

Abstract
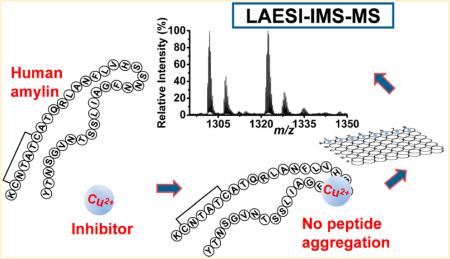
Native electrospray ionization (ESI) mass spectrometry (MS) is often used to monitor noncovalent complex formation between peptides and ligands. The relatively low throughput of this technique, however, is not compatible with extensive screening. Laser ablation electrospray ionization (LAESI) MS combined with ion mobility separation (IMS) can analyze complex formation and provide conformation information within a matter of seconds. Islet amyloid polypeptide (IAPP) or amylin, a 37-amino acid residue peptide, is produced in pancreatic beta-cells through proteolytic cleavage of its prohormone. Both amylin and its precursor can aggregate and produce toxic oligomers and fibrils leading to cell death in the pancreas that can eventually contribute to the development of type 2 diabetes mellitus. The inhibitory effect of the copper(II) ion on amylin aggregation has been recently discovered, but details of the interaction remain unknown. Finding other more physiologically tolerated approaches requires large scale screening of potential inhibitors. Here, we demonstrate that LAESI-IMS-MS can reveal the binding stoichiometry, copper oxidation state, and the dissociation constant of human amylin–copper(II) complex. The conformations of hIAPP in the presence of copper(II) ions were also analyzed by IMS, and preferential association between the β-hairpin amylin monomer and the metal ion was found. The copper(II) ion exhibited strong association with the –HSSNN– residues of the amylin. In the absence of copper(II), amylin dimers were detected with collision cross sections consistent with monomers of β-hairpin conformation. When copper(II) was present in the solution, no dimers were detected. Thus, the copper(II) ions disrupt the association pathway to the formation of β-sheet rich amylin fibrils. Using LAESI-IMS-MS for the assessment of amylin–copper(II) interactions demonstrates the utility of this technique for the high-throughput screening of potential inhibitors of amylin oligomerization and fibril formation. More generally, this rapid technique opens the door for high-throughput screening of potential inhibitors of amyloid protein aggregation.
Noncovalent supramolecular self-assembly of proteins and other biological macromolecules is an essential event in many important biochemical and cellular processes, such as cell signaling, cell growth and differentiation, cell-to-cell adhesion, and inflammation.1 The characterization of protein–ligand interactions is needed for understanding certain biochemical processes and the structural properties of protein complexes.1–3 Finding inhibitors of protein complex formation or aggregation, a common task in the screening of potential pharmaceuticals, requires information on the affinity and binding selectivity between numerous drug candidates and their protein targets.
Islet amyloid polypeptide (IAPP) or amylin, a 37-residue polypeptide, cosecreted with insulin from pancreatic β-cells, functions as a partner to insulin in regulating glucose homeostasis.4–7 Under normal physiological conditions, amylin exists as a soluble monomer in a random coil state. In type 2 diabetes mellitus (T2DM), however, this peptide is a major component of amyloid plaques in the pancreatic tissue of patients.8–10 High resolution electron and atomic force microscopy revealed that amylin can aggregate into insoluble β-pleated IAPP amyloid fibrils in a time and concentration dependent manner.11–13 CD spectroscopy studies demonstrated that amylin self-association into oligomers and fibrils is strongly conformation dependent, and it is triggered by the transition of the peptide from random coil to predominantly β-sheet conformation.11,12,14 The viability assays further reveal that amylin fibril formation always accompanies amylin toxicity.15 Such findings contribute to the hypothesis that the formation of toxic amylin fibrils induces β-cell death, resulting in the relative insulin deficiency in type 2 diabetes.6,15,16
Ion mobility separation (IMS) and molecular modeling studies reveal that fibril formation and the aggregation of amylin are conformation dependent.17 Nuclear magnetic resonance (NMR) measurements on this peptide in a membrane environment show an α-helix conformation for the core residues,18,19 whereas the synthesized amylin fibrils exhibit parallel β-sheet layers with the formation of steric zipper, a structural moiety common to all aggregating amyloid polypeptides.20,21
Recent studies have found that the presence of divalent copper ions reduces the cellular toxicity of hIAPP.22 This is thought to be related to the inhibition of amylin aggregation by Cu(II) ions.22–24 Copper is involved in the pathological mechanisms of certain neurodegenerative diseases, and its role has been extensively studied for amyloidogenic proteins in Alzheimer's, Parkinson's, and prion-protein diseases.25 However, the properties of amylin–copper interactions, the effect of amylin conformation on its interaction with Cu(II), and the conformational changes of amylin induced by Cu(II) remain relatively unknown.
Various established analytical techniques, such as fluorescence,26 surface plasmon resonance spectroscopy,27 and UV–visible spectroscopy28,29 have been employed to identify and quantitatively assess the noncovalent peptide–protein interactions. With high sensitivity, accuracy, and speed, mass spectrometry (MS) plays an important role in observing peptide–ligand structures.30 Extensive reviews indicate that MS has an increasing role in gauging the composition, conformation, binding strength, and dissociation constants of noncovalent interactions.1,31–33
Native electrospray ionization (ESI) is a widely used MS technique for studying protein–ligand interactions that offers the advantages of high sensitivity, specificity, and unprecedented accuracy for the mass of the complex.1,2,30,34 However, there is an ongoing debate on the relationship between the gas phase complex ion and the corresponding species found in the solution phase. Other concerns include the need for more physiological buffers (most studies use ammonium acetate), the interfering effect of other macromolecules in the system, and general throughput limitations related to the need for buffer exchange that takes 1 to 2 h.1,35,36 Although ESI arrays can accelerate the measurement, lengthy buffer exchange is still required. As experiments often involve titration assays to determine binding constants, a rapid method applicable to systems under conditions closer to their biological state would benefit this effort.
Laser ablation electrospray ionization (LAESI), an emerging ambient ionization method for MS, has been used for studying biological tissues and cells.37,38 Ablation by nanosecond mid-IR laser pulses results in rapid sampling of unprocessed biological systems in their natural environment.37 The ejected neutral particulates in the ablation plume are ionized by charged droplets from an electrospray. High throughput capabilities are greatly enhanced by automated sample handling in a 96-well plate format. Integration of LAESI-MS with IMS provides additional insight by revealing conformational information,39 a potential benefit for studying noncovalent complexes.
Native-ESI and LAESI appear to have complementary advantages. According to earlier studies,36 the optimal concentration range for native-ESI detection is between 1 and 5 μM and it exhibits a limit of detection in the picomolar range. They also recommend a protein concentration below 25 μM. In contrast, limits of detection for LAESI are in the femtomole range and concentrations ranging from submicromolar to millimolar can be interrogated. Compared to native-ESI, there is a throughput advantage for LAESI stemming from the ability to move from sample to sample within seconds without having to change the electrospray. In comparison, native-ESI requires one to establish a stable electrospray for every sample.
An important advantage of native-ESI compared to LAESI is the simplicity of instrumentation. A conventional ESI source can be easily operated in native-ESI mode, whereas LAESI requires retrofitting the system with a different ion source.
In a recent study, the efficacy of DESI was demonstrated for the study of protein ligand interactions in a high throughput fashion.40 In this arrangement, the ligand was supplied through the electrospray solution, which enabled the high throughput screening of ligands through segmented flow. This method achieved a throughput of 1 min/ligand, a significant improvement over native-ESI.
In this study, LAESI-MS is used in combination with IMS to investigate the stoichiometric ratio, copper oxidation state, and the conformation and dissociation constant of human amylin–copper(II) complexes. In addition, the domain of the possible coordination site for the metal ion is identified using collision-induced dissociation (CID). High throughput determination of the peptide–ligand complex properties by LAESI-IMS-MS can facilitate rapid screening of potential inhibitors of amylin association and fibril formation.
MATERIALS AND METHODS
Sample Preparation
Lyophilized C-terminal amidated synthetic human amylin (hIAPP), H18A mutated amylin (hIAPPH18A), and hIAPP fragment 8-37 (hIAPP8-37) (American Peptide Co., Sunnyvale, CA) were prepared as 500 μM stock solutions by dissolving them in hexafluoroisopropanol (HFIP) (Sigma-Aldrich, St. Louis, MO). The stock solutions were incubated overnight to completely dissolve any preformed amylin oligomers and aggregates, in accordance to a previously described protocol.41,42 Prior to an experiment, the required amount of amylin was aliquoted, and the HFIP solvent was evaporated with a gentle stream of nitrogen gas. The peptide was then reconstituted in deionized water with or without freshly dissolved 40 μM copper(II) chloride (Sigma-Aldrich, St. Louis, MO) to yield a final peptide monomer concentration of 20 μM. Samples were incubated at room temperature for 8 h prior to analysis by mass spectrometry. To determine the binding stoichiometry and dissociation constant of hIAPP–Cu(II) complex, copper(II) chloride solutions at different concentrations were mixed with 20 μM hIAPP.
Standard ubiquitin from bovine erythrocytes was purchased from Sigma-Aldrich (St. Louis, MO). It was dissolved and diluted in 50% methanol for further use. For matrix-assisted laser desorption ionization (MADI), 6-aza-2-thiothymine (Alfa Aesar, Ward Hill, MA) was used as a matrix. A saturated solution (10 mg/mL) was prepared in 70% acetonitrile prior to use.
LAESI-IMS-MS
The instrumentation for LAESI-IMS-MS has been described previously.39 Briefly, a 5 ns Q-switched mid-IR laser (IR Opolette 100, Opotek Inc., Carlsbad, CA) beam at 2940 nm wavelength and ~1 mJ/pulse energy was focused onto the sample through a plano-convex lens (Infrared Optical Products, Farmingdale, NY) to achieve ablation of the liquid samples. The neutral droplets in the ablation plume were ionized by multiply charged droplets from an electrospray. A spray solution of 50% methanol (v/v) with 0.1% acetic acid was supplied by a syringe pump at a flow rate of 400 nL/min. To produce an electrospray, 3300 V was applied to a 320 μm OD and 50 μm ID tapered tip metal emitter (MT320-50-5-5, New Objective, Woburn, MA). The pH of the ablated sample solution was 6.9 before and during the analysis. This value is close to biological conditions. The electrospray solution does not interact with the sample until the droplets from the sample ablation coalesce with the electrospray droplets. This is a very fast process on the time scale of milliseconds leaving little time for changes in the native-like structure.
The LAESI-generated ions then entered a quadrupole time-of-flight mass spectrometer (Synapt G2 S, Waters Co., Milford, MA) for detection in a mode optimized to exhibit a mass resolution of m/Δm ≈ 13 000 in the m/z range of the amylin ions. The instrument incorporated a traveling wave (T-wave) ion mobility separation (IMS) system, in which the gas phase ions became separated through the interaction with the nitrogen buffer gas as they were moved by an advancing sinusoidal potential distribution. The collision cross section (CCS), Ω, resolution of the traveling wave IMS system in the Ω range of the amylin ions was Ω/ΔΩ ≈ 30.
For high-throughput analysis, a commercial LAESI source (DP-1000, Protea Biosciences, Morgantown, WV) was integrated with the IMS-MS system. Small aliquots of sample (1 to 5 μL) were deposited into the wells of a 96-well plate. The analysis time for each sample was less than 10 s, yielding ~2 min in total for the analysis of ten samples.
For structural studies, collision-induced dissociation (CID) was implemented. The collision energy was ramped from 25 to 40 eV for the selected hIAPP ions in the trap ion guide, and after IMS, collision energies from 40 to 60 eV were applied for the fragments in the transfer ion guide.
To explore the utility of MALDI-MS for the detection of the human amylin copper complex, experiments were carried out on a MALDI LTQ Orbitrap XL mass spectrometer (Thermo Scientific, San Jose, CA) at 6 μJ laser energy, the threshold for the observation of the complex.
Data Processing
The mass spectra were collected and processed using the MassLynx V4.1 software (Waters, Milford, MA). The spectra were normalized to the highest ion count and smoothed using Savitzky-Golay filtering. The theoretical isotope distributions of hIAPP and its copper adducts were calculated using ProteinProspector V5.12.2 (University of California, San Francisco, CA) and the Molecular Weight Calculator (Pacific Northwest National Laboratory, Richland, WA) software. The tandem MS spectra were analyzed by comparing the fragment peaks with the fragmentation pattern generated by the MS-Product calculator in ProteinProspector. The DriftScope V2.4 (Waters, Milford, MA) software was used to review the drift time (DT) of isobaric ions from the sample.
The protocol for CCS calculation in a T-wave IMS-MS system has been reported previously.39,43 Our CCS calibration curve was established on the basis of a ubiquitin (8565 Da) peptide standard. The CCS values (Ω) for the analyte ions were then interpolated from the calibration curve using the measured drift time. Standard Ω values of ubiquitin were obtained from the Clemmer Group Cross Section Database (http://www.indiana.edu/~clemmer, last accessed: 02/09/2015).
RESULTS AND DISCUSSION
hIAPP–Cu Complex and Copper Oxidation State Probed by LAESI-IMS-MS
In 20 μM hIAPP solution, the formation of multiply charged protonated, sodiated, and potassiated ions were detected by LAESI-IMS-MS, with maximum ion intensities for m/z 788.772, m/z 976.233, and m/z 1301.297 ions, corresponding to amylin in the form of [hIAPP + 4H + K]5+, [hIAPP + 4H]4+, and [hIAPP + 3H]3+, respectively (see Figure S1a). The isotope distribution patterns in the insets reveal the charge states for the hIAPP-related ions. The deconvoluted mass of the observed triply charged peaks is 3900.867, which is 4 mDa or 0.0001% (1 ppm) apart from the calculated 3900.863 monoisotopic mass of hIAPP with a disulfide bridge between the cysteines at positions two and seven (see Figure S2a).
Aliquots of 20 μM hIAPP were treated with 40 μM copper(II) chloride. In the LAESI mass spectrum, we observed the quintuply, quadruply, and triply charged hIAPP–Cu adducts (see Figure S1b). A single copper(II) ion was incorporated into the adducts. The mass difference between the detected monoisotopic peaks of hIAPP (e.g., m/z 976.233 at z = 4) and its copper adducts (e.g., m/z 991.459 at z = 4) correspond to Δm/z 60.9 indicating the displacement of two protons by 63Cu2+. The 65Cu2+ addition was not identified separately because the mass difference between 63Cu and 65Cu was twice of that between the 12C and 13C isotopes resulting in interference. This can be observed as a shift between the isotope distributions for hIAPP and its copper adduct in Figure 1. Copper complexes can also be found using LAESI-MS without IMS, but in the DT vs m/z plots, better signal-to-noise ratios can be achieved.
Figure 1.

Isotope peak distributions of (a) [hIAPP + 4H]4+, (b) [hIAPP + Cu(II) + 2H]4+, (c) [hIAPP + 3H]3+, and (d) [hIAPP + Cu(II) + H]3+. The isotope pattern of copper adducts is shifted by Δm/z 60.9 for both charge states. The acquisition time for each spectrum is 4 s.
The isotope peak distributions of the hIAPP–Cu complex ions also assist in determining the copper oxidation state. Depending on the charge carried by the copper ions, Cu(I) or Cu(II), it can produce quadruply charged amylin ions in the form of [hIAPP + Cu(I) + 3H]4+ or [hIAPP + Cu(II) + 2H]4+, respectively. We calculated the isotope peak distribution patterns for the quadruply and triply charged ions of the two copper oxidation states in the complex, hIAPP–Cu(I) and hIAPP–Cu(II), and compared them with the measured peaks (see Table S1). The much smaller Δm/z error for hIAPP–Cu(II) (Δm/z < 17 mDa for the 4+ hIAPP–Cu(II) compared to >235 mDa for the 4+ hIAPP–Cu(I), Δm/z < 8 mDa for the 3+ hIAPP–Cu(II) versus >334 mDa for the 3+ hIAPP–Cu(I)) suggested that Cu(II), rather than Cu(I), was present in the complex (see Table S1). The result that Cu(II) was not reduced to Cu(I) in the binding interaction was consistent with a previous study of amylin copper interactions22 and the reported oxidation state of copper binding to β-amyloid peptides.44
In addition, we also analyzed human amylin incubated with copper(II) using MALDI-MS (see Figure S6). In the amylin copper complex, the copper was detected as Cu(I), indicating that in the MALDI measurement the Cu(II) ions were reduced. This is in agreement with a previous study.45
Binding Stoichiometry and Dissociation Constant
To determine the binding stoichiometry of hIAPP–Cu(II) complexes as a function of Cu(II) concentration, we incubated 20 μM hIAPP with copper(II) chloride solutions in the 5–400 μM range. After 8 h of incubation, LAESI mass spectra were recorded for each sample. The spectra of hIAPP–Cu(II) complex for all charge states (see Figure S1b) indicated a binding stoichiometry of 1:1 between the peptide and the ligand for all of the concentrations, even for a 20-fold excess of Cu(II) ions.
The dissociation constant, Kd, for the ML peptide–ligand complex is Kd = [M][L]/[ML], where M is the peptide and L is the free ligand. Since amylin binds one Cu(II) ion, the saturation fraction, Y, of the peptide–ligand complex is described as Y = [ML]/([M] + [ML]) = [L]/(Kd + [L]).46 To determine Kd, we plotted the saturation fraction of the hIAPP–Cu(II) complex as a function of the copper(II) concentration, [L] (see Figure 2). In the low ligand concentration limit, [L] ᄑ Kd, the saturation fraction is simplified to Y = [L]/Kd, and Kd can be easily determined. Fitting the low [L] data points yielded Kd = 22.1 ± 0.7 μM for the 3+ hIAPP–Cu(II) and Kd = 41.3 ± 1.5 μM for the 4+ hIAPP–Cu(II). At Cu(II) concentrations above ~60 μM, the measured saturation fractions (Y = ~0.55 for the 3+ hIAPP–Cu(II) and Y = ~0.48 for the 4+ hIAPP–Cu(II)) are significantly below the values predicted by the Y = [L]/(Kd + [L]) equation. This can be interpreted as an inhibitory effect of the Cu(II) ions for the formation of the complex.
Figure 2.

Saturation fraction Y, defined as the ratio of hIAPP–Cu(II) to the total hIAPP ions, increases with the concentration of Cu(II) ions. The inset shows the determination of the dissociation constant, Kd, in the 5 to 30 μM copper concentration range using equation Y = [L]/(Kd + [L]). The values for Kd are determined as 22.1 ± 0.7 μM for the 3+ hIAPP–Cu(II) and 41.3 ± 1.5 μM for the 4+ hIAPP–Cu(II). At Cu(II) concentrations above ~60 μM, the saturation fraction is much lower than the values predicted by the equation.
Complex Coordination Site
To investigate the binding site between the metal and the peptide ions, LAESI-MS/MS was performed using CID of both the quadruply charged hIAPP (m/z 976.233) and hIAPP–Cu(II) (m/z 991.459). Figure 3a,b displays the tandem MS spectra of the quadruply charged hIAPP and hIAPP–Cu(II), respectively. The fragments, b17, b25, and b26, were identified by comparing their measured m/z values to the predicted fragmentation patterns generated by the MS-Product program in ProteinProspector. A copper adduct was detected for fragment b25 and the subsequent b26, but it was absent for b17, suggesting a possible copper coordination site within residues 18–25 (–HSSNNFGA–) for the gas phase 4+ hIAPP–Cu(II) complex.
Figure 3.

Zoomed tandem mass spectrum of [hIAPP + 4H]4+ at m/z 976.233 (a) is compared with that of [hIAPP + Cu(II) + 2H]4+ at m/z 991.459 (b). The remaining parent ions are shown in the insets. The copper adduct is observed for the fragments b25 and b26, but it is absent for b17.
Two modified hIAPP samples were also examined using LAESI-MS and tandem MS to further explore the copper binding site. Histidine is an amino acid with high copper binding affinity,47 and it is considered to be a key residue in amylin folding and aggregation. Given that Cu(II) inhibits amylin aggregation,22 it is possible that Cu interacts with histidine to impose this effect. To test this idea, a human amylin mutant with histidine 18 substituted by alanine (hIAPPH18A) (see Figure S2b) was synthesized and incubated with copper(II) chloride to assess if residue18 was essential for the binding reaction. Both quadruply and triply charged hIAPPH18A–Cu(II) ions were observed in the collected LAESI mass spectrum (see Figure S3b); however, the relative peak abundance of the copper adduct was much lower as compared to that in the original hIAPP mass spectrum (see Figure S3a). To evaluate the copper binding activity, we calculated the peak abundance ratio of the amylin copper adducts to the total intensity of amylin related ions. For the original hIAPP, the ratio for quadruply charged ions (R4+) is 0.38 ± 0.08, and the ratio for triply charged ions (R3+) is 0.47 ± 0.06. For the alanine substituted hIAPPH18A, the R4+ is 0.13 ± 0.04 and the R3+ is 0.13 ± 0.04, both of which are significantly lower than the original values. This indicates that the substitution of histidine 18 by alanine weakens the binding between copper and amylin, and histidine 18 is likely to play an important role in this peptide–ligand interaction.
Additionally, to probe the role of the amylin segment containing the intramolecular disulfide bond, the complexation of the fragment 8-37 (hIAPP8-37) was studied by incubating it with Cu(II) ions. The obtained LAESI mass spectra indicated the presence of the copper adducts (see Figure S3c). The intensity ratio of the copper adducts to the sum of the amylin-related ions is 0.48 ± 0.06 for quadruply charged ions, slightly higher than the R4+ of hIAPP (0.38 ± 0.08). The ratio for triply charged hIAPP8-37 is 0.25 ± 0.05, somewhat lower than that of wild type hIAPP (0.47 ± 0.06). Also, the forms of cationization for hIAPP8-37–Cu(II) is more complex. Instead of being dominated by the protonated ions, the mass spectra reveal strong sodiated, potassiated peaks and a 4+ hIAPP8-37–2Cu(II) adduct. The altered intensity ratios of the copper adduct to the sum of the amylin-related ions suggest that the seven-residue segment, hIAPP1–7, with the disulfide bond affects the conformation of the amylin structure and exerts an effect on the binding between the copper and amylin ion.
In the tandem mass spectrum of the triply charged hIAPP8-37–Cu(II), we identified copper adducts for fragments b18 and possibly b15 (see Figure S4), indicating that in the triply charged complex the copper binds to positions before residue 16 of hIAPP8-37 (corresponding to residue 23 of hIAPP). Combining this finding with the previous results for the 4+ hIAPP–Cu(II) ion and assuming that the copper coordination site is conserved, it further narrows the possible binding site to residues 18–22 (–HSSNN–). Interestingly, Kallay et al. also reported the tetrapeptide fragment –SSNN– as the shortest sequence contributing to the copper(II) binding in rat amylin solutions.28 However, using ESI-MS, Kim and Kim suggested that residues 22–26 (–NFGAI–) form the binding site for the 3+ hIAPP–Cu(II) gas phase complex.48 Studies using alternative methods are needed to further clarify the copper coordination site in hIAPP ions. However, both potential coordination sites for hIAPP fall into the “core mutation region” (residue 18–29) of hIAPP, the six amino acids different from that in the rat amylin sequence. The human form of IAPP aggregates while rat amylin does not. The assigned copper binding site suggests that the copper's interaction with the “core mutation region” results in the inhibition of hIAPP aggregation. The initial seven-residue segment, hIAPP1–7, can modulate binding by affecting the conformation of the binding domain.
Favorable hIAPP Ion Conformations for Cu(II) Binding
Amylin fibril formation is conformation dependent.17,49,50 We probed the conformational states of gas phase ions of hIAPP–Cu(II) and elucidated their CCSs using IMS. The measured CCS values for the 3+ and 4+ hIAPP ions were compared to replica exchange molecular dynamics (REMD) simulations in the literature.17 The synthesized hIAPP at pH 6.9 (the measured pH of the hIAPP solution) possesses net 3+ charges in the liquid phase. The hIAPP and its Cu(II) complex detected in the gas phase showed both the 3+ and 4+ charge states.
The collision cross section values measured for each charge state of the hIAPP and hIAPP–Cu(II) are listed in Tables 1 and 2. The triply charged hIAPP ions exhibited two conformers with CCSs of 611.2 and 646.5 Å2. The quadruply protonated hIAPP species were present in two conformations with CCSs of 594.2 and 747.8 Å2, respectively. The measured CCS values are within a range of 0.3% to 3.5% of the previous experimental and simulation results.17 According to the REMD simulations, the 3+ conformer with CCS of 611.2 Å2 corresponds to a solvent-free structure, whereas the one with 646.5 Å2 can be assigned as a β-sheet rich compact dehydrated solution structure. “Solvent-free structure” refers to the simulated structure under vacuum condition with no solvent molecules included. “Solution phase structure” describes the conformation obtained by including water molecules in the simulation, whereas “dehydrated solution structure” resulted from the solution phase structure by removing all the water molecules followed by further energy minimization. The latter was needed to access local energy minima for structures produced by sudden evaporation of the solvent molecules.17 The 594.2 Å2 CCS of the 4+ conformer matches well with that of the calculated solvent-free structure, whereas the one with CCS of 747.8 Å2 correlates well with the calculated CCS of a long β-hairpin (dehydrated solution) structure.17
Table 1.
Measured Collision Cross Sections (CCSmeas) of the Triply and Quadruply Charged hIAPP Conformers Are Compared to Experimental (CCSref-exp) and Simulation (CCSref-sim) Values from the Literature17
| m/z | charge state | CCSmeas (Å2) | CCSref-exp (Å2) | ΔCCS (%) | CCSref-sim (Å2) | ΔCCS (%) | |
|---|---|---|---|---|---|---|---|
| [hIAPP + 3H]3+ | 1301.297 | 3+ | 611.2 ± 4.8 | 598 | 2.2 | 604 ± 14 | 1.2 |
| [hIAPP + 3H]3+ | 1301.297 | 646.5 ± 4.8 | 630 | 2.6 | 635 ± 11 | 1.8 | |
| [hIAPP + 4H]4+ | 976.233 | 4+ | 594.2 ± 6.7 | 616 | 3.5 | 615 ± 25 | 3.4 |
| [hIAPP + 3H + K]4+ | 985.712 | 683.8 ± 6.6 | |||||
| [hIAPP + 4H]4+ | 976.233 | 747.8 ± 6.5 | 770 | 2.9 | 750 ± 10 | 0.3 |
Table 2.
Measured Collision Cross Sections (CCSmeas) of the Triply and Quadruply Charged hIAPP–Cu(II) Complex Conformers
| m/z | charge state | CCSmeas (Å2) | |
|---|---|---|---|
| [hIAPP + Cu(II) + H]3+ | 1321.596 | 3+ | 611.2 ± 4.8 |
| [hIAPP + Cu(II) + H]3+ | 1331.596 | 646.4 ± 4.7 | |
| [hIAPP + Cu(II) + 2H]4+ | 991.459 | 4+ | 594.1 ± 6.7 |
| [hIAPP + Cu(II) + 2H]4+ | 991.459 | 747.7 ± 6.5 | |
| [hIAPP + Cu(II) + H + K]4+ | 1000.952 | 594.0 ± 6.7 | |
| [hIAPP + Cu(II) + H + K]4+ | 1000.952 | 683.6 ± 6.6 | |
| [hIAPP + Cu(II) + H + K]4+ | 1000.952 | 747.6 ± 6.5 |
For triply charged hIAPP, the deconvoluted DT distribution (top panel of Figure 4a) shows that the dehydrated solution phase conformer (CCS of 646.5 Å2) accounts for 68% and the solvent-free conformer (CCS of 611.2 Å2) constitutes 32% of the ions. In earlier ion funnel based experiments, the lowest energy solvent-free conformation of hIAPP is found to be the most stable.17 The higher amount of the solution phase conformer produced by LAESI suggests that the laser ablation process did not significantly increase its internal energy.51 This is consistent with earlier results indicating that mid-IR laser irradiation does not affect the internal energies of the ablated species, as the ions generated by ESI and LAESI are indistinguishable. In contrast, among the quadruply charged hIAPP ions, 98% exhibit the solvent-free structure (CCS of 594.2 Å2) and only 2% displays the extended solution phase conformation (CCS of 747.8 Å2) (top panel of Figure 4b). This could be due to the 26% larger CCS of the extended solution conformer, resulting in reduced stability leading to refolding into the more stable solvent-free conformation.
Figure 4.

Drift time (DT) distribution of hIAPP and hIAPP–Cu(II) ions reveal the differentiated conformers for the triply charged and the quadruply charged states. (a) The spectrum of [hIAPP + 3H]3+ was deconvoluted using two Gaussian functions. The smaller shoulder peak corresponds to a conformation with CCS of 611 Å2, whereas the larger peak matches a structure with CCS of 647 Å2. Likewise, the spectrum of [hIAPP + Cu(II) + H]3+ has two peaks with nearly identical CCS values as those of [hIAPP + 3H]3+. (b–d) The quadruply charged hIAPP is present as (b) protonated, (c) potassiated, and (d) sodiated ions. The DT distribution for [hIAPP + 4H]4+ and [hIAPP + Cu(II) + 2H]4+ reveal two distinct conformations with CCSs of 594 and 748 Å2. The spectrum of [hIAPP + 3H + K]4+ displays a major peak with CCS of 748 Å2 and a minor peak with CCS of ~684 Å2. The spectrum of [hIAPP + Cu(II) + H + K]4+ reveals three peaks, with a dominant one at CCS of 748 Å2, a small one at CCS of 684 Å2, and an almost negligible peak at CCS of 594 Å2. hIAPP was detected with the sodiated ions in the form of [hIAPP + 3H + Na]4+. Using Gaussian functions, the DT distribution spectrum was deconvoluted into two peaks with CCSs of 594 and 748 Å2.
To explore the favored conformation for copper binding, the DT distributions were compared for hIAPP ions without and with Cu(II) complexation. In the bottom panel of Figure 4a, the two conformers of the triply charged hIAPP–Cu(II) ions exhibit nearly identical CCS values with the 3+ hIAPP ions (top panel). This indicates that the conformations of hIAPP ions are not altered significantly by the presence of copper. The abundance of the conformer with CCS of 611.2 Å2, however, increased from 32% (top panel) to 46% (bottom panel) upon copper coordination. A possible explanation of this result is that the copper ion prefers to bind with the slightly more compact solvent-free structure (CCS of 611.2 Å2). Alternatively, the conformation of the binding site in the more compact structure is more favorable for the attachment of copper.
The DT distributions for the quadruply charged hIAPP and its copper adduct (see Figure 4b,c,d) are presented for three different forms of cationization. For the protonated hIAPP, the long β-hairpin conformer (CCS of 747.8 Å2) was enhanced from 2% (top panel) to 11% (bottom panel), and the solvent-free conformer (CCS of 594.2 Å2) was reduced from 98% to 89% upon the copper binding. The preferential binding of copper to the extended β-hairpin conformer might disrupt amylin aggregation and the formation of fibrils rich in β sheets. According to the simulated structure of the β-hairpin monomer, the proposed copper binding site, i.e., the –HSSNN– segment, is located at the loop of the β-hairpin. Visualization of this structure in the 3+ charge state indicates a negatively charged pocket that can potentially accommodate the copper ion (see Figure S5).
In addition to the dominant protonated hIAPP, the potassiated hIAPP also reveals conformer abundance changes upon copper addition. The corresponding CCS values are almost identical to those of the protonated ions (see Figure 4c), but the conformations cannot be verified without additional simulation studies. A new conformer with CCS of 683.6 Å2 was also observed for the potassiated hIAPP–Cu(II) ions. Upon copper coordination, the abundance of the potassiated amylin conformer with CCS of 747.8 Å2 decreased from 92% to 77%, whereas the contribution of the conformers with CCSs of 594.0 and 683.6 Å2 increased. For the sodiated hIAPP, two conformers with CCSs of 594.1 and 747.8 Å2 were found (see Figure 4d); however, no copper adducts were detected.
Dimer Formation and Its Inhibition by Copper
The human form of IAPP can aggregate into oligomers that eventually form amyloid fibrils.52,53 We observed [2hIAPP + 5]5+ ions at m/z 1562 resulting from the dimerization of amylin incubated for at least 3 h. The abundance of the dimer peak continued to increase during a 24 h period (top panel in Figure 5a). When incubated with copper(II) chloride, no amylin dimer peak was detected (bottom panel in Figure 5a) indicating inhibited aggregation due to the presence of copper.
Figure 5.

(a) Without copper(II) ions in the solution, the hIAPP dimer peak [2hIAPP + 5H]5+ at m/z 1562 is present in the spectrum. The dimer is absent when hIAPP is incubated with copper(II) chloride. The abundance of hIAPP dimer increases with time (3, 12, and 24 h). (b) The DT distribution of the dimer reveals a CCS of 1140 Å2.
The DT distribution of the detected hIAPP dimer (see Figure 5b) suggests a conformer with collision cross section of 1139.6 Å2. Assuming the oligomerization follows the isotropic self-assembly pathway, the CCS of neutral oligomers, Ωn, can be estimated from the monomer CCS, Ωmono, using the equation: Ωn = Ωmono × n2/3, where n is the number of monomers in the oligomer.49,54 It is shown that adding charges to the monomers and oligomers only results in a small increment in the CCS values.54 On the basis of the measured CCSs of the three 4+ monomers, the CCSs of the corresponding hIAPP dimer were estimated to be 943.2, 1085.5, and 1187.1 Å2 (see Table S2). The measured CCS of 1139.6 Å2 of the detected 5+ dimer is closest to the estimated CCS of 1187.1 Å2 for the dimer formed from β-hairpin monomers. Although the β-hairpin structure has a negatively charged pocket, the charged groups in the solution phase are shielded by counterions. This can facilitate the assembly of dimers. This result is consistent with the hypothesis that amylin fibrils are constructed through the direct assembly of β-hairpin monomers. The absence of dimer peaks in hIAPP in the presence of Cu(II) indicates that copper bound to the extended β-hairpin conformer disrupts amylin oligomerization and consequently fibril formation.
CONCLUSIONS
In this paper, we have demonstrated a rapid, high-throughput analysis of amylin–copper complex formation and amylin dimerization using LAESI-IMS-MS. The binding stoichiometry, complex dissociation constant, and the oxidation state of the copper were established for the amylin–copper interaction. The copper coordination site has been tentatively assigned to residues 18–22 (–HSSNN–) of hIAPP based on the CID of the gas phase complex ions. With IMS, the conformational states of hIAPP were explored for each charge state and the measured CCS values showed good agreement with previous results. We demonstrated that copper inhibits the formation of amylin dimers thereby disrupting fibril formation. This effect probably involves a complex between copper and the extended β-hairpin conformer of amylin. The LAESI-IMS-MS approach described here enables the sensitive and high-throughput screening of potential inhibitors for amylin oligomerization. More generally, this rapid technique opens the door for high-throughput screening of potential inhibitors of amyloid forming protein aggregation.
To verify the copper coordination site in the hIAPP–Cu(II) adduct, alternative fragmentation methods, e.g., electron capture dissociation, can be implemented to more systematically fragment the complexions. Additional simulation studies on the hIAPP–Cu(II) complex ion might further elucidate the location of the binding site and improve the assessment of the favorable conformation for copper binding. To illuminate aggregation and inhibition pathways, it would also be beneficial to detect higher-order amylin oligomers.
Supplementary Material
ACKNOWLEDGMENTS
Support for the LAESI-IMS-MS work was provided by Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, Office of Science of the U.S. Department of Energy through Grant DE-FG02-01ER15129 to A.V. One of the authors acknowledges financial support from the National Institutes of Health (Grant R01 DK091845; A.M.J.) and the Luther Rice Undergraduate Research Fellowship of the George Washington University (to E.H.). The simulated structure of the amylin ion was kindly provided by Professor Michael T. Bowers of the Department of Chemistry and Biochemistry at the University of California, Santa Barbara.
Footnotes
Supporting Information The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.5b02217.
The authors declare no competing financial interest.
REFERENCES
- (1).Kitova EN, El-Hawiet A, Schnier PD, Klassen JS. J. Am. Soc. Mass Spectrom. 2012;23:431–441. doi: 10.1007/s13361-011-0311-9. [DOI] [PubMed] [Google Scholar]
- (2).Potier N, Rogniaux H, Chevreux G, Van Dorsselaer A. Methods Enzymol. 2005;402:361–389. doi: 10.1016/S0076-6879(05)02011-2. [DOI] [PubMed] [Google Scholar]
- (3).Loo JA. Mass Spectrom. Rev. 1997;16:1–23. doi: 10.1002/(SICI)1098-2787(1997)16:1<1::AID-MAS1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- (4).Cooper GJS, Day AJ, Willis AC, Roberts AN, Reid KBM, Leighton B. Biochim. Biophys. Acta, Mol. Cell Res. 1989;1014:247–258. doi: 10.1016/0167-4889(89)90220-6. [DOI] [PubMed] [Google Scholar]
- (5).Gebre-Medhin S, Mulder H, Pekny M, Westermark G, Tornell J, Westermark P, Sundler F, Ahren B, Betsholtz C. Biochem. Biophys. Res. Commun. 1998;250:271–277. doi: 10.1006/bbrc.1998.9308. [DOI] [PubMed] [Google Scholar]
- (6).Ludvik B, KautzkyWiller A, Prager R, Thomaseth K, Pacini G. Diabet. Med. 1997;14:S9–S13. doi: 10.1002/(sici)1096-9136(199706)14:2+<s9::aid-dia397>3.3.co;2-4. [DOI] [PubMed] [Google Scholar]
- (7).Reidelberger RD, Arnelo U, Granqvist L, Permert J. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001;280:R605–R611. doi: 10.1152/ajpregu.2001.280.3.R605. [DOI] [PubMed] [Google Scholar]
- (8).Cooper GJS, Willis AC, Clark A, Turner RC, Sim RB, Reid KBM. Proc. Natl. Acad. Sci. U. S. A. 1987;84:8628–8632. doi: 10.1073/pnas.84.23.8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Westermark P, Wernstedt C, Wilander E, Hayden DW, O'Brien TD, Johnson KH. Proc. Natl. Acad. Sci. U. S. A. 1987;84:3881–3885. doi: 10.1073/pnas.84.11.3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Westermark P, Wernstedt C, Wilander E, Sletten K. Biochem. Biophys. Res. Commun. 1986;140:827–831. doi: 10.1016/0006-291x(86)90708-4. [DOI] [PubMed] [Google Scholar]
- (11).Cho W-J, Jena BP, Jeremic AM. Methods Cell Biol. 2008;90:267–286. doi: 10.1016/S0091-679X(08)00813-3. [DOI] [PubMed] [Google Scholar]
- (12).Cho W-J, Trikha S, Jeremic AM. J. Mol. Biol. 2009;393:765–775. doi: 10.1016/j.jmb.2009.08.055. [DOI] [PubMed] [Google Scholar]
- (13).Westermark P, Engstrom U, Johnson KH, Westermark GT, Betsholtz C. Proc. Natl. Acad. Sci. U. S. A. 1990;87:5036–5040. doi: 10.1073/pnas.87.13.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Westermark P, Andersson A, Westermark GT. Physiol. Rev. 2011;91:795–826. doi: 10.1152/physrev.00042.2009. [DOI] [PubMed] [Google Scholar]
- (15).Lorenzo A, Yankner BA. In: The Neurobiology of Alzheimer's Disease. Wurtman RJ, Corkin S, Growdon JH, Nitsch RM, editors. New York Academy of Sciences; New York: 1996. pp. 89–95. [Google Scholar]
- (16).Hoppener JWM, Ahren B, Lips CJM. N. Engl. J. Med. 2000;343:411–419. doi: 10.1056/NEJM200008103430607. [DOI] [PubMed] [Google Scholar]
- (17).Dupuis NF, Wu C, Shea JE, Bowers MT. J. Am. Chem. Soc. 2009;131:18283–18292. doi: 10.1021/ja903814q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Nanga RPR, Brender JR, Vivekanandan S, Ramamoorthy A. Biochim. Biophys. Acta, Biomembr. 2011;1808:2337–2342. doi: 10.1016/j.bbamem.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Patil SM, Xu S, Sheftic SR, Alexandrescu AT. J. Biol. Chem. 2009;284:11982–11991. doi: 10.1074/jbc.M809085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJW, McFarlane HT, Madsen AO, Riekel C, Eisenberg D. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- (21).Luca S, Yau WM, Leapman R, Tycko R. Biochemistry. 2007;46:13505–13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lee EC, Ha E, Singh S, Legesse L, Ahmad S, Karnaukhova E, Donaldson RP, Jeremic AM. Phys. Chem. Chem. Phys. 2013;15:12558–12571. doi: 10.1039/c3cp44542a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mirhashemi SM, Shahabaddin ME. Pak. J. Biol. Sci. 2011;14:288–292. doi: 10.3923/pjbs.2011.288.292. [DOI] [PubMed] [Google Scholar]
- (24).Ward B, Walker K, Exley C. J. Inorg. Biochem. 2008;102:371–375. doi: 10.1016/j.jinorgbio.2007.09.010. [DOI] [PubMed] [Google Scholar]
- (25).Miller Y, Ma B, Nussinov R. Coord. Chem. Rev. 2012;256:2245–2252. [Google Scholar]
- (26).Maiti NC, Jiang D, Wain AJ, Patel S, Dinh KL, Zhou F. J. Phys. Chem. B. 2008;112:8406–8411. doi: 10.1021/jp802038p. [DOI] [PubMed] [Google Scholar]
- (27).Jecklin MC, Schauer S, Dumelin CE, Zenobi R. J. Mol. Recognit. 2009;22:319–329. doi: 10.1002/jmr.951. [DOI] [PubMed] [Google Scholar]
- (28).Kallay C, David A, Timari S, Nagy EM, Sanna D, Garribba E, Micera G, De Bona P, Pappalardo G, Rizzarelli E, Sovago I. Dalton Transactions. 2011;40:9711–9721. doi: 10.1039/c1dt10835b. [DOI] [PubMed] [Google Scholar]
- (29).Tougu V, Karafin A, Palumaa P. J. Neurochem. 2008;104:1249–1259. doi: 10.1111/j.1471-4159.2007.05061.x. [DOI] [PubMed] [Google Scholar]
- (30).Robinson CV, Chung EW, Kragelund BB, Knudsen J, Aplin RT, Poulsen FM, Dobson CM. J. Am. Chem. Soc. 1996;118:8646–8653. [Google Scholar]
- (31).Daniel JM, Friess SD, Rajagopalan S, Wendt S, Zenobi R. Int. J. Mass Spectrom. 2002;216:1–27. [Google Scholar]
- (32).Schmidt C, Robinson CV. FEBS J. 2014;281:1950–1964. doi: 10.1111/febs.12707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Erba EB, Zenobi R. Annu. Rep. Prog. Chem., Sect. C: Phys. Chem. 2011;107:199–228. [Google Scholar]
- (34).Di Marco VB, Bombi GG. Mass Spectrom. Rev. 2006;25:347–379. doi: 10.1002/mas.20070. [DOI] [PubMed] [Google Scholar]
- (35).Lin H, Kitova EN, Klassen JS. Anal. Chem. 2013;85:8919–8922. doi: 10.1021/ac401936x. [DOI] [PubMed] [Google Scholar]
- (36).Rosati S, Yang Y, Barendregt A, Heck AJR. Nat. Protoc. 2014;9:967–976. doi: 10.1038/nprot.2014.057. [DOI] [PubMed] [Google Scholar]
- (37).Nemes P, Vertes A. Anal. Chem. 2007;79:8098–8106. doi: 10.1021/ac071181r. [DOI] [PubMed] [Google Scholar]
- (38).Shrestha B, Sripadi P, Walsh CM, Razunguzwa TT, Powell MJ, Kehn-Hall K, Kashanchi F, Vertes A. Chem. Commun. 2012;48:3700–3702. doi: 10.1039/c2cc17225a. [DOI] [PubMed] [Google Scholar]
- (39).Shrestha B, Vertes A. Anal. Chem. 2014;86:4308–4315. doi: 10.1021/ac500007t. [DOI] [PubMed] [Google Scholar]
- (40).Liu PY, Zhang J, Ferguson CN, Chen H, Loo JA. Anal. Chem. 2013;85:11966–11972. doi: 10.1021/ac402906d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Trikha S, Jeremic AM. PLoS One. 2013;8:e73080. doi: 10.1371/journal.pone.0073080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Trikha S, Jeremic AM. J. Biol. Chem. 2011;286:36086–36097. doi: 10.1074/jbc.M111.240762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ruotolo BT, Benesch JLP, Sandercock AM, Hyung S-J, Robinson CV. Nat. Protoc. 2008;3:1139–1152. doi: 10.1038/nprot.2008.78. [DOI] [PubMed] [Google Scholar]
- (44).Jiang D, Men L, Wang J, Zhang Y, Chickenyen S, Wang Y, Zhou F. Biochemistry. 2007;46:9270–9282. doi: 10.1021/bi700508n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zhang J, Frankevich V, Knochenmuss R, Friess SD, Zenobi R. J. Am. Soc. Mass Spectrom. 2003;14:42–50. doi: 10.1016/S1044-0305(02)00807-3. [DOI] [PubMed] [Google Scholar]
- (46).Campbell MK, Farrell SO. Biochemistry. 4th ed. Thomson/Brooks/Cole; Belmont, CA: 2002. [Google Scholar]
- (47).Bluhm BK, Shields SJ, Bayse CA, Hall MB, Russell DH. Int. J. Mass Spectrom. 2001;204:31–46. [Google Scholar]
- (48).Kim MJ, Kim HT. Eur. Mass Spectrom. 2012;18:51–58. doi: 10.1255/ejms.1167. [DOI] [PubMed] [Google Scholar]
- (49).Dupuis NF, Wu C, Shea JE, Bowers MT. J. Am. Chem. Soc. 2011;133:7240–7243. doi: 10.1021/ja1081537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Young LM, Cao P, Raleigh DP, Ashcroft AE, Radford SE. J. Am. Chem. Soc. 2014;136:660–670. doi: 10.1021/ja406831n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Nemes P, Huang HH, Vertes A. Phys. Chem. Chem. Phys. 2012;14:2501–2507. doi: 10.1039/c2cp23411d. [DOI] [PubMed] [Google Scholar]
- (52).Jaikaran ETAS, Clark A. Biochim. Biophys. Acta, Mol. Basis Dis. 2001;1537:179–203. doi: 10.1016/s0925-4439(01)00078-3. [DOI] [PubMed] [Google Scholar]
- (53).Haataja L, Gurlo T, Huang CJ, Butler PC. Endocr. Rev. 2008;29:303–316. doi: 10.1210/er.2007-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Bleiholder C, Dupuis NF, Wyttenbach T, Bowers MT. Nat. Chem. 2011;3:172–177. doi: 10.1038/nchem.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.