

Abstract
New strategies for combining conventional photo- and soft-lithographic techniques with high-resolution patterning and etching strategies are needed in order to produce multiscale fluidic platforms that address the full range of functional scales seen in complex biological and chemical systems. The smallest resolution required for an application often dictates the fabrication method used. Micromachining and micropowder blasting yield higher throughput, but lack the resolution needed to fully address biological and chemical systems at the cellular and molecular scales. In contrast, techniques such as electron beam lithography or nanoimprinting allow nanoscale resolution, but are traditionally considered costly and slow. Other techniques such as photolithography or soft lithography have characteristics between these extremes. Combining these techniques to fabricate multiscale or hybrid fluidics allows fundamental biological and chemical questions to be answered. In this study, a combination of photolithography and electron beam lithography are used to produce two multiscale fluidic devices that incorporate porous membranes into complex fluidic networks in order to control the flow of energy, information, and materials in chemical form. In the first device, materials and energy were used to support chemical reactions. A nanoporous membrane fabricated with e-beam lithography separates two parallel, serpentine channels. Photolithography was used to pattern microfluidic channels around the membrane. The pores were written at 150 nm and reduced in size with silicon dioxide deposition from plasma enhanced chemical vapor deposition and atomic layer deposition. Using this method, the molecular weight cutoff of the membrane can be adapted to the system of interest. In the second approach, photolithography was used to fabricate 200 nm thin pores. The pores confined microbes and allowed energy replenishment from a media perfusion channel. The same device can be used for study of intercellular communication via the secretion and uptake of signal molecules. Pore size was tested with 750 nm fluorescent polystyrene beads and fluorescein dye. The 200 nm polydimethylsiloxane pores were shown to be robust enough to hold 750 nm beads while under pressure, but allow fluorescein to diffuse across the barrier. Further testing showed that extended culture of bacteria within the chambers was possible. These two examples show how lithographically defined porous membranes can be adapted to two unique situations and used to tune the flow of chemical energy, materials, and information within a microfluidic network.
I. INTRODUCTION
Water filtration can be traced back to 12th century Greece when water would be passed through a cloth sack to purify it. While the efficiency of commercial filters has improved, the basic concept of using an interwoven sheet of fibers as a filter membrane is a common method of filtration to this day. The random alignment of fibers creates tortuous paths through the membrane, limiting what molecules and particles can pass through. Studying this type of filter, Holdich et al.1 found that although spaces between some fibers were greater than 50 μm, the membrane blocked 99% of particles larger than 3 μm. The packing of the fibers as well as fouling of the filter controlled the effective pore size. Large particulates filled the void spaces of the filter and caused a reduction in the effective pore size. The effective pore sizes in this case ranged from 2.5 to 5 μm, 10% of the actual pore size.1 Beginning with track etching of cellulose nitrate membranes for filtration studies,2 micro- and nanofabrication techniques have been used to fabricate porous membranes with well controlled permeability.3 Microfabrication allows improved control of pore size as well as pore density. These techniques can be categorized as “bottom up” approaches or “top down” approaches.
Bottom up approaches rely upon self-assembling structures to create pores that restrict transport by creating tortuous paths through the membrane. Such membranes have been made using self-assembled nanowires,4 colloidal self-assemblies,5 amorphous silicon (a-Si) to porous nanocrystalline silicon (pnc_Si) crystallization,6 and vertically aligned carbon nanofibers.7–11 These membranes still rely on restricting transport with implementation of tortuous paths that molecules must take to pass through the membrane. The distribution of effective pore size depends on the thickness of the membrane. Required membrane thickness is an important property to take into account when incorporating these types of membranes into microfluidic devices. Top-down approaches are able to more tightly control the distribution and size of pores within a nano- or microporous membrane. This has been accomplished using lithographic steps to produce a predefined pattern on a membrane using track etching,2,12,13 sacrificial oxide layers,14 focused ion beam milling,15 reactive ion etching (RIE),3 and e-beam lithography.16 Top down approaches produce membranes that have a pore size dependent upon the pore design rather than on fouling of the membrane to create tortuous paths.
Top down fabricated filters have a range of biological applications centered around systems that are regulated by semipermeable membranes by limiting species transport based on size. Accurately replicating these systems requires the spatial control that is afforded by the use of microfluidics. Membranes have been incorporated into microfluidics by using slits etched into silicon membranes.16–20 These devices have been shown to provide control over the transport of materials and energy to support cell-free protein synthesis (CFPS) reactions.16,20 Other applications addressed with embedded membrane in microfluidic architectures include dialysis,21 cell-free exchange reactors,19 and even for experimental DNA sequencing techniques.22
In addition to molecular transport control, microfluidics have been used in a number of ways to manipulate, grow, and analyze cells by leveraging their fluid flow control and microstructure environmental advantages.23 Many microfluidic cell culture devices contain membranes to provide nutrients to the cells or control movement of cells through the device.24–30 Microhabitat patches developed by the Austin group were used to limit transport of nutrients in order to study bacterial competition.27,31–33 However, a majority of current microfluidic cell culture chambers either deal with larger mammalian cells rather than smaller bacterial cells,25 or are fabricated in silicon,27,29 which are slower to fabricate and limit the use of transmitted light microscopy compared to polydimethylsiloxane (PDMS) devices.
In the first approach, e-beam lithography was used to define pores that were etched into a silicon substrate and monolithically integrated into a microfluidic network using RIE. Oxide deposition in the pores was used to tune the molecular weight cutoff (MWCO), creating a well-defined nanoporous membrane separating microfluidic channels. These pores were able to tune the exchange of energy and materials to support biochemical reactions. The second approach consisted of a square cell culture chamber and two nutrient channels separated by a microporous membrane. Flat, 200 nm deep pores isolated microbial cells in a culture chamber, but allowed for transport of nutrients and chemical signals. These larger pores were fabricated in PDMS to facilitate imaging via live-cell microscopy. Green fluorescent protein (GFP) expressing Escherichia coli cells were grown in the culture chambers with nutrient transport to demonstrate operation. These two applications for incorporation of top down fabricated porous membranes show how fabrication techniques can be adapted to control the transport of energy, materials, and information within a microfluidic network in a manner that is tailored to the scale of the biological system of interest.
II. EXPERIMENT
Two multiscale fluidic devices were fabricated and tested. The first device incorporated a membrane with nanoscale pores to control transport of chemical species between microfluidic channels, retaining larger molecular weight (MW) molecules and allowing exchange of small MW molecules. Top-down fabrication techniques allowed for tuning of the pore size to control MWCO of the membrane and pore density to influence total exchange. The MWCO of the membrane was tuned by controlled coating of pores to adjust pore size. The second multiscale device incorporated a microporous membrane designed to confine cell colonies in individual chambers while allowing communication and nutrient transfer. Both devices used porous membranes to control the flow of energy, materials, and information, selectively renewing chemical species critical to the long-term function of the biochemical and biological systems of interest.
A. Nanoporous exchange device
The nanoporous exchange device consists of two parallel, serpentine channels separated by nanopores. A microporous device was first fabricated using photolithography shown in Fig. 1(a).
Fig. 1.

(Color online) Nanoporous exchange device. (a) An early iteration of the exchange device with microporous membrane with an inset of the channels. (b) (1) A 500 nm thick silicon dioxide layer is patterned onto a bare silicon substrate. (2) Nanopores are then patterned with e-beam lithography and etched into the oxide layer with RIE. (3) Microchannels are patterned onto the wafer using conventional photolithography. (4) Microchannels are etched through the oxide layer followed by deep RIE etching into the silicon substrate. (5) Photoresist is removed from the wafer and (6) the nanopores are subsequently etched into the substrate. (7) Pore sizes are reduced with PECVD and ALD oxide deposition.
Fluidic devices with nanoporous membranes, having the same design but narrower pores, were created using a combination of e-beam lithography and photolithography to define a silicon dioxide etch mask. Anisotropic silicon etching was used to pattern the microchannel network, and then, both the network and nanopores were etched with the same process. The pore size was decreased via silicon dioxide deposition using plasma enhanced chemical vapor deposition (PECVD) and atomic layer deposition (ALD) to tune the MWCO. The device was designed with a 200 μm wide primary channel and a 75 μm wide secondary channel separated by a 25 μm thick nanoporous membrane. The pores were 8.5 μm deep while the channels are 60 μm deep.
1. Electron beam lithography
Pore features were written directly to each device using e-beam lithography. Silicon dioxide of 500 nm was deposited onto a bare 4 in. silicon wafer with a thermal oxide process (Temperature: 1000 °C, O2: 3000 sccm, H2O: 3 ml/min, pressure: 1 atm, and time: 80 min). ZEP520A (ZEON, Tokyo, Japan) e-beam resist was spin-coated onto the wafer at 1000 rpm for 45 s. Pre-exposure bake was done on a hot plate at 180 °C for 45 s. The nanopore pattern was written with a JEOL JBX-9300FS Electron Beam Lithography system (Peabody, MA) (shot size: 4 nm, voltage: 100 kV, and current: 2 nA) and developed with Xylenes for 30 s. Samples were rinsed with isopropyl alcohol (IPA) and dried with nitrogen. The exposed oxide was etched with an Oxford Instruments Plasmalab System 100 Reactive Ion Etcher (Abington, Oxfordshire, UK) (RF: 200 W, inductively coupled plasma (ICP): 2000 W, C4F8: 45 sccm, O2: 2sccm, pressure: 7 mTorr, and temperature: 15 °C) at a rate of approximately 300 nm/min.
2. Microchannels
A photolithography mask with the fluidic network was written on a Heidelberg DWL 66 (Heidelberg, Germany) with a 20 mm write head. The Si wafer with etched pores was coated with MicroPrime P20 adhesion promoter (Shin-Etsu Microsci, Phoenix, AZ) at 3000 rpm for 45 s. Rohm and Haas Electronic Materials Megaposit SPR 220–4.5 Positive Photoresist (Marlborough, MA) was spin-coated onto the wafer at 3000 rpm for 45 s. The wafer was baked on a hot plate at 90 °C for 90 s. After cooling, the wafer and microchannel mask were aligned with a Neutronix, Inc., NxQ 7500 mask aligner (Morgan Hill, CA) with a dose of 165 mJ/cm2. The device was held at room temperature for 30 min to ensure no bubbling of the resist occurred during subsequent baking. Postexposure bake was done at 115 °C for 90 s. The wafer was then developed in Microposit MF CD-26 developer (Marlborough, MA) until clear.
3. Reactive ion etching of fluidic network and membrane
The exposed oxide was etched using the same recipe as described above. The channels were then etched 75 loops of a Bosch etch process (Deposition: RF 10 W, ICP 1750W, C4F8 140 sccm, SF6 1 sccm, pressure 20 mTorr, time 3 s, and temperature 15 °C. Etching: RF 7 W, ICP 1750 W, C4F8 2 sccm, SF6 120 sccm, pressure 20 mTorr, time 10 s, and temperature 15 °C). The wafer was then sonicated in an acetone bath for 5 min to remove the resist. A PVA TePla IoNWave10 oxygen plasma (RF 6000 W, O2 250 sccm, Ar 25 sccm, pressure 200 mTorr, and time 20 min) was used to remove any remaining resist. An additional 30 loops of the same Bosch etching process etched the pores and channel simultaneously to final depth of 13 and 52 μm, respectively. The ratio between these two etch steps determined the ratio of pore depth to total channel height.
4. Silicon dioxide coating of nanopores
The etched silicon nanopores were coated with an oxide layer in order to reduce the gap size in a controllable way. Silicon dioxide was deposited with an Oxford Instruments Plasmalab System 100 PECVD (Abington, Oxfordshire, UK) tool (RF 20 W, 5%SiH4/Ar 85 sccm, N2O 157 sccm, pressure 1000 mTorr, time 14 min, and temperature 350 °C). The nonconformal deposition decreased the amount of scalloping left by the Bosch process. Oxford Instruments FlexAL ALD System [plasma: RF 400 W, O2 60 sccm, pressure 15 mTorr, time 2 s, temperature 150 °C precursor: bis(diethylamino)silane time 0.7 s] with a deposition rate of 3 Å/cycle was used for 27 cycles to further decrease the pore size by 8 nm and tune the MWCO of the nanomembrane. After the bioreactor fabrication was complete, the devices were sealed by air plasma bonding with a Harrick Plasma PDC-001 air plasma cleaner (Ithaca, NY) a 5 mm thick PDMS cover over the device. Inlets and outlets were punched using a Ted Pella 0.75 mm biopsy punch (Redding, CA) and removed with tweezers.
5. Device testing
Testing of the nanopores was done with fluorescein dye. Quantifying the MWCO of the membrane was done by loading one channel of the device with a protein ladder and the other with a buffer solution. Incubation overnight allowed proteins to diffuse across the membrane. Fluorescein dye (Sigma-Aldrich, St. Louis, MO) was suspended in phosphate buffered saline (PBS) to promote dissolution at a concentration of 10 μM. The dye was loaded into one channel while PBS was loaded into the other. The channels were set to flow rates of 15 and 5.6 μl/h to maintain a constant velocity in the two differently sized channels. After coming to equilibrium, pictures were taken of the device with an Olympus IX51 microscope (Shinjuku, Tokyo) at loops 1, 20, and 150 of the device, which corresponded to 0%, 5%, and 66% of the total channel length to show diffusion across the membrane. Images were taken in epi-fluorescence using a Chroma 41001FITC (Bellows Falls, VT) filter cube (480 nm excitation band pass filter with a 40 nm band width and 535 nm emission band pass filter with a 50 nm band width). To determine the molecular weight cutoff of the nanoporous membrane, an ultralow range molecular weight marker ladder (Sigma-Aldrich, St. Louis, MO) with molecular weights of 1.060, 6.500, 14.200, 17.000, and 26.600 kDa was used. One channel of the device was filled with the protein ladder, and the other with the accompanying sample buffer. Devices were covered in water soaked wipes and placed in 30 °C incubator overnight. Flushing each channel with water, samples were collected from the device. Samples were run on 16.5% Mini-PROTEAN Tris-Tricine Gel (BioRad, Hercules, CA) using the BioRad Mini-PROTEAN Tetra Cell (Hercules, CA) electrophoresis system. The gel was fixed in 4% paraformaldehyde diluted in PBS, rinsed in water, and stained with SimplyBlue SafeStain (Life Technologies, Carlsbad, CA). Gel image was analyzed for relative protein content at each molecular weight using gel analysis tool in Fiji.
B. Cell culture device
Devices to be used in cell culture studies were replicated in PDMS from multiscale silicon masters. Two photolithography steps were used to assemble masters starting with a 200 nm silicon dioxide layer used to form 1 μm wide micropores. The fluidic network was formed over the pores in a second photolithography step. The device consisted of a 1 mm square culture chamber flanked by two 100 μm wide nutrient channels to supply nutrients to the cells. Pores 200 nm thick were used to mimic the function of a sterile biological filter. The thickness can be altered based on the type of cells used by adjusting the thermal oxidation process. SU-8 photoresist was then used to form the culture chamber area over the pores. These are each separated by a 25 μm membrane with pores at a pitch of 25 μm. Figure 2 shows a micrograph of the culture device along with the steps involved in the fabrication process.
Fig. 2.
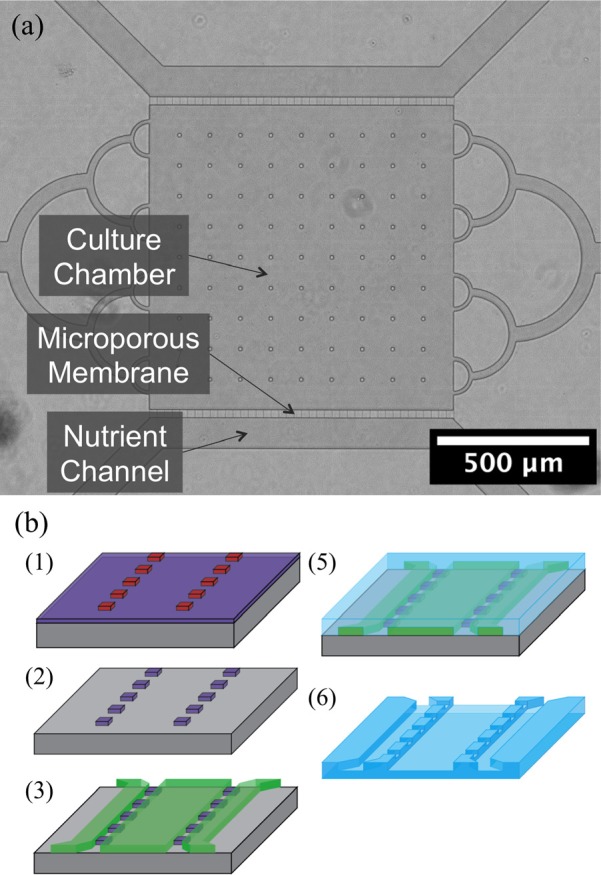
(Color online) Microporous cell culture device. (a) The device is made up of a central cell culture chamber flanked by two nutrient exchange channels. (b) (1) Lines defining the pore width are patterned into a 200 nm oxide using photolithography and (2) reactive ion etching. (3) Fluidic network are then aligned and patterned over the pores in SU-8. (5) PDMS casting is then used to replicate the patterns. (6) The PDMS casting is removed and later plasma bonded to a glass slide.
1. Photolithography
For PDMS reactor masters, 200 nm of oxide were deposited onto a silicon wafer with a thermal oxide process (temperature: 1000 °C, O2: 3000sccm, H2O: 3 ml/min; pressure: 1 atm, and time: 18 min). MicroPrime P20 adhesion promoter (Shin-Etsu Microsci, Phoenix, AZ) was spin-coated onto a wafer at 3000 rpm for 45 s. JSR Micro Microphotoresist (negative) NFR 016D2–55cP (Sunnyvale, CA) was spin-coated on the P20 at 3000 rpm for 45 s. The wafer was baked on a hot plate at 90 °C for 90 s. After cooling, the pores and microchannel mask were aligned with a Neutronix Inc NxQ 7500 mask aligner (Morgan Hill, CA) with a dose of 36 mJ/cm2, and baked at 115 °C for 90 s. The wafer was developed using Microposit MF CD-26 developer (Marlborough, MA). The patterned wafers were then etched with an Oxford Instruments Plasmalab System 100 Reactive Ion Etcher (Abington, Oxfordshire, UK) with the same oxide etch parameters as used with the nanoporous device at a rate of 300 nm/min through the oxide leaving 1 μm pores. Devices were sonicated in acetone to remove the photoresist.
Micro-Chem SU-8 2010 positive photoresist (Newton, MA) was spin-coated on the wafer at 3000 rpm for 45 s and baked at 90 °C for 2.5 min. An exposure dose of 132 mJ/cm2 was used on a Neutronix, Inc., NxQ 7500 mask aligner (Morgan Hill, CA). Postexposure bake was done at 95 °C for 3.5 min. The wafer was developed with the spray-puddle method with SU-8 developer (Micro-Chem, Newton, MA) until clear. The wafer was then baked at 250 °C for 5 min to promote adhesion. The master wafer was plasma cleaned with a Harrick Plasma PDC-001 air plasma cleaner (Ithaca, NY) and silanized with trichloro(1H,1H,2H,2H-perfluorooctyl) silane (Sigma-Aldrich, St. Louis, MO) by storing in a closed glass container with 20 μl of silane, at 85 °C for 2 h.
2. PDMS casting and device bonding
Sylgard 184 PDMS from Dow Corning (Midland, MI) prepolymer and crosslinker components were used in a 5:1 prepolymer to crosslinker ratio to form devices. Doubling the amount of crosslinker from the base 10:1 ratio stiffens the resulting PDMS. This increases the yield for the nanopores, which can otherwise flex and bond to the glass, resulting in reduced or no transport across the membrane. The PDMS mixture was poured over the silanized master, degassed, and baked at 75 °C for 1 h. The devices were removed from the master with a razor blade and placed in a dish with the features facing up. The molded devices were baked at 75 °C for an additional 48 h to fully cure the elastomer and evaporate any remaining solvents in the PDMS. This further stiffens the PDMS, improving the number of open pores in the device. Replication of the pores was consistent when using these techniques. Inlets and outlets were punched with a Ted Pella 0.75 mm biopsy punch (Redding, CA). The device and a glass slide were plasma cleaned using a Harrick Plasma PDC-001 air plasma cleaner (Ithaca, NY) for 2 min, and brought into contact to form a permanent bond. The devices were baked at 75 °C for 15 min to anneal the polymer and improve bonding. Devices were used the same day; otherwise, pores were liable to collapse after a couple of days.
3. Device testing and cell culture
Fluorescent species that were larger (fluorescent microspheres) and smaller (fluorescein dye) than the pore size were loaded into the culture chamber and monitored over a 30 min period. Seven hundred and fifty nanometers fluoresbrite yellow green carboxylate microspheres (PolyScience, Niles, IL) in deionized water, and fluorescein dye (Sigma-Aldrich, St. Louis, MO) in PBS were loaded into separate devices with their respective solvents loaded into the support channels using a New Era NE-1800 syringe pump (Farmingdale, NY). Fluorescence images of each device were taken on an Olympus IX70 (Shinjuku, Tokyo) inverted epi-fluorescence microscope. Image overlay was performed in image j.
One Shot TOP10 chemically competent E. coli cells (Invitrogen, Waltham, MA) were transformed with a pUC19 vector containing enhanced GFP (EGFP) and ampicillin resistance. The constitutively expressed EGFP was used to quantify cell growth under continuous perfusion of media. Lysogeny Broth (LB) media made with 10 g/l tryptone, 5 g/l yeast extract, and 10 g/l sodium chloride (Sigma-Aldrich, St. Louis, MO) with 100 μg/ml ampicillin (Sigma-Aldrich, St. Louis, MO) was inoculated with cell culture from an LB agar plate [LB media with 15 g/l agar (Fisher, Pittsburgh, PA)] with a similar concentration of ampicillin. The liquid culture was incubated at 37 °C in a shaker incubator for 2 h. The culture was then spun down in a centrifuge at 2500 rpm for 5 min. The media was poured off and replaced with M9 minimal media broth (Amresco, Solon, OH) in order to minimize autofluorescence when imaging. Cells were then loaded into the culture chamber, and blank M9 media was loaded into the nutrient channels via syringes. Media was perfused through the nutrient channels with a Harvard Apparatus Pump II Elite (Holliston, MA) at a rate of 5 μl/h over 72 h. Fluorescent images were taken every hour with a Nikon Eclipse Ti-U inverted epifluorescent microscope (Tokyo, Japan). Image analysis was done with Image J.
III. RESULTS AND DISCUSSION
A. Nanoporous exchange device
Verification of the fabrication process described in Sec. II began with SEM images of the device. The tested device had two channels each 52 μm deep and 1.8 m long. The channels were 75 or 200 μm wide. The pore design, patterned by e-beam lithography, was 150 nm wide. Pore width, measured by SEM, was 180 nm at the oxide mask and decreased to a point. The depth of the pores was between 12.9 and 14.2 nm. Figure 3 shows SEM micrographs of the completed device prior to closing the pores with silicon dioxide and after PECVD treatment.
Fig. 3.

Nanoporous membrane images. (a) Two microfluidic channels, 200 and 75 μm wide, separated by a 25 μm wide nanoporous membrane with an inset of the nanoporous membrane. (b) Nanopores prior to silicon dioxide deposition. (c) Nanopores coated with silicon dioxide to reduce pore size.
Transport of fluorescein dye across the membrane was measured to verify the operation of the membrane. Fluorescent images were taken after the device had reached a steady state. Images were taken at the inlet of the device, after ten loops, and after 150 loops or 5% and 66% of the serpentine channel. Figure 4 shows a diagram of the device with flow direction through the device and where on the device images were taken. At the inlet, fluorescein is in the large channel only. Fluorescein begins to diffuse across the membrane within 5% of the channel, and the two channels are at a similar concentration at 66% of the channel.
Fig. 4.

(Color online) Diffusion of fluorescein dye under constant flow. Top image shows the flow direction through the serpentine channel and the dots represent sampling points. (a) At the inlet fluorescein is in one channel only. (b) Diffusion begins within 92 μm of the inlet—5% of the total length. (c) At 1.2 m from the inlet, 1.2 m, 66% of the channel, signals from each channel are similar.
Diffusion of the components of a small molecular weight protein ladder through the membrane was measured to allow estimation of the MWCO of the nanoporous membrane. Samples collected from each channel were run on a protein gel, which showed that the ratios of feeder to reactor concentrations for the 1.06 and 6.5 kDa ladder components were higher in the feeder channel than with the larger 14.2, 17, and 26.6 kDa proteins. The slope of the graph is steepest, indicating a rapid change in permeability, between 6.5 and 17 kDa. This range is the transition between restricted and unrestricted proteins due to size. The graph in Fig. 5 shows the fraction of each of the proteins found in the feeder channel. This transition indicates that below the threshold MW of the membrane, diffusion of proteins is hindered by the pore size. Higher molecular weight proteins found in the feeder channel can result from defects in the membrane. Rather than diffusing through the pores of the membrane, it is possible that the proteins were moving through larger gaps between the PDMS lid and membrane.
Fig. 5.
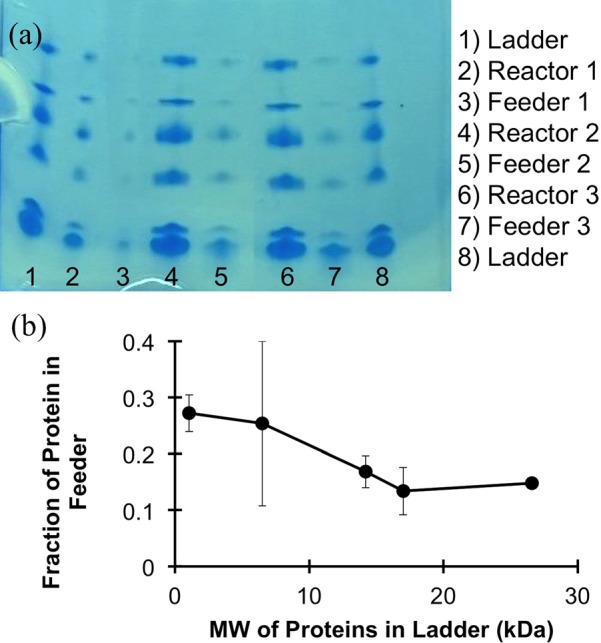
(Color online) Protein diffusion across the membrane of the nanoporous exchange device. (a) A protein gel run with effluent from the reaction and feeder channels of the device after a 24 h incubation. (b) Analysis of the gel shows that the steepest slope of the graph and the transition between restricted proteins and nonrestricted proteins based on molecular weight occurs between 6.5 and 17 kDa.
The MWCO can be tuned further depending on the application by altering the number of ALD cycles performed. For biological applications, chemical energy and material transport across the membrane can be controlled based on MW. Multiscale fluidic networks allow the channels to be controlled individually. The large MW components of a CFPS reaction can be contained on one side of the membrane while Adenosine triphosphate (ATP) and amino acids can be replenished from a support channel. The resulting protein can be contained in the reaction channel or allowed to diffuse into the support channel for purification while the reaction continues.
B. Cell culture device
The second fluidic device is designed to control transport of information and energy between nutrient channels and a cell culture chamber. The microporous membrane limits the movement of cells within the fluidic network. Operation of the microporous membrane was evaluated with fluorescent species. Figure 6 shows SEM images of the device and micropores on the silicon and SU-8 master. The pores were measured to be 27 μm long, 240 nm tall, and 1.38 μm wide.
Fig. 6.

(Color online) Culture device (a) SEM image of the entire device with two culture chambers and two nutrient exchange channels. (b) Expanded view of the membrane separating the nutrient channel and the culture chamber. (c) The pores are 200 nm deep by 1 μm wide and 25 μm long.
Transferring these small features to PDMS with conventional soft lithography procedures resulted in a large portion of the pores being sealed to the glass slide. Higher fidelity replication required stiffer PDMS to improve pore yield. Twice as much crosslinker was used in the PDMS formulation and extending baking times to a minimum of 48 h resulted in a stiff PDMS. Using these techniques, the 200 nm features of the pores could be consistently reproduced. To verify operation of the pores, 750 nm beads approximating the size of bacterial cells were flowed through the device. Aggregation of beads at the pores as shown in Fig. 7 indicated that there was flow through the pores, but the beads were unable to pass through. Small molecules such as signaling molecules and nutrients were represented with fluorescein dye. Over a period of 30 min, the dye was able to diffuse across the membrane as shown in Fig. 7.
Fig. 7.
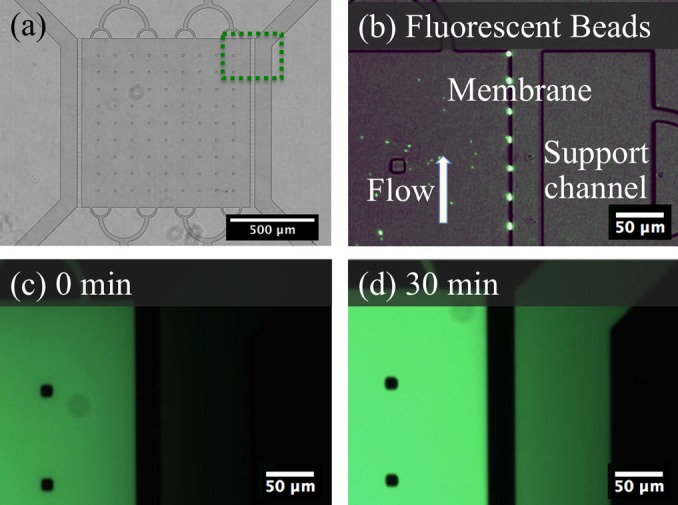
(Color online) (a) Overview of the culture chamber device with a dotted outline of the expanded section in (b)–(d). (b) Beads of 750 μm are isolated in the culture chamber and aggregate at the pores under positive pressure. (c) Fluorescein dye loaded into the culture chamber. (d) Fluorescent signal within the support channel increases over 30 min due to transport of fluorescent dye.
Top ten E. coli cells expressing GFP were cultured in the device over a 48 h period. Media was supplied to the culture chamber by the nutrient channel. Figure 8 shows fluorescence images of the device after loading, after 24, and after 48 h. The cells grew rapidly within the first 24 h and slowed as they reached a high density. Growth was not uniform across the device, showing higher concentrations of cells at the microporous membrane interface where nutrients were being replenished. This device makes long-term studies possible by providing nutrients to the microbial cultures.
Fig. 8.

E. coli growth in the culture chamber. (a) Bright field image of the culture chamber. Fluorescent images show the chamber after (b) loading, (c) 24 h of incubation, and (d) 48 h of incubation.
The membrane within the device allows cultures to be addressed dynamically through the fluidic network. Cell culture within microfluidic devices can be used to address a number of biological questions pertaining to nutrient replenishment, chemical dosing, stress responses, or cell signaling. Spatial and temporal control over these problems requires the control afforded by multiscale fluidic networks and specifically, microporous membranes.
IV. SUMMARY AND CONCLUSIONS
Presently, we have shown that by combining fabrication techniques into multiscale architectures, complex biological questions can be addressed. The two devices presented represent unique applications of microfluidic membranes, but have common elements in that they address the transport of energy, materials, or information within a fluidic network. The nanoporous membrane device is able to control transport of chemical species with a tunable MWCO. This device has applications in CFPS systems to prolong reactions with ATP and amino acid replenishment. The second device provides a method for interfacing and culturing cells. Larger microporous membranes confine bacterial cells to culture chambers where they can be addressed via a nutrient channel or other culture chambers. Replenishment of nutrients for long term studies and chemical species can be dosed without perturbing the culture. Each device has unique applications, but the fundamentals of controlling transport are similar.
ACKNOWLEDGMENTS
A portion of this work was supported by DARPA Award No. HR001134005. The views expressed are those of the authors and do not reflect the official policy or position of the Department of Defense or the U.S. Government. A portion of this work was supported by NIH Award No. 1R01DE024463-01 Culturing of the Uncultured. This research was performed at Oak Ridge National Laboratory (ORNL). ORNL is managed by UT-Battelle, LLC, for the U.S. Department of Energy Under Contract No. DE-AC05-00OR22725. The fabrication of nano- and microfluidic devices was conducted at the Center for Nanophase Materials Sciences, which is DOE Office of Science User Facilities.
References
- 1. Holdich R., Kosvintsev S., Cumming T., and Zhdanov S., Philos. Trans. R. Soc. A 364, 161 (2006). 10.1098/rsta.2005.1690 [DOI] [PubMed] [Google Scholar]
- 2. Fleischer R. L., Price P. B., and Symes E. M., Science 143, 249 (1964). 10.1126/science.143.3603.249 [DOI] [PubMed] [Google Scholar]
- 3. Russo A. P., Retterer S. T., Spence A. J., Isaacson M. S., Lepak L. A., Spencer M. G., Martin D. L., MacColl R., and Turner J. N., Sep. Sci. Technol. 39, 2515 (2004). 10.1081/SS-200026706 [DOI] [Google Scholar]
- 4. Rahong S. et al. , Anal. Sci. 31, 153 (2015). 10.2116/analsci.31.153 [DOI] [PubMed] [Google Scholar]
- 5. Zeng Y. and Harrison D. J., Anal. Chem. 79, 2289 (2007). 10.1021/ac061931h [DOI] [PubMed] [Google Scholar]
- 6. Striemer C. C., Gaborski T. R., McGrath J. L., and Fauchet P. M., Nature 445, 749 (2007). 10.1038/nature05532 [DOI] [PubMed] [Google Scholar]
- 7. Zhang L., Melechko A. V., Merkulov V. I., Guillorn M. A., Simpson M. L., Lowndes D. H., and Doktycz M. J., Appl. Phys. Lett. 81, 135 (2002). 10.1063/1.1490142 [DOI] [Google Scholar]
- 8. Fletcher B. L., Hullander E. D., Melechko A. V., McKnight T. E., Klein K. L., Hensley D. K., Morrell J. L., Simpson M. L., and Doktycz M. J., Nano Lett. 4, 1809 (2004). 10.1021/nl0493702 [DOI] [Google Scholar]
- 9. Fowlkes J. D., Fletcher B. L., Hullander E. D., Klein K. L., Hensley D. K., Melechko A. V., Simpson M. L., and Doktycz M. J., Nanotechnology 16, 3101 (2005). 10.1088/0957-4484/16/12/063 [DOI] [PubMed] [Google Scholar]
- 10. Fowlkes J. D., Hullander E. D., Fletcher B. L., Retterer S. T., Melechko A. V., Hensley D. K., Simpson M. L., and Doktycz M. J., Nanotechnology 17, 5659 (2006). 10.1088/0957-4484/17/22/021 [DOI] [PubMed] [Google Scholar]
- 11. Retterer S. T., Melechko A., Hensley D. K., Simpson M. L., and Doktycz M. J., Carbon 46, 1378 (2008). 10.1016/j.carbon.2008.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beck R. E. and Schultz J. S., Science 170, 1302 (1970). 10.1126/science.170.3964.1302 [DOI] [PubMed] [Google Scholar]
- 13. Apel P., Radiat. Meas. 34, 559 (2001). 10.1016/S1350-4487(01)00228-1 [DOI] [Google Scholar]
- 14. Chu W. H., Chin R., Huen T., and Ferrari M., J. Microelectromech. Syst. 8, 34 (1999). 10.1109/84.749400 [DOI] [Google Scholar]
- 15. Tong H. D., Jansen H. V., Gadgil V. J., Bostan C. G., Berenschot E., van Rijn C. J. M., and Elwenspoek M., Nano Lett. 4, 283 (2004). 10.1021/nl0350175 [DOI] [Google Scholar]
- 16. Siuti P., Retterer S. T., Choi C. K., Fowlkes J. D., and Doktycz M. J., 2009 First Annual ORNL Biomedical Science and Engineering Conference: Exploring the Intersections of Interdisciplinary Biomedical Research ( IEEE, Oak Ridge, TN, 2009), p. 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Srijanto B. R., Retterer S. T., Fowlkes J. D., and Doktycz M. J., J. Vac. Sci. Technol. B 28, C6P48 (2010). 10.1116/1.3518911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Retterer S. T., Siuti P., Choi C. K., Thomas D. K., and Doktycz M. J., Lab Chip 10, 1174 (2010). 10.1039/b921592a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Siuti P., Retterer S. T., and Doktycz M. J., Lab Chip 11, 3523 (2011). 10.1039/c1lc20462a [DOI] [PubMed] [Google Scholar]
- 20. Siuti P., Retterer S. T., Choi C. K., and Doktycz M. J., Anal. Chem. 84, 1092 (2012). 10.1021/ac202726n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kurita R., Yabumoto N., and Niwa O., Biosens. Bioelectron. 21, 1649 (2006). 10.1016/j.bios.2005.07.016 [DOI] [PubMed] [Google Scholar]
- 22. Branton D. et al. , Nat. Biotechnol. 26, 1146 (2008). 10.1038/nbt.1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. El-Ali J., Sorger P. K., and Jensen K. F., Nature 442, 403 (2006). 10.1038/nature05063 [DOI] [PubMed] [Google Scholar]
- 24. Mehling M. and Tay S., Curr. Opin. Biotechnol. 25, 95 (2014). 10.1016/j.copbio.2013.10.005 [DOI] [PubMed] [Google Scholar]
- 25. Lee K. S., Boccazzi P., Sinskey A. J., and Ram R. J., Lab Chip 11, 1730 (2011). 10.1039/c1lc20019d [DOI] [PubMed] [Google Scholar]
- 26. Hung P. J., Lee P. J., Sabounchi P., Lin R., and Lee L. P., Biotechnol. Bioeng. 89, 1 (2005). 10.1002/bit.20289 [DOI] [PubMed] [Google Scholar]
- 27. Keymer J. E., Galajda P., Muldoon C., Park S., and Austin R. H., Proc. Natl. Acad. Sci. U.S.A. 103, 17290 (2006). 10.1073/pnas.0607971103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kovarik M. L., Gach P. C., Ornoff D. M., Wang Y. L., Balowski J., Farrag L., and Allbritton N. L., Anal. Chem. 84, 516 (2012). 10.1021/ac202611x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang P., Robert L., Pelletier J., Dang W. L., Taddei F., Wright A., and Jun S., Curr. Biol. 20, 1099 (2010). 10.1016/j.cub.2010.04.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ma L., Datta S. S., Karymov M. A., Pan Q. C., Begolo S., and Ismagilov R. F., Integr. Biol. 6, 796 (2014). 10.1039/C4IB00109E [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lambert G., Liao D., Vyawahare S., and Austin R. H., J. Bacteriol. 193, 1878 (2011). 10.1128/JB.01430-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Austin R. H., Tung C. K., Lambert G., Liao D., and Gong X. Q., Chem. Soc. Rev. 39, 1049 (2010). 10.1039/b911230h [DOI] [PubMed] [Google Scholar]
- 33. Keymer J. E., Galajda P., Lambert G., Liao D., and Austin R. H., Proc. Natl. Acad. Sci. U.S.A. 105, 20269 (2008). 10.1073/pnas.0810792105 [DOI] [PMC free article] [PubMed] [Google Scholar]