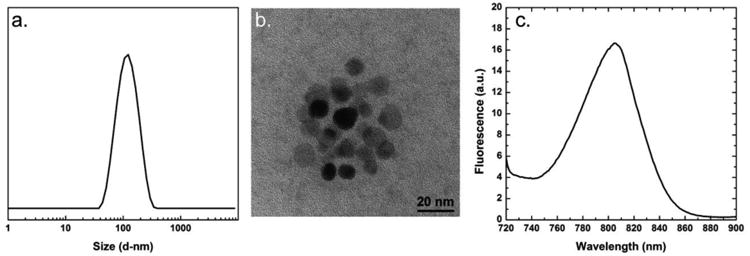
(a.) The intensity weighted size distribution of the 121 ± 2 nm (33 nm) multi-modal composite nanocarriers (CNC) is plotted. (b.) A representative TEM image of the multi-modal CNC is shown. (c.) The emission spectrum of the multi-modal CNC when excited at 700 nm is plotted. The apex of the emission peak is at 806 nm.
The PZn3 loading is at approximately the optimum concentration for fluorescence intensity; at higher loadings, self-quenching reduces fluorescence.[32] The emission spectrum of the multi-modal CNC, when excited at 700 nm, is plotted in Figure 5c. The apex of the emission peak is at 806 nm. The presence of IONCs did not affect the emission spectra of PZn3 (SI Figure S9). Of note is the dye photo-stabilization afforded by trapping the dye in a solid state within the CNC core, which minimizes photo-induced degradation.[1] Previous work has demonstrated that fluorophores in CNC cores undergo only minor photo-bleaching when exposed to intense UV or blue-green light.[1, 33] This robustness ensures minimal degradation of the CNC fluorescence signal during extended imaging experiments.
When injected intravenously into mice, the particles successfully targeted the liver and were detectable by both MRI and fluorescence imaging. Representative MRI mouse images are shown in Figure 6. Prior to tail vein administration (100 mL of a 2.6 mg/mL CNC solution), the liver exhibited normal MRI contrast (Figure 6a). 24 hours post-administration, the healthy liver tissue was “dark” due to the strong T2-active CNC (Figure 6b). The fluorescent images pre-dose exhibited a low level of auto-fluorescence (Figure 6c). 24 hours post-administration, a strong fluorescence signal was observed in the abdomen of the mouse indicating the co-localization of the MRI and fluorescence signals (Figure 6d). After whole animal imaging, the mouse was sacrificed and the individual organs were imaged to confirm the source of the fluorescence. Fluorescence tissue images are in the Supplementary Information. A strong fluorescence signal was observed in the liver confirming the co-localization of imaging signals in the liver. There was also some fluorescence observed in the spleen, which was not unexpected. CNCs are known to accumulate in the spleen, which like the liver is part of the reticuloendothelial system.[34]
Figure 6.
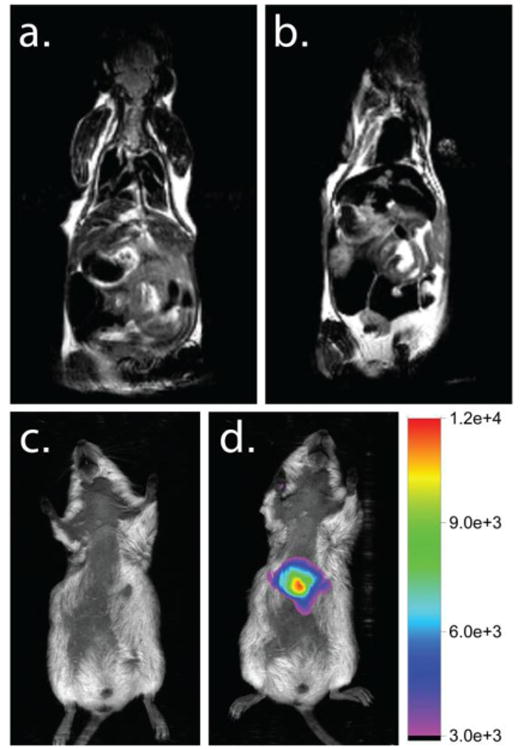
The MRI and fluorescent images of a mouse before and after administration of a multi-modal imaging CNC. (a.) MRI image of a mouse pre-dose with liver tissue exhibiting normal, grey contrast. (b.) 24 hours post-dose, the healthy liver tissue appears black, indicating a change in contrast due to the presence of CNCs. (c.) A fluorescence image of a mouse pre-dose. No auto-fluorescence is observed. (d.) 24 hours post-dose, fluorescence is observed in the abdomen of the mouse. The fluorescence signal was confirmed in the liver and spleen via ex-vivo tissue analysis. This demonstrates the co-localization of MRI and fluorescence signal in the liver and the multi-modal imaging capabilities of the CNC. The fluorescent images of the ex-vivo tissues can be found in the Supplementary Information.
3. Conclusion
FNP is a powerful method for making a variety of composite particles for imaging applications. The one-step process was used to make MRI active CNCs by encapsulating cobalt ferrite-based IONCs in the CNC core. The particle size was readily tuned between 80 and 360 nm by adjusting the core-to-block copolymer ratio. The particle relaxivity varied linearly with IONC content at constant CNC size ranging from 66 to 533 mM-1s-1 over the tested formulations. The ability to tune the relaxivity is important when designing CNCs for specific applications. Here we encapsulated cobalt ferrite nanoparticles as an example of a mixed ferrite that may be of interest for MRI contrast. Because the assembly process is dominated by particle size and surface chemistry FNP should be equally applicable to formation of CNCs encapsulating other mixed ferrites, and mixtures of IONCs, enabling systematic studies of the effects of IONC composition and interactions on T2 contrast properties. By functionalizing the CNC surface with hydroxyl groups, the CNCs were able to target the liver. A strong T2 contrast agent, the CNCs darkened healthy liver tissue, which enabled the detection of small NSCLC metastases in vivo. Because of the modularity of the FNP process, using other targeting ligands is readily feasible. Examples of targeted FNP CNCs include CNCs decorated with mannose to target lung macrophages and LHRH peptide sequences to target cancer cells.[1, 10] By using a hydrophobic NIR dye, PZn3, CNCs with multi-modal imaging capabilities were created. In vivo imaging revealed the ability of these particles to act both as MRI and fluorescent imaging agents. Future work will investigate how the identity of the core material affects the CNC relaxivity and if using smaller IONCs can maximize co-core excipient loading, while retaining T2 contrast.
4. Experimental Section
Materials
Iron (III) acetylacetonate (99.9 % metal basis), and cobalt (II) acetylacetonate (97%), oleic acid (99%), oleylamine (>70 %), hexane (reagent grade), ethanol (99%), poly(lactic acid) (PLA) 106k and sodium chloride (reagent grade) were purchased from Sigma Aldrich. Chloroform (99%), tetrahydrofuran (THF, HPLC grade), methanol (HPLC grade), trifluoroacetic acid (TFA, HPLC grade), phosphate buffered saline powder concentrate (PBS, biotech grade), fetal bovine serum (research grade) were purchased from Fisher Scientific. Poly(styrene) (PS) 1.6k, and poly(styrene)-b-poly(ethylene glycol) (PS-PEG) 1.6k-b-5.0k, were purchased from Polymer Source. Poly(lactic acid)-b-poly(ethylene glycol) (PLA-PEG) 3.7k-b-5.0k and poly(ε-caprolactone)-b-poly(ethylene glycol) (PCL-PEG) 9.0k-b-5.0k were provided by Evonik Industries (Birminghan Alabama). Hostasol Yellow was purchased from Clariant, Inc. Deuterium oxide (99.9%) was purchased from Cambridge Isotope Laboratories, Inc. Poly(lactic acid) 11k was synthesized using previously described protocols.[35] A NANOpure Diamond UV ultrapure water system (Barnstead International, Germany) was used to generate deionized water (DI water) (18.2 MΩ·cm). PZn3 was synthesized using previously described protocols.[17] All materials were used without further purification.
IONC Synthesis and Characterization
The cobalt substituted iron oxide nanocrystals (IONCs) were synthesized by the thermodecomposition method using iron (III) acetylacetonate, cobalt (II) acetylacetonate, octadecene, oleic acid and oleylamine as previously described.[36] The details of the synthesis can be found in the Supplementary Information. A Zeiss 922 200 kV Transmission Electron Microscope was used to determine the shape and size of the nanocrystals. A Varian 800 Scimitar series FTIR was used to verify the presence of oleic acid on the nanoparticle surfaces. Magnetic properties of the particles suspended in hexane were determined using a Quantum Design MPMS XL-7SQUID magnetometer. Elemental analysis was done in an Optima 2000 DV inductively coupled plasma optical emission spectrometer (ICP-OES). The dry sample was digested in a 2% (v/v) HNO3 solution prior to ICP-OES analysis. All results of IONC characterization can be found in the Supplementary Information.
Composite Nanocarrier Formation and Characterization
CNCs were formed via FNP using a confined impinging jet mixer, described by Han et al.[37] The stabilizing polymer (PS-PEG or PLA-PEG) and co-core excipient (PLA, PS, HOS or PZn3) were dissolved in a THF solution containing the IONCs. Representative concentrations for each component are 10.0 mg/mL for the stabilizing polymer, 8.4 mg/mL for the co-core excipient and 1.6 mg/mL for the IONCS. The THF solution was rapidly mixed against an equal volume of DI water in a confined impinging jet mixer. The resulting CNC solution was collected in a DI water reservoir to further dilute the solution to 10 vol% THF. Using a 6-8k MWCO regenerated cellulose Spectra/Por dialysis membrane (Spectrum Laboratories), the CNC solution was dialyzed against DI water for 6 hours to remove the THF. It should be noted that at larger scales, the removal of THF would be done using tangential flow filtration[38] or flash evaporation,[39] both scalable processes, or via lyophilization,[23] a common technique used in the pharmaceutical industry. The use of magnetic columns, as described later in this section, is another simple technique that can be used for solvent switching or rapid particle concentration.
To determine the particle size, samples were analyzed via dynamic light scattering (DLS) using a Zetasizer Nano-ZS (Malvern Instruments). The reported particle size is the intensity average diameter as determined by the Malvern deconvolution software using the Normal Mode analysis. The standard deviation reported is the deviation of the intensity weighted average diameter across samples. The PDI width (PW) is noted in parenthesis. For zeta potential measurements, CNC samples were diluted 1:3 vol:vol with a sodium chloride solution (4 mM). Samples were analyzed on a Zetasizer Nano-ZS (Malvern Instruments) in zeta potential mode. Transmission Electron Microscopy (TEM) was performed using a Philips CM200 FEG-TEM. Fluorescence measurements were performed on a Hitachi F-7000 Fluorescence Spectrophotometer. Samples were diluted 1:19 vol:vol with DI water to minimize particle scattering and excited at 700 nm and analyzed from 710 to 900 nm.
Composite NanoCarrier Composition Characterization
To separate IONC-free micelles from the magnetic CNCs, samples were run on magnetic columns (MACS LS Column, Miltenyi Biotec, Inc.) in a single step. The magnetic CNCs were collected on the column and washed with excess DI water to remove non-magnetic components. The purified CNCs were eluted with DI water and analyzed for size. Both purified and unpurified CNC samples were lyophilized using a VirTis Benchtop lyophilizer (SP Scientific). The composition of the dry CNCs was determined via TGA analysis (Perkin-Elmer Pyris 1 TGA). Samples were heated in ultra-pure nitrogen from 28°C to 500°C at 10 °C/min and held for 20 minutes at 500°C. Representative thermograms can be found in the Supplementary Information.
Composite NanoCarrier Relaxivity Characterization
CNC samples were transferred to deuterium oxide (D2O) and concentrated using magnetic columns (MACS LS Column, Miltenyi Biotec, Inc.). Samples were loaded onto the magnetic columns and washed with excess D2O. Samples were eluted with D2O. CNC concentration was determined via TGA analysis (Perkin-Elmer Pyris 1 TGA) and iron concentration via inductively coupled plasma mass spectrometer (ICP-MS) (Thermo Scientific iCAP Q ICP-MS). For ICP-MS, samples were digested in concentrated nitric acid.
T2 measurements at various CNC concentration were carried out using a Carr-Purcell-Meiboom-Gill (CPMG) pulse sequence[40] with a Bruker Avance 500 MHz nuclear magnetic resonance (NMR) spectrometer. Typical experimental conditions were: on-resonance at D2O, single-scan, delay of 10 s, acquisition time of 0.6816 s (8k points were acquired, spectral width of 6 kHz), 90° pulse width of 8.2 μs (at -1 dB), spin-echo period from 0.4 to 2.4 ms covered with 10 points. NMR data processing was performed with MNova version 8.0.2 software: LB = 30 Hz, zero-filling to 64k points, and non-linear fitting with mono-exponential decay function. The precision of the T2 data was measured (6 repetitions): ± 0.1 ms.
In Vivo Studies
All CNC samples for in vivo and in vitro samples were made via FNP under sterile conditions. For the MRI-only active CNC, the following formulation was used for the THF stream: 10.0 mg/mL 1.6k-5k PS-PEG, 6.8 mg/mL 1.6k PS and 3.2 mg/mL IONC. For the multi-modal CNC, the following formulation was used: 10 mg/mL 1.6k-5k PS-PEG, 6.8 mg/mL 1.6k PS, 3.2 mg/mL IONC, and 0.5 mg/mL PZn3. Under sterile conditions, CNC samples were transferred to 10 mM PBS and concentrated using magnetic columns (MACS LS Column, Miltenyi Biotec, Inc.). CNC concentration was determined via TGA (Perkin-Elmer Pyris 1 TGA).
A549, a human NSCLC cell line, was purchased from ATCC (Manassas, VA). The cell line was maintained in Dulbecco's modified eagle's medium supplemented with 10% fetal bovine serum (FBS), penicillin (200 units/mL), and streptomycin (200 mg/mL) at 37 °C in 5% CO2 atmosphere. Cells with confluency less than 80% were used for animal inoculation. A549 cells were cotransfected with the firefly luciferase (Luc2) and TurboFP635, a red fluorescent protein (RFP), using Lipofectamine™ 2000 (Invitrogen, Grand Island, NY) to deliver the vectors. Briefly, the cells were first transfected with pTurboFP635-N (Evrogen, Moscow, Russia) according to the manufacturer's instructions. Stably transfected clones were obtained by selection with neomycin. The RFP transfected cells were further transfected with pGL4.50[luc2/CMV/Hygro] vector (Promega, Madison, WI). Cells were thereafter selected with neomycin and hydromycin to obtain stably cotransfected clones (A549RL). A subline of A549RL was isolated from a brain metastasis in NSG mouse injected intravenously with A549RL cells.
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (NSG mice, stock number 005557) were purchased as breeders from The Jackson Laboratory (Bar Harbor, Maine). The mice were bred in Laboratory Animal Services at Rutgers, The State University of New Jersey, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC). Mice at age of 10-12 weeks were used in the study. The animals were housed in a pathogen-limited animal facility with free access to food and water. The light cycle was 12 hours in light and 12 hours in dark and the room temperature was maintained between 68 and 74°F. All procedures involving mice in this study were approved by the Institutional Animal Care and Use Committee.
The A549RL cells (2×106 cells/mouse) were injected intravenously into NSG mice. Optical imaging and MRI scans were conducted to monitor the tumor growth in the animals. Mice with tumor growths in liver that were observable via MRI without contrast agent (growths larger than 60 mm3) were subjected to the present study.
For in vivo imaging, mice were dosed intravenously with 100 μl of CNC solution (4.6 mg/mL CNC for the MRI-active CNC or 2.6 mg/mL CNC for the dual MRI and fluorescence-active CNC). Mouse MR images were acquired with a 1T M2‐High Performance MRI System (Aspect Magnet Technologies) using the following scan: Fast Spin Echo (FSE) 2D, 15 slices, slice thickness 1.5 mm, field of view 100 mm, sample encoding 256/250, TE/TR: 80/2840, flip angle 180°, 2 excitations, 3 averages, scan time 7 m 23 s. All imaging procedures were performed under inhalation anesthesia with isoflurane at a concentration of 4-5% for induction of anesthesia and 1–2% for maintenance. The mouse was secured within the animal bed and placed in the coil with a compatible physiological monitoring system. Isoflurane was supplied via a nose cone. Coronal images were evaluated using the VivoQuant Pre-clinical Image Postprocessing Software. Fluorescence images were acquired using a Carestream MS FX PRO (Bruker/CareStream, Rochester, NY). Mice were kept under isoflurane anesthesia. Bright field and fluorescence images were used in the imaging protocol. The following instrument settings were used: excitation/emission at 730/790 nm and 1 minute acquisition time. The images were processed with Carestream Molecular Imaging Software MI (5.3.3).
Supplementary Material
Acknowledgments
This work was funded by the National Institutes of Health (Award No. 1RO1CA155061-1), the National Institutes of Health CounterACT Program through the National Institute of Arthritis and Musculoskeletal and Skin Diseases (AwardU54AR055073) and the National Science Foundation-MRSEC program through the Princeton Center for Complex Materials (DMR-0819860). Also support was provided by the Princeton SEAS Old Guard research fund, the Blair Pyne fund and the Helen Shipley research fund. N. M. Pinkerton would like to acknowledge support from the Department of Defense through the National Defense Science & Engineering Graduate Fellowship (NDSEG) Program (32 CFR 168a) and the National Science Foundation through the NSF Graduate Research Fellowship Program (NSF GRFP). R. F. Pagels would like to acknowledge support from the National Science Foundation through the NSF Graduate Research Fellowship Program (NSF GRFP). R. Wang and M. J. Therien acknowledge financial support from Department of Defense (W81XWH-13-1-0086). We would like to thank Gerald Poirier (Princeton University) for his TEM and XRD expertise. We would like to thank Dr. George W. Scherer (Princeton University) for TGA access. We want to also thank Dr. Howard Bowman (Evonik Industries) for his collaboration on PEG-b-PLA polymers.
Footnotes
Supporting Information: Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Nathalie M. Pinkerton, Department of Chemical and Biological Engineering, Princeton University, Princeton, New Jersey 08544, United States
Dr. Marian E. Gindy, Department of Chemical and Biological Engineering, Princeton University, Princeton, New Jersey 08544, United States
Dr. Victoria L. Calero-DdelC, Department of Chemical Engineering, University of Puerto Rico, Mayagüez, 00681, Puerto Rico
Theodore Wolfson, Department of Chemical and Biological Engineering, Princeton University, Princeton, New Jersey 08544, United States.
Robert F. Pagels, Department of Chemical and Biological Engineering, Princeton University, Princeton, New Jersey 08544, United States
Derek Adler, College of Pharmacy, Rutgers University, Piscataway, New Jersey 08854, United States.
Dr. Dayuan Gao, College of Pharmacy, Rutgers University, Piscataway, New Jersey 08854, United States
Dr. Shike Li, College of Pharmacy, Rutgers University, Piscataway, New Jersey 08854, United States
Ruobing Wang, Department of Chemistry, Duke University, Durham, North Carolina 27708, United States.
Margot Zevon, Department of Biomedical Engineering, Rutgers University, Piscataway, New Jersey 08854, United States.
Dr. Nan Yao, Princeton Materials Institute, Princeton University, Princeton, New Jersey 08540, United States
Dr. Carlos Pacheco, Department of Chemistry, University of Connecticut, Storrs, Connecticut 06269, United States
Prof. Michael J. Therien, Department of Chemistry, Duke University, Durham, North Carolina 27708, United States
Prof. Carlos Rinaldi, J. Crayton Pruitt Family Department of Biomedical Engineering, University of Florida, Gainesville, Florida 32605, United States
Prof. Patrick J. Sinko, College of Pharmacy, Rutgers University, Piscataway, New Jersey 08854, United States
Prof. Robert K. Prud'homme, Email: prudhomm@princeton.edu, J. Crayton Pruitt Family Department of Biomedical Engineering, University of Florida, Gainesville, Florida 32605, United States.
References
- 1.Akbulut M, Ginart P, Gindy ME, Theriault C, Chin KH, Soboyejo W, Prud'homme RK. Adv Funct Mater. 2009;19:718. [Google Scholar]
- 2.Cho HS, Dong Z, Pauletti GM, Zhang J, Xu H, Gu H, Wang L, Ewing RC, Huth C, Wang F. ACS Nano. 2010;4:5398. doi: 10.1021/nn101000e. [DOI] [PubMed] [Google Scholar]
- 3.a) Tromsdorf UI, Bigall NC, Kaul MG, Bruns OT, Nikolic MS, Mollwitz B, Sperling RA, Reimer R, Hohenberg H, Parak WJ, Forster S, Beisiegel U, Adam G, Weller H. Nano Lett. 2007;7:2422. doi: 10.1021/nl071099b. [DOI] [PubMed] [Google Scholar]; b) Cheng W, Ping Y, Zhang Y, Chuang KH, Liu Y. Journal of healthcare engineering. 2013;4:23. doi: 10.1260/2040-2295.4.1.23. [DOI] [PubMed] [Google Scholar]; c) Prashant C, Dipak M, Yang CT, Chuang KH, Jun D, Feng SS. Biomaterials. 2010;31:5588. doi: 10.1016/j.biomaterials.2010.03.070. [DOI] [PubMed] [Google Scholar]
- 4.Pöselt E, Kloust H, Tromsdorf U, Janschel M, Hahn C, Maβülo C, Weller H. ACS Nano. 2012;6:1619. doi: 10.1021/nn204591r. [DOI] [PubMed] [Google Scholar]
- 5.Lu J, Ma S, Sun J, Xia C, Liu C, Wang Z, Zhao X, Gao F, Gong Q, Song B. Biomaterials. 2009;30:2919. doi: 10.1016/j.biomaterials.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 6.Quint LE, Tummala S, Brisson LJ, Francis IR, Krupnick AS, Kazerooni EA, Iannettoni MD, Whyte RI, Orringer MB. The Annals of thoracic surgery. 1996;62:246. doi: 10.1016/0003-4975(96)00220-2. [DOI] [PubMed] [Google Scholar]; Kurata T, Oguri T, Isobe T, Ishioka Si, Yamakido M. Cancer Science. 1999;90:1238. doi: 10.1111/j.1349-7006.1999.tb00702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poeckler-Schoeniger C, Koepke J, Gueckel F, Sturm J, Georgi M. Magnetic resonance imaging. 1999;17:383. doi: 10.1016/s0730-725x(98)00180-5. [DOI] [PubMed] [Google Scholar]
- 8.Bu L, Xie J, Chen K, Huang J, Aguilar ZP, Wang A, Sun KW, Chua MS, So S, Cheng Z, Eden HS, Shen B, Chen X. Contrast Media & Molecular Imaging. 2012;7:363. doi: 10.1002/cmmi.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O'Neil CP, Lee LK, Swartz MA, Hubbell JA. Nat Biotechnol. 2007;25:1159. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 10.D'Addio SM, Baldassano S, Shi L, Cheung L, Adamson DH, Bruzek M, Anthony JE, Laskin DL, Sinko PJ, Prud'homme RK. J Controlled Release. 2013;168:41. doi: 10.1016/j.jconrel.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hubbell JA, Reddy ST, Swartz MA, van der Vlies A. Google Patents. 2011 [Google Scholar]
- 12.Yoon TJ, Lee H, Shao H, Hilderbrand SA, Weissleder R. Adv Mater. 2011;23:4793. doi: 10.1002/adma.201102948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim HM, Lee H, Hong KS, Cho MY, Sung MH, Poo H, Lim YT. ACS Nano. 2011;5:8230. doi: 10.1021/nn202912b. [DOI] [PubMed] [Google Scholar]
- 14.Calero-DdelC VL, Rinaldi C. J Magn Magn Mater. 2007;314:60. [Google Scholar]; Sun S, Zeng H, Robinson DB, Raoux S, Rice PM, Wang SX, Li G. J Am Chem Soc. 2004;126:273. doi: 10.1021/ja0380852. [DOI] [PubMed] [Google Scholar]
- 15.Pansare VJ, Hejazi S, Faenza WJ, Prud'homme RK. Chem Mater. 2012;24:812. doi: 10.1021/cm2028367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vonarbourg A, Passirani C, Saulnier P, Benoit JP. Biomaterials. 2006;27:4356. doi: 10.1016/j.biomaterials.2006.03.039. [DOI] [PubMed] [Google Scholar]
- 17.Duncan TV, Susumu K, Sinks LE, Therien MJ. J Am Chem Soc. 2006;128:9000. doi: 10.1021/ja061897o. [DOI] [PubMed] [Google Scholar]
- 18.Ghoroghchian PP, Frail PR, Susumu K, Blessington D, Brannan AK, Bates FS, Chance B, Hammer DA, Therien MJ. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:2922. doi: 10.1073/pnas.0409394102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson BK, Prud'homme RK. Aust J Chem. 2003;56:1021. [Google Scholar]
- 20.Kumar V, Prud'homme RK. J Pharm Sci. 2008;97:4904. doi: 10.1002/jps.21342. [DOI] [PubMed] [Google Scholar]
- 21.D'Addio SM, Prud'homme RK. Advanced drug delivery reviews. 2011;63:417. doi: 10.1016/j.addr.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 22.Johnson BK, Prud'homme RK. AlChE J. 2003;49:2264. [Google Scholar]
- 23.Kumar V, Hong SY, Maciag AE, Saavedra JE, Adamson DH, Prud'homme RK, Keefer LK, Chakrapani H. Molecular pharmaceutics. 2009;7:291. doi: 10.1021/mp900245h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gindy ME, Panagiotopoulos AZ, Prud'homme RK. Langmuir. 2008;24:83. doi: 10.1021/la702902b. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Tong Z, Prud'homme RK. Pest management science. 2008;64:808. doi: 10.1002/ps.1566. [DOI] [PubMed] [Google Scholar]
- 26.Pinkerton NM, Grandeury A, Fisch A, Brozio J, Riebesehl BU, Prud'homme RK. Molecular pharmaceutics. 2012;10:319. doi: 10.1021/mp300452g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dobrovolskaia MA, Aggarwal P, Hall JB, McNeil SE. Molecular pharmaceutics. 2008;5:487. doi: 10.1021/mp800032f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hawley AE, Illum L, Davis SS. Pharm Res. 1997;14:657. doi: 10.1023/a:1012117531448. [DOI] [PubMed] [Google Scholar]
- 29.D'Addio SM, Saad W, Ansell SM, Squiers JJ, Adamson DH, Herrera-Alonso M, Wohl AR, Hoye TR, Macosko CW, Mayer LD. J Controlled Release. 2012;162:208. doi: 10.1016/j.jconrel.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar V, Adamson DH, Prud'homme RK. small. 2010;6:2907. doi: 10.1002/smll.201001199. [DOI] [PubMed] [Google Scholar]
- 31.Rubenstein M, Colby R. 2003 [Google Scholar]
- 32.Ghoroghchian PP, Lin JJ, Brannan AK, Frail PR, Bates FS, Therien MJ, Hammer DA. Soft Matter. 2006;2:973. doi: 10.1039/b604212k. [DOI] [PubMed] [Google Scholar]
- 33.Pinkerton NM, Zhang SW, Youngblood RL, Gao D, Li S, Benson BR, Anthony J, Stone HA, Sinko PJ, Prud'homme RK. Biomacromolecules. 2014;15:252. doi: 10.1021/bm4015232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Owens DE, III, Peppas NA. Int J Pharm. 2006;307:93. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]; Jokerst JV, Lobovkina T, Zare RN, Gambhir SS. Nanomedicine. 2011;6:715. doi: 10.2217/nnm.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kricheldorf HR, Boettcher C, Tönnes KU. Polymer. 1992;33:2817. [Google Scholar]
- 36.Calero-DdelC VL, Gonzalez AM, Rinaldi C. Journal of manufacturing science and engineering. 2010;132 [Google Scholar]
- 37.Han J, Zhu Z, Qian H, Wohl AR, Beaman CJ, Hoye TR, Macosko CW. J Pharm Sci. 2012;101:4018. doi: 10.1002/jps.23259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dalwadi G, Benson HA, Chen Y. Pharm Res. 2005;22:2152. doi: 10.1007/s11095-005-7781-z. [DOI] [PubMed] [Google Scholar]
- 39.Kumar V, Prud'homme RK. Chem Eng Sci. 2009;64:1358. [Google Scholar]
- 40.Carr HY, Purcell EM. Physical Review. 1954;94:630. [Google Scholar]; Meiboom S, Gill D. Rev Sci Instrum. 1958;29:688. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.