

Abstract
Amyloidosis is a biological event in which proteins undergo structural transitions from soluble monomers and oligomers to insoluble fibrillar aggregates that are often toxic to cells. Exactly how amyloid proteins, such as the pancreatic hormone amylin, aggregate and kill cells is still unclear. Islet amyloid polypeptide, or amylin, is a recently discovered hormone that is stored and co-released with insulin from pancreatic islet β-cells. The pathology of type 2 diabetes mellitus (T2DM) is characterized by an excessive extracellular and intracellular accumulation of toxic amylin species, soluble oligomers and insoluble fibrils, in islets, eventually leading to β-cell loss. Obesity and elevated serum cholesterol levels are additional risk factors implicated in the development of T2DM. Because the homeostatic balance between cholesterol synthesis and uptake is lost in diabetics, and amylin aggregation is a hallmark of T2DM, this chapter focuses on the biophysical and cell biology studies exploring molecular mechanisms by which cholesterol and phospholipids modulate secondary structure, folding and aggregation of human amylin and other amyloid proteins on membranes and in cells. Amylin turnover and toxicity in pancreatic cells and the regulatory role of cholesterol in these processes are also discussed.
Keywords: Atomic force microscopy, Phospholipids, Cholesterol, Membranes, Islet amyloid, Type-2 diabetes mellitus
4.1 Amylin Biology and Function
Amylin, also known as islet amyloid polypeptide (IAPP), is a 37 amino acid hormone produced and co-secreted with insulin from pancreatic β-cells (Hoppener and Lips 2006; Clark and Nilsson 2004). IAPP is also expressed in pancreatic islet δ-cells in rat and mouse, in gastrointestinal tract of rat, mouse, cat, and human, as well as in sensory neurons of rat and mouse. In chicken, IAPP is mainly expressed in the brain and intestine and, at much lower levels, in the pancreas (Fan et al. 1994; Miyazato et al. 1991; Mulder et al. 1996). Amylin is expressed in both human and rat placenta primarily during early pregnancy (Piper et al. 2004). Placental amylin mRNA expression is highest in the third trimester of pregnancy in humans and 16 days of gestational age in rats, and gets lower as the gestation progresses (Caminos et al. 2009).
Although the exact hormonal function of amylin is still unclear, it has been proposed that amylin controls food intake and energy homeostasis (Lutz 2006, 2010). Amylin primarily regulates nutrient fluxes by acting as a potent satiation signal that reduces secretion of gastric juices and the glucagon hormone, and also reduces the rate of gastric emptying (Young and Denaro 1998). Peripheral amylin regulates satiation signal by directly binding to area postrema neurons, which are rich in amylin receptors and subsequently convey this signal to other brain areas (Lutz 2006). Interestingly, the central regulatory pathway by which other gastrointestinal peptides like cholecystokinin, glucagon-like peptide 1 and peptide YY 3–36 suppress eating widely overlaps with that of amylin (Lutz 2006; Riediger et al. 2004). In addition, amylin is also involved in adiposity signaling and, similar to leptin, in body weight regulation all through adult life (Lutz 2010). Studies with animal and human subjects showed that combinational application of leptin and amylin increases leptin responsiveness in anti-obesity treatments, which suggests the synergistic function of these hormones (Lutz 2010). Recent studies suggest that amylin-mediated regulation of energy balance is not limited to the control of nutrient flux but also involves the body’s energy expenditure (Lutz 2010). However, the exact mechanism and the physiological relevance are still under scrutiny (Lutz 2010).
As the first two trimesters of pregnancy in humans and the equivalent period in rats are considered to be highly anabolic, and placental amylin expression is highest during this time, amylin could potentially play important roles in anabolic control of food intake in both the mother and the fetus during pregnancy (Caminos et al. 2009). Amylin also plays a developmental role by contributing to the development of bone, kidney and pancreas (Wookey et al. 2006). Finally, amylin regulates the early postnatal development of hindbrain in mouse though its positive neurotropic effects (Lutz 2010). In addition to its hormonal role, amylin also imposes important paracrine and autocrine effects in islets by regulating glucagon and insulin release from α- and β-cells, respectively (Trikha and Jeremic 2013; Wagoner et al. 1993).
Amylin and insulin genes share common promoter elements, and the transcription factor PDX1 regulates glucose-stimulated secretion of both these genes (German et al. 1992). It has been reported that in rodent models, glucose stimulation of pancreatic β-cells results in parallel expression patterns of insulin and IAPP, although in experimental diabetic models of rodents this parallel expression pattern is altered (Mulder et al. 1996). Amylin is synthesized in cells as an 89-residue pre-pro-protein (Nakazato et al. 1990; Nishi et al. 1989). The 22-residue signal peptide of immature form is cleaved off in the endoplasmic reticulum (ER). Further processing of pro-IAPP, along with pro-insulin, takes place in the Golgi and the secretory vesicles in a pH-dependent manner using two endoproteases: pro-hormone convertase 2 (PC2) and prohormone convertase 1/3 (PC1/3) (Westermark et al. 2011). PC2 and PC1/3 cleave pro-IAPP after Lys10 and/ or Arg11 (Wang et al. 2001) and after Lys 50 and Arg51, respectively (Marzban et al. 2004). After PC1/3-mediated cleavage, the two C-terminal amino acid residues are then removed by carboxypeptidase E, which results in an exposed glycine residue at the C-terminus of pro-IAPP (Westermark et al. 2011). This glycine is used as a signal for C-terminal amidation; finally, a disulfide bridge is formed between Cys2 and Cys7. Both C-terminal amide and this disulfide bridge are important for full biological activity of IAPP (Westermark et al. 2011). Fully processed IAPP is a 37-residue polypeptide stored in secretory granules of pancreatic islet β-cells along with fully processed insulin. Upon physiological stimulation such as glucose spike in serum, insulin and amylin are co-secreted at a molar ratio of 20:1 (Martin 2006).
The hormone amylin, which is similar to the neuropeptide calcitonin gene-related peptide, should have specific receptors to mediate its physiological function. However, efforts to identify specific amylin receptors were futile for a long time until the identification of a family of single-domain proteins called “receptor activity-modifying proteins”, or RAMPs, which do not function as receptors by themselves (McLatchie et al. 1998). Amylin receptor utilizes a novel principle that has so far been detected only among the family of calcitonin receptors. RAMPs bind to the calcitonin receptors, and heterodimerization of RAMP with calcitonin receptor yields a unique high-affinity amylin receptor (AM-R) phenotype (Poyner et al. 2002). The three known AM-R isoforms discovered so far have been shown to exhibit distinct pharmacological and functional properties (Morfis et al. 2008). AM-R expression in different organs and tissues, particularly in the brain and in the pancreas, suggests its regulatory role in glucose homeostasis, hormone and neurotransmitter release and signaling (Martinez et al. 2000; Trikha and Jeremic 2013).
4.2 Amylin Aggregation, Islet Amyloidosis and Type 2 Diabetes Mellitus
Islet amyloid was first reported in 1901 (Opie 1901) as thick proteinaceous deposits in the pancreas of diabetics, and was initially named “islet hyalinization” because of its hyaline-like or glassy appearance. It was later renamed “amyloid”, which means “starch-like”, because islet amyloids were initially believed to be carbohydrates as they could take up dyes which are typically used to stain starch (Clark and Nilsson 2004). Despite numerous studies, the origin and nature of islet amyloid remained enigmatic for a long time (Westermark et al. 2011). Purification and characterization of amyloid aggregates from the amyloid-rich insulinoma cells and islets of human and feline origin identified amylin as the main component (Cooper et al. 1987; Westermark et al. 1986). It is now known that amylin-derived amyloid aggregates often associate with apolipoprotein E (apoE) and heparan sulfate proteoglycans (Ancsin 2003; Clark and Nilsson 2004; Hoppener et al. 2000).
Type 2 diabetes mellitus, one of the most common metabolic diseases in the world, is characterized by “insulin resistance” in the target organs, mainly muscle and liver, and by the decline in the production and secretion of insulin, loss of β-cell mass and formation of islet amyloid (Clark and Nilsson 2004; Hoppener et al. 2000). The role of islet amyloidosis in the pathogenesis of T2DM is supported by several studies showing the presence of amylin-derived amyloid plaques in over 90 % of diabetics (Clark and Nilsson 2004; Hoppener et al. 2000). While amylin has been detected in monkeys and cats, species known to develop T2DM, it is absent in rodents and mice, species which do not develop T2DM (Clark and Nilsson 2004; Hoppener et al. 2000). This is strong yet indirect evidence correlating T2DM and islet amyloidosis. Whether islet amyloidosis is a cause or a consequence of the disease is still unclear.
In vitro studies revealed that human but not rat amylin undergoes rapid aggregation in physiological buffers and that insulin, but not pro-insulin, inhibits IAPP aggregation by forming heteromolecular complexes (Clark and Nilsson 2004; Westermark et al. 1999; Kayed et al. 1999). Therefore, faulty insulin processing in diabetics could partially explain amylin aggregation in T2DM. Defective processing of pro-IAPP into IAPP is another candidate for amylin aggregation in T2DM, as N-terminal intact pro-IAPP has been identified in islet β-cells of diabetics (Clark and Nilsson 2004). In fact, pro-peptides have strong self-association properties and are capable of forming amyloid aggregates (Krampert et al. 2000). However, compared to fully processed IAPP, proIAPP has less amyloidogenicity and less toxicity (Jha et al. 2009; Krampert et al. 2000) This suggests that pro region of pro-peptide may play a protective role in amyloidogenic and toxic potentials of fully processed IAPP (Krampert et al. 2000). Increased accumulation of amyloid aggregates inside and outside the cells accounts for downstream pathological events such as calcium overload, cell membrane disruption, ER stress, mitochondrial dysfunction, defects in autophagy, oxidative stress and activation of JNK and caspase-3 death signaling pathways (Abedini and Schmidt 2013 ; Cao et al. 2013a; Costes et al. 2014; Huang et al. 2011; Konarkowska et al. 2006; Rivera et al. 2014; Zhang et al. 2003). Since the ability of IAPP to penetrate through lipid membranes depends on the lipid-to-peptide ratio, the toxicity of IAPP is thought to be enhanced due to an increase in its local concentration (Cao et al. 2013b; Clark and Nilsson 2004).
4.3 Amylin Misfolding, Aggregation, and Toxicity: A Dangerous Trio
The primary sequences of mature (fully processed) rat (rIAPP) and human amylin (hIAPP) are depicted in Fig. 4.1a. Although human and rat amylin share high sequence homology, the presence or absence of just a few key amino acids in the amyloidogenic region of the peptide (residues 18–29, Fig. 4.1a) may drastically alter protein’s aggregation and cytotoxic properties. Computational and mutational studies confirmed that 18–29 aa segment of mature hIAPP is highly amyloidogenic (Chiu et al. 2013; Moriarty and Raleigh 1999; Westermark et al. 1990). For instance, the presence of His at position 18 in human amylin is required for amylin-plasma membrane interactions, aggregation and toxicity (Abedini and Raleigh 2005; Brender et al. 2008a; Tu and Raleigh 2013). The presence of three Pro residues in positions 25, 28 and 29 renders rat amylin soluble (non-amyloidogenic) and non-toxic (Fig. 4.1b, c) (Westermark et al. 2011). Likewise, substitutions of Asn22, Gly24, and residues 26–28 with Pro markedly reduced aggregation of 20–29 hIAPP fragment (Moriarty and Raleigh 1999). Thus, an absence of His and the presence of Pro in the sensitive residue segment 18–29 of rat as compared to human amylin is believed to prevent its aggregation and toxicity in rodents.
Fig. 4.1.
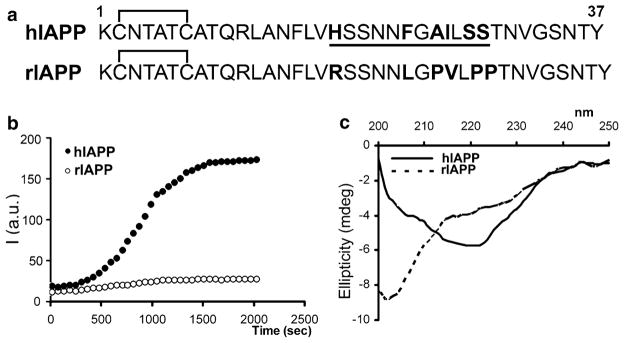
Aggregation of human amylin and changes of its secondary structure coincides in time. (a) Primary structures of mature human (hIAPP) and rat (rIAPP) amylin are depicted. Species-specific aminoacids within the amyloidoigenic region (underlined) of the polypeptide chain are bolded for clarity. (b) Kinetics and extent of aggregation of human and rat amylin in PBS as a function of time. Thioflavin-T fluorescent assay reveals fibrilogenesis of 20 μM human amylin in solution (closed circles) and lack of aggregation of non-amyloidogenic rat amylin (20 μM; open circles). (c) Far-UV CD spectra of human amylin (solid line) and rat amylin (dashed line) taken after 20 min. in PBS solution in the presence of 2 % HFIP. Note the absorption minimum at ~220 nm for human but not rat amylin, typical for peptides and proteins adopting β-sheet conformation
In addition to His and Pro, other polar amino acids from the amyloidogenic region (Fig. 4.1a), such as Ser20, may also play a regulatory role in human amylin aggregation and islet amyloid formation. In fact, Ser20 to Gly mutation in mature human amylin was observed in a small subset of Chinese and Japanese populations who are at an increased risk of developing T2DM. Interestingly, in vitro studies revealed that Ser20Gly substitution accelerated amylin aggregation in solution (Cao et al. 2012), which may help explain increased incidence of diabetes in these two ethnic groups. Notably, amylin has a characteristic intramolecular disulfide bond between Cys2 and Cys7, which does not initially contribute to the aggregation (nucleation) process, although its absence reduces fibril formation (Khemtemourian et al. 2008; Koo and Miranker 2005). The rate of amylin fibrillization parallels the onset and the extent of membrane damage in vitro (Engel et al. 2008). These findings support the fibril hypothesis of amylin’s toxicity in pancreatic islets. However, recent studies point to an important role of pre-fibrillar, soluble oligomeric species in human amylin-induced membrane damage and β-cell death (Cao et al. 2013a; Haataja et al. 2008; Janson et al. 1999; Konarkowska et al. 2006; Ritzel et al. 2007; Trikha and Jeremic 2011; Zhang et al. 2014). This process, commonly referred as toxic oligomer hypothesis, together with the fibril hypothesis, will be addressed in this chapter.
4.4 Conformation Changes and Aggregation of Amylin: A Causal Link
Because the dynamics and the extent of amylin oligomerization and aggregation were shown to be important parameters of amylin’s toxicity (Cao et al. 2013a; Engel et al. 2008; Ritzel et al. 2007), implementation of biophysical methods such as fluorescence and circular dichroism (CD) spectroscopy and high-resolution microscopy capable of tracking these changes in real time have become a norm in recent years. Such biophysical studies are essential to understand amylin aggregation at the molecular level and to determine how certain cellular factors such as pH, ionic content and temperature may contribute to the formation of amyloid plaques in the pancreas and other organs.
Thioflavin (ThT) fluorescence assay (Fig. 4.1b) is a commonly used method to monitor the extent and the kinetics of aggregation of various amyloid peptides and proteins in vitro in cell-free environment (Munishkina and Fink 2007). In the absence of amyloid, the diagnostic dye ThT is weakly fluorescent in solution. However, during amyloid formation, the ThT molecules intercalate into the growing amyloid fibers, rendering the probe more fluorescent (for details see Gorbenko et al., Chap. 6, this volume). Thus, increase in ThT fluorescence over time reflects the fibrillization process that is amenable for experimental manipulations. Lag (nucleation) phase followed by sigmoidal (fibril growth) phase are two common traits shared by amyloid proteins undergoing aggregation (Fig. 4.1b). The ThT assay was previously used by many investigators to understand how changes in pH, temperature or presence or absence of certain metals affect the rate and the extent of amylin aggregation. For example, increasing the salt concentrations in the incubation medium to screen out electrostatic interactions in solution decreased both the rate and the extent of human amylin aggregation (Cho et al. 2008). Thus, amylin aggregation inversely correlates to the solvent ionic strength, which suggests that intra- and intermolecular non-covalent interactions among certain residues play a major role in self-association and polymerization of human amylin in solution. Aromatic and hydrophobic interactions were proposed to play a major role in amylin polymerization in solution (Gazit 2002; Tu and Raleigh 2013). These two non-covalent interactions also play an important role in self-assembly (oligomerization) of peptides into channel-like structures in the membrane, the efficacy of which inversely correlates to ionic strength of the solution (Zhao et al. 2008). In this study formation of protein pores was inhibited when ionic strength of solution increased, whereas both hydrophobic and aromatic interactions were retarded with the increase of salt concentration (Zhao et al. 2008). Thus, it is highly conceivable that amylin oligomerization, the first step in amylin aggregation, is retarded in solution with increased ionic strength due to the inhibitory effect of salts on aromatic and hydrophobic interactions, two major driving forces in amylin polymerization (Gazit 2002; Tu and Raleigh 2013). This eventually would diminish aggregation of human amylin, as showed recently (Cho et al 2008).
Together with ThT assay, the structural studies revealed a causal link between conformational changes in amylin and its propensity to aggregate (Fig. 4.1c) (Brender et al. 2008b; Cho et al. 2008, 2009; Wiltzius et al. 2008). Similar to many other small proteins and peptides (described by Uversky in Chap. 2 of this volume), amylin is natively unfolded in solution. However, amylin can polymerize in a cross-β-sheet conformation upon aggregation in amyloid fibers. CD analysis revealed that aggregation of human amylin is accompanied by secondary structural changes, from random coil in the monomeric from to the β-sheet-enriched fibrillar form characterized by a single minimum at ~220 nm (Fig. 4.1c). In contrast, rat amylin retains its random coil conformation in solution, characterized by a minimum at 202 nm (Fig. 4.1c), which prevents its aggregation (Fig. 4.1b). The likely reason for this difference is that rat amylin contains three structure-breaking prolines, Pro25, Pro28 and Pro29, in the residue segment that probably initiates amyloid formation of human amylin (Fig. 4.1a); these three prolines are expected and observed to prevent β-aggregation (Fig. 4.1b, c) (Moriarty and Raleigh 1999). Inhibition of human amylin transition towards β-sheet conformation by certain inhibitors (divalent metals, insulin or cholesterol) also prevents its aggregation (Cho et al. 2008, 2009; Salamekh et al. 2011; Susa et al. 2014). Collectively, these biophysical studies reveal that aggregation of amylin, like other amyloid proteins, is strongly conformation-dependent and that transition to β-sheet is a requirement for the formation of fibrils.
Although the aforementioned bulk spectroscopy studies provided important information on the dynamics and conformational changes associated with protein misfolding and aggregation, they could neither provide information on the nature and architecture of pre-aggregated species, nor explain how they assemble into fibrils. Without this information, the process of amylin aggregation and amyloid formation in tissues cannot be fully understood. Therefore, visualization of amylin aggregation became imperative. Given the small size of aggregated species, and in order to visualize peptide/protein transition from monomers to oligomers to large aggregates, a new real-time imaging tool capable of imaging at nm-resolution was needed. The development of atomic force microscope (AFM), a 3D lens imaging instrument, allowed investigators to examine, for the first time, the process of amyloid formation with unprecedented clarity and specificity. Formation and growth (extension) of a single fibril has been monitored using this technology (Fig. 4.2) (Cho et al. 2008; Goldsbury et al. 1999; Green et al. 2004). The unique capability of AFM to directly monitor changes in the conformation or aggregation state of macromolecules, and to study dynamic aspects of molecular interactions in their physiological buffer environment has allowed examination of amylin aggregates at ~5 nm lateral and ≤1 nm vertical resolution (Figs. 4.2, 4.4, and 4.5) (Cho et al. 2008, 2009; Green et al. 2004). This novel imaging technology has provided new insights into the molecular mechanism of amyloid assembly.
Fig. 4.2.

Dynamics of amylin aggregation on solid surface. (a) Structural intermediates, oligomers and fibrils, are resolved during amylin aggregation on mica by time-lapse AFM (tapping mode amplitude images). Note a time-dependent transition of human amylin from small round oligomers (0–10 min) into fibrils during early-mid stage of amylin aggregation (10–25 min). Late–stage of amylin aggregation (25–35 min) is characterized by accumulation of massive peptide deposits on the mica surface. All micrographs are 5 × 5 μm. (b) Fibril growth curves reveal two phases of amylin aggregation, an early oligomeric phase (0–10 min) characterized by oligomers formation, followed by oligomers incorporation into growing fibrils (10–25 min, second phase or fibril maturation). Note the significant increase in oligomer heights (inset) and widths during the first phase of amylin aggregation, and an abrupt increase in fibrils length following formation of full-size oligomers. Data represents mean particle size at each time point (mean ± SEM), obtained from three independent time-lapse AFM experiments. (c) 3-D AFM image of a single full-grown fibril on mica showing arrangement of several amylin oligomers and their bi-directional extension into a fibril (depicted by arrowheads). Micrograph is 800 × 800 nm scale
Fig. 4.4.

Dynamics and organization of amylin aggregates on planar membranes. (a) Amylin (20 μM) at time zero was injected into the imaging chamber and the peptide membrane assembly was monitored in real time by time-lapse AFM. All micrographs are 5 × 5 μm, and are taken at the same time intervals of 5 min. (b) Section analysis of amylin aggregates on anionic membranes. Channel-like topology of two amylin supramolecular complexes featuring a central pore is shown (b, PC:PS, 2.8:1.2 mol:mol, 10 min, inset). High-resolution 2D AFM micrographs of several amylin supramolecular complexes on PC:PS membranes are shown. Tetrameric and pentameric subunits are outlined (b, right panel). Bar is 50 nm. (c) Quantitative analysis of cholesterol-regulated amylin assembly on anionic membranes. Presence of cholesterol in planar membranes (PC:PS:Chol,2.3:1:0.8 mol:mol:mo l, circles) stimulates a significant increase in the size (height) of amylin aggregates over time when compared with cholesterol-depleted membranes (PC:PS, 2.8:1.2 mol:mol, filed squares, c, height plot). Cholesterol abrogates amylin deposition and overall membrane surface coverage with amylin (c, surface plot). Cholesterol also inhibits seeding of amylin aggregates on PC:PS membranes (c, particle plot). The mean (Vm) and the total particle volume (Vt) of amylin aggregates on PC:PS membranes are significantly different in the presence of cholesterol after 20 min (*p < 0.05 and **p < 0.01, n = 3 Student’s t-test) (c, volume plot)
Fig. 4.5.
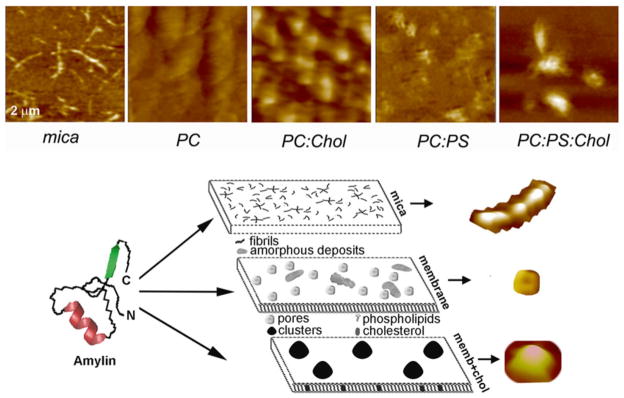
AFM analysis of membrane-directed amylin self-assembly. High-resolution 2D AFM micrographs (top panel) reveal distinct patterns of amylin aggregation and deposition on different surfaces. Note the clustering of amylin aggregates on cholesterol-containing membranes, PC:Chol (3.2:0.8 mol:mol) and PC:PS:Chol (2.3:1:0.8 mol:mol:mol) and their homogenous distribution/aggregation on cholesterol-free membranes, PC and PC:PS (2.8:1.2 mol:mol). In contrast to mica, no fibrils were detected on either membranes. Micrographs are 2 × 2 μm. Bottom panel: Proposed pathway of amylin polymerization and accumulation on different surfaces. The form and amount of amylin deposits correlate with the physicochemical properties of the supporting surface. On stiffpolar mica surface, amylin monomers (left) associate into spherical oligomers that align and elongate overtime to produce mature fibrils that randomly distribute across the surface (mica). On soft planar membranes, amylin self-assembles into globular highly symmetrical supramolecular structures featuring a central pore. Unstructured amorphous amylin aggregates are also formed on this surface (membrane). Incorporation of cholesterol into planar membranes redirects amylin surface deposition by stimulating formation of larger, but fewer amylin clusters (memb+chol). Consequently, the membrane surface area free of amylin deposits increases significantly, which diminishes amylin accumulation. Three major polymorphic forms, a fibril, a pore and a single cluster formed during amylin polymerization on different surfaces, are presented top to bottom (bottom panel)
In our studies, time-lapse AFM operating either in contact or tapping mode was used to investigate the organization of amylin aggregates on solid surface such as mica (Fig. 4.2) and on planar lipid membranes (Figs. 4.4 and 4.5), two surfaces bearing distinct physicochemical properties. With the scanner speed set at 1 Hz and image acquisition time of ~5 min/image, and using high-resolution scanning parameters (512 × 512 lines per image), the dynamics, polymorphism and the extent of amylin fibrillization can be obtained (“the fibril growth” in Fig. 4.2b). Time-lapse amplitude AFM micrographs revealed structural transitions of amylin on mica, from small spherical oligomers to extended fibrils, over a 30 min time period (Fig. 4.2a).
After acquiring micrographs, the size of individual fibrils and oligomers (i.e. radius, length and height) that were deposited on mica (Fig. 4.2b) or on planar membranes (Fig. 4.4) could be determined using a section analysis tool (Veeco, Santa Barbara, CA). Cross-sectional analysis revealed that amylin fibrils varied by length and consistently measured 90–110 nm in width and 5–6 nm in height (Fig. 4.2a, b) (Cho et al. 2008). In addition to amplitude AFM images (Fig. 4.2a), the height-AFM micrographs revealed changes in fibril height during amylin aggregation (Figs. 4.2c and 4.4). Changes in particle height are more visible in the height imaging mode as compared to amplitude images, which are better suited for imaging the fine morphological details of amylin aggregates. Some fibrils were relatively short (less than 200 nm), whereas others extended over 500 nm in length (Fig. 4.2a, c). To construct fibril growth curves, the average size of oligomers and fibrils was determined and plotted for each time point (Figs. 4.2b and 4.4c). In the presence of 1–2 % hexafluoride isopropanol, which accelerates amylin aggregation, massive amyloid-like amylin deposits generally developed after 30 min of incubation (Fig. 4.2a, 30–35 min), thus precluding the monitoring of fibril growth for an extended period of time. However, AFM resolved amylin structural intermediates prior to amyloid accumulation. Formation of fibrils occurred in two distinct phases. The initial phase involved deposition of small spherical oligomers with diameter (width) of 47 ± 7 nm and height of 3.4 ± 0.3 nm (Fig. 4.2a, b, 0–5 min). During the next 5 min, oligomers almost doubled in size (diameter 89 ± 13 nm, height 5.5 ± 0.4 nm; Fig. 4.2a, b), followed by the oligomer’s bi-directional extension into a fibril (Fig. 4.2c) at an average fibrillization rate of 21 nm/min (Fig. 4.2a, b, 10–25 min). Growth curves revealed two distinct phases in amylin aggregation: the first phase, or oligomer growth, was characterized by the large change in the oligomer’s height and width within the first 10 min of aggregation, reaching ~90 % of their maximal value (Fig. 4.2b, inset) but accounting for only ~20 % of maximum fibril length during that period; and second phase or fibril growth, when fibrils rapidly elongated by doubling their extension rate from 9 nm/min (0–10 min interval, Fig. 4.2b) to 21 nm/min (10–25 min interval, Fig. 4.2b). Taken together, these results suggest that fibrils are formed on mica by longitudinal extension of full-grown oligomers. Hence, amylin fibrillization depends on formation of “building block” oligomers, or nuclei, measuring approximately ~6 nm in height and ~90 nm in diameter. Once formed, these nuclei align and elongate into a fibril (Fig. 4.2a–c), a scenario originally proposed by Aebi and co-workers (Green et al. 2004).
4.5 Amylin Folding and Aggregation in Solution Are Strongly Modulated by Anionic Lipids and Cholesterol
The above-mentioned spectroscopy and microscopy studies revealed species and molecular mechanism of amylin aggregation in solution and on solid surface. However, amylin aggregates were also found in close proximity to islet β-cells, with some fibrils integrated into the β-cell plasma membranes (PM) (MacArthur et al. 1999). This finding suggests that amylin-membrane interactions may be important for both amylin aggregation on the cell surface and for the integrity and function of the β-cell PM. The regulatory role and involvement of the membrane’s main constituents, phospholipids and cholesterol, in amylin aggregation were explored during the last decade, prompted by findings that amylin toxicity stems, at least in part, from its ability to disrupt fluidity and organization of cellular membranes (Brender et al. 2008a, b; Khemtemourian et al. 2008). Thus, understanding the process of amylin aggregation on membranes has a direct implication for the etiology of islet amyloidosis and T2DM.
Given that human amylin is a positively charged (cationic) peptide, one can expect that lipids bearing strong negative charge, such as phosphatidylserine (PS) and other anionic lipids, interact with human amylin to modulate its aggregation in solution and/or on membranes. This idea, first tested by Miranker and colleagues (Knight and Miranker 2004), was later verified by several independent biochemical studies showing accelerated amylin aggregation and conformational changes in solution following addition of anionic lipids such as PS (Cho et al. 2008, 2009; Jayasinghe and Langen 2005, 2007; Knight et al. 2006). Supporting this notion, the presence of negatively charged liposomes composed of phosphatidylcholine (PC) and PS (PC:PS, 2.8:1.2 mol:mol) in the incubation solution potentiated amylin aggregation by increasing both the extent (Fig. 4.3a) and the rate of amylin aggregation (Cho et al. 2008). Not only do the PS-enriched liposomes increase the rate of amylin aggregation, but they also shorten the lag phase, suggesting that electrostatic interactions between the peptide and the lipid accelerate nucleation, which is a rate-limiting step in aggregation (Fig. 4.3a).
Fig. 4.3.

Membrane cholesterol and anionic phospholipids oppositely regulate amylin aggregation and misfolding in solution. (a) Thioflavin-T fluorescent assay reveals slow aggregation of 10 μM human amylin in solution (circles). Presence of 100 μM anionic liposomes (PC:PS, 2.8:1.2 mol:mol, squares) in incubating solution (PBS, 1 % HFIP) accelerates amylin aggregation, the effect of which was reversed by inclusion of cholesterol in the lipid vesicles (PC:PS:Chol, 2.3:1:0.8 mol:mol:mol, triangles). (b) Dynamics of amylin secondary structural transitions in solution are regulated by membranes. CD spectras of human amylin (10 μM) incubated with 100 μM PC:PS liposomes (2.8:1.2 mol:mol, black trace) or PC:PS:Chol liposomes (2.3:1:0.8 mol:mol:mol, gray trace) were continuously acquired at 220 nm to monitor appearance of β-sheet conformation. Note that the onset of amylin aggregation and the transition from random coil to β-sheet coincide (hA+PC:PS, Fig. 4.3a, b). Inclusion of cholesterol prolonged amylin’s transition to β-sheets evoked by anionic liposomes (Fig. 4.3b), ultimately reducing kinetics and the extent of amylin aggregation (Fig. 4.3a)
In contrast to anionic liposomes, zwitterionic (neutral) PC liposomes did not significantly affect the rate or the extent of amylin aggregation in solution, further implicating electrostatic interactions as a culprit in amylin aggregation (Cho et al. 2008). Interestingly, inclusion of cholesterol, another essential component of cellular membranes, into anionic liposomes (PC:PS:Chol, 2.3:1:0.8 mol:mol:mol) attenuated the stimulatory effect of PS on amylin aggregation in solution by ~30 % (PC:PS versus PC:PS:Chol, Fig. 4.3a), a decrease comparable to the inhibitory effect of cholesterol on amylin deposition across planar membranes (PC:PS versus PC:PS:Chol, Fig. 4.4a). By applying the calculated rate constants to the Arrhenius equation it was inferred that the presence of negatively charged PC:PS vesicles decreases the activation energy (Ea) of aggregation by ΔEa = −3.7 kJ/mol as compared to amylin alone, which in turn increases the rate of amylin aggregation by more than four times (Cho et al. 2008). In contrast, inclusion of cholesterol in PC:PS vesicles reversed their stimulatory effect on amylin aggregation by increasing the activation energy by ΔEa = 845 J/mol. The ΔEa values were calculated for amylin aggregation at room temperature (25 °C) at which the experiment was performed (Cho et al. 2008). Remarkably, despite marked difference in their aggregation rates, all three reactions exhibited first-order kinetics (Cho et al. 2008), indicating that anionic lipids and cholesterol do not change the mechanism of amylin aggregation but act as catalyst and inhibitor, respectively. This finding also suggests that phospholipids and cholesterol in membranes oppositely modulate amylin aggregation by changing the activation energy of the amylin transition from random coil to β-sheets. In support of this conclusion, inclusion of cholesterol reduced the stimulatory effect of PS-containing liposomes on the conformational transition of amylin from random coil to β-sheets (Fig. 4.3b) (Cho et al. 2008).
4.6 Cholesterol Regulates Amylin Aggregation on Planar Lipid Membranes
We used AFM to investigate the supramolecular organization and dynamics of amylin aggregates on model membranes (Cho et al. 2009) that resemble the cell PM in composition and fluidity. Amylin aggregation on neutral and negatively-charged planar membranes that contained or lacked cholesterol was investigated by time-lapse AFM followed by single-particle analysis as described above. AFM revealed transition of small 25–35 nm diameter spherical oligomers (formed during the first 5 min) into larger 90–130 nm supramolecular complexes on anionic PC:PS (2.8:1.2 mol:mol) membranes (Fig. 4.4a, 10 min, arrowheads). Amylin oligomers formed during the first 5 min (Fig. 4.4a, left panel) and were morphologically similar to the oligomers initially assembled on mica (Fig. 4.2a). In marked contrast to amylin fibrillization on mica, amylin oligomers did not align and elongate into fibrils on anionic membranes but rather assembled into channel-or pore-like structures (Fig. 4.4a, 10 min, arrowheads). Interestingly, AFM revealed that amylin oligomers preferentially deposited on planar PC:PS membranes (Fig. 4.4a, 5 min) and much less frequently (<3 % of all particles) on mica surfaces (Fig. 4.2a, 5 min). This result demonstrates that amylin interacts with anionic membranes earlier during the oligomeric stage of aggregation, which most likely serve as a catalyst for amylin oligomerization. 3D image analysis revealed a characteristic fourfold rotational symmetry of self-assembled supramolecular complexes of amylin featuring a central pore (Fig. 4.4b, PC:PS, 10 min; inset). The majority of self-assembled amylin complexes on planar PC:PS membranes exhibited tetrameric and, at times, pentameric globular organization (Fig. 4.4b, right panels). Tetrameric amylin complexes accounted for ~95 % of all supramolecular amylin structures assembled on the membranes. Less than 5 % of amylin complexes were pentamers (7 out of 163 particles examined, n = 3).
A twofold symmetrical organization of amylin and other amyloid proteins, incorporated first into liposomes and subsequently into planar membranes, has been previously demonstrated (Quist et al. 2005). In our study, which replicates amylin interactions with the pancreatic β-cells, we showed that amylin oligomers in solution can also directly assemble into symmetrical channel-like structures on preformed planar membranes. This may be relevant for the pathology of diabetes, as amylin and other amyloid proteins interact with cellular membranes and are cytotoxic when assembled into oligomers (Haataja et al. 2008; Ritzel et al. 2007; Trikha and Jeremic 2011). Interestingly, the sizes of amylin globular particles assembled on planar membranes (Fig. 4.4b) were in the same range (20–40 nm) as the soluble intermediate-sized cytotoxic amylin particles reported earlier (Janson et al. 1999). As expected from the peptide’s amphiphilic nature, cytotoxic amylin oligomers readily form ion-permeable channels in bilayers and in cell membranes (Mirzabekov et al. 1996; Quist et al. 2005; Trikha and Jeremic 2011; Zhao et al. 2014). Besides forming channel-like structures, amylin also accumulates on membranes as unstructured amorphous aggregates (Fig. 4.4a, left panel; arrows), resembling in size (300–500 nm) and morphology the amyloid deposits often associated with T2DM (Jaikaran and Clark 2001). Incorporation of cholesterol into anionic membranes (PC:PS:Chol, 2.3:1:0.8 mol:mol:mol) re-directed the surface distribution of amylin aggregates (Fig. 4.4a, right panel). While oligomers were observed again during the first 5 min of aggregation on cholesterol-containing membranes, amylin further aggregated and concentrated in discrete areas measuring 300–500 nm in diameter (Fig. 4.4a, 10–20 min; PC:PS:Chol). Presence or absence of channel-like structures on these membranes could not be verified due to a strong clustering effect of cholesterol from the very early stages of amylin aggregation.
To further understand how phospholipids and cholesterol modulate amylin surface deposition and to learn more about the regulatory mechanisms driving aggregation, we measured amylin accumulation on planar membranes using single-particle analysis as explained above (see Fig. 4.2b). The regulatory effect of cholesterol was quite obvious from the early stages of amylin aggregation (10 min and thereafter; Fig. 4.4a–c). Over time there was a significant increase in the height of amylin aggregates due to the large clustering effect of cholesterol (Fig. 4.4c, height analysis). This was accompanied by an overall decrease in amylin deposition across the planar membranes (Fig. 4.4c, surface analysis). As amylin aggregated and accumulated in some membrane areas, other regions of the membrane were virtually devoid of the protein aggregates (Fig. 4.4a, right panel). Consequently, amylin’s capacity to form an extensive network of amyloid aggregates on the membrane was diminished in membranes that contained cholesterol (Fig. 4.4a, c, surface analysis).
Similar to most amyloid proteins, amylin aggregation is nucleation-dependent (Khemtemourian et al. 2008; Padrick and Miranker 2002). Consistent with the “nucleation” hypothesis, amylin seeding was diminished in the presence of cholesterol: a sevenfold decrease in the number of amylin particles was observed on planar anionic membranes that contained cholesterol as compared to those that lacked cholesterol (Fig. 4.4c, particle analysis). This phenomenon may also account for the observed inhibitory effect of membrane cholesterol on initial (nucleation) phase of amylin polymerization in solution evoked by anionic liposomes (Fig. 4.3a). As the number of amylin particles diminished in the presence of cholesterol (Fig. 4.4a), their size and average volume increased over time (Fig. 4.4a, c, volume analysis). The mean volume Vm of amylin particles was larger on lipid membranes than on mica. A further, much larger increase in particle size was detected on membranes containing cholesterol (PC:PS:Chol. Fig. 4.4c, volume analysis). However, the total volume Vt of amylin aggregates on the cholesterol-containing membranes significantly decreased when compared to the cholesterol-free membranes (Fig. 4.4c volume analysis, inset) due to a large decrease in the amylin seeding capacity (Fig. 4.4c, particle analysis). Thus, in the presence of cholesterol, amylin aggregated and accumulated on planar membranes as submicron-sized protein clusters, which served as templates for the ongoing amylin binding and aggregation.
Comparisons of AFM micrographs and amylin growth curves on surfaces bearing different physicochemical properties (Fig. 4.5, upper panel) revealed that amylin monomers polymerize via two distinct mechanisms: on stiffand polar mica, amylin formed fibrils by longitudinal bi-directional extension of full-grown spherical oligomers, or nuclei, measuring ~6 nm in height and ~90 nm in diameter (Fig. 4.2), and on soft negatively-charged PC:PS planar membranes amylin formed pore-like supramolecular structures that self-assembled from ~25 to 35 nm diameter globular subunits or oligomers (Figs. 4.4 and 4.5, lower panel). AFM revealed another important feature of amylin aggregates on planar membranes. Amorphous deposits and channel-like structures that were formed during the early (5–10 min) stages of amylin aggregation did not transition into new structures, although the total amount and size of amorphous aggregates increased over time (Figs. 4.4 and 4.5). These findings signify the important contribution and long-lasting effect of lipids and cholesterol in the regulation of amylin aggregation, summarized in Fig. 4.5 (lower panel).
To test if the regulatory effect of cholesterol is charge-specific, amylin aggregation was also studied on neutral PC membranes that lack or include cholesterol (Fig. 4.5, upper panel). As in anionic PC:PS-membranes (Figs. 4.4c and 4.5), the presence of cholesterol in neutral PC membranes (PC:Chol, 3.2:0.8 mol:mol) stimulated an increase in the height of amylin aggregates by almost twofold, from 6.2 ± 0.9 to 11.9 ± 1.7 nm (mean ± SEM). Similar to anionic membranes (Fig. 4.4, PC:PS:Chol), the presence of cholesterol in neutral membranes resulted in heterogeneous amylin distribution and clustering over the membrane surface with a mean particle (cluster) radius of 196 ± 19 nm based on section analysis reported previously (Fig. 4.5, PC:Chol upper panel) (Cho et al. 2009). Thus, the amylin clusters on neutral membranes (PC:Chol) were 20–30 % smaller compared to those formed on cholesterol-containing anionic (PC:PS:Chol) membranes (Fig. 4.5, upper panel). Collectively, our results demonstrate the intrinsic ability of cholesterol to regulate amylin aggregation and deposition on membranes, irrespective of the chemical composition or charge of the membrane (Cho et al. 2009).
Amylin’s self-assembly into non-fibrillogenic, channel-like structures on neutral and anionic membranes (Figs. 4.4 and 4.5) suggests that physical properties, rather than specific chemical properties of the membrane, determine its susceptibility to distinct amyloid forms. For example, β-amyloid oligomers, but not fibrils, were found to be enriched in neuronal membrane lipid rafts (Schneider et al. 2006), which are discrete membrane segments with phospholipids in a liquid-ordered phase. Cholesterol is a known regulator of membrane fluidity. It is found in lipid rafts and many endomembranes, where it establishes a sorting platform for a scaffold of various protein-lipid complexes important for cell signaling, endocytosis and other essential physiological processes (Simons and Toomre 2000). It is therefore quite possible that, by modulating membrane fluidity and/or membrane curvature (Chen and Rand 1997; Smith et al. 2009), cholesterol also regulates amylin-lipid interactions and amylin aggregation observed in our studies (Cho et al. 2008, 2009; Trikha and Jeremic 2011).
Another possibility is that amylin directly interacts with cholesterol in the membrane. Amylin, as a monomer, has a strong tendency to insert into phospholipid monolayers (Engel et al. 2006). This event may set a stage for direct peptide-cholesterol interactions in the membrane core. To test if cholesterol can directly interact with amylin, we evaluated their interaction by ThT aggregation assay and CD spectroscopy (Cho et al. 2008, 2009). The presence of soluble cholesterol (1:1 and 1:2 mol/mol peptide/sterol ratios) impeded amylin aggregation in solution in two major ways: it prolonged the lag (nucleation) phase and it decreased the fibrillization rate by 16-fold, from k = 8.32 × 10−3 s−1 (amylin) to k = 5.2 × 10−4 s−1 (amylin plus cholesterol) (Cho et al. 2008, 2009). These results are consistent with inhibition of both fibril nucleation and elongation. These reconstituted studies have demonstrated that cholesterol may directly affect amylin conformational changes in solution and possibly in the membranes. However, whether human amylin directly interacts with cholesterol in plasma and/or endomembranes still remains to be confirmed.
4.7 Plasma Membrane Cholesterol Restricts Aggregation of Human Amylin on Cellular Membranes and Amylin’s Toxicity
Experiments performed with synthetic liposomes and planar membranes provided important although indirect evidence for the role of membranes in aggregation of amylin and other amyloid proteins. To confirm that native membranes modulate amylin’s turnover and toxicity in situ, we resorted to cellular studies (Trikha and Jeremic 2011, 2013). In our experimental setup, monomeric human amylin was added to pancreatic rat and human islet cells in which we systematically varied plasma membrane cholesterol levels using cholesterol biosynthesis inhibitor lovastatin (Lov) and/or cholesterol-depleting agent, beta-cyclodextrin (BCD). The extent of human amylin aggregation (Fig. 4.6) and toxicity (Fig. 4.7) in cholesterol-containing and cholesterol-depleted cells was assessed over 24 h by confocal microscopy (Trikha and Jeremic 2011). Oligomers were detected with the oligomer-specific A11 antibody (Fig. 4.6a) that does not react with either monomers or fibrils (Kayed et al. 2003). To detect human amylin monomer distribution on the PM and inside the cells (Fig. 4.6b), we used a human-specific amylin antibody that does not cross-react with the rat isoform or large oligomers/aggregates (Trikha and Jeremic 2011). In addition to human amylin, we used the lipid raft marker, cholera toxin (CTX), and the clathrin endocytotic marker, transferrin (not shown), to determine the specificity of amylin monomer and oligomer binding to the cell PM (Fig. 4.6).
Fig. 4.6.

Binding and clustering of amylin oligomers into microdomains on the cell PM requires cholesterol. (a) Confocal microscopy analysis of amylin oligomer and cholera toxin (CTX) distribution on the cell PM. Characteristic binding profiles of amylin oligomers on the cell PM for each treatment (within boxes, right panel) are rendered in gray tones for easier particle comparisons, which are presented side by side with the original fluorescence images (left panels). Note the time-dependent increase in the number of internalized amylin oligomers (control, 30 min vs. 24 h, left panel), which prevents accumulation of amylin oligomers on the cell PM (control, 30 min vs. 24 h, right box). Single particle analysis demonstrates a threefold increase in the number of amylin oligomer clusters, or puncta, on the PM (particle no.), and their dispersion across the PM in cholesterol-depleted cells (mean particle area). Significance established at *p < 0.05, **P < 0.01 control vs. BCD/Lov, and #p < 0.05, ##p < 0.01 BCD/Lov vs. BCD/Lov/Chol. (b) Amylin monomer internalization is not blocked by cholesterol depletion. Confocal microscopy demonstrates internalization of amylin monomers both in controls (hA) and cholesterol-depleted cells (hA + BCD/Lov). No significant (NS) change in PM-binding pattern of amylin monomers (right boxes) was noticed upon cholesterol depletion. PM cholesterol does not modulate amylin deposition on the PM. The number of amylin puncta on PM (particle no.) and their area (mean particle area) did not change significantly upon depletion of PM cholesterol by BCD/Lov. Bars are 5 μm
Fig. 4.7.
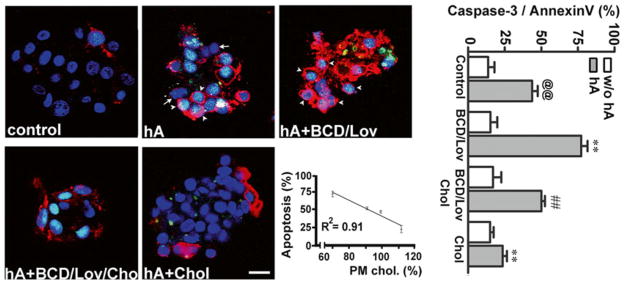
PM cholesterol prevents toxicity of soluble amylin oligomers in cultured human islet cells. Confocal microscopy analysis of phosphatidylserine (PS) externalization and caspase-3 proteolytic activation by amylin in cells with normal, depleted and enriched cholesterol levels (micrographs, left panel). Arrows depict non-apoptotic nuclei (blue) in viable cells, whereas arrowheads depict fluorogenic caspase-3 substrate found in the nuclei of apoptotic cells, giving these nuclei a green/blue appearance. The majority of PS-positive cells (red) show shrinkage and nuclear condensation (arrowheads) indicative of apoptosis (hA and hA+BCD/Lov). Bar is 10 μm. Linear regression analysis shows an inverse relationship between amylin-induced cell death and PM cholesterol levels. The extent of cell death evoked by amylin was plotted as a function of variable PM cholesterol levels in controls and treatments (apoptosis vs. cholesterol, graph). Quantitative analysis of amylin-induced apoptosis in human islets (caspase-3/annexin, graph) reveals a significant increase in amylin toxicity in cholesterol-depleted cells as compared to controls (@@p < 0.01 hA vs. control, **p < 0.01 hA vs. treatments, right panel). Replenishment of PM cholesterol levels significantly decreased amylin toxicity (##p < 0.01 hA+BCD/lov vs. hA+BCD/Lov/Chol)
Amylin and CTX were sequentially (Fig. 4.6) or concurrently (Trikha and Jeremic 2011) incubated with cultured pancreatic insulinoma RIN-m5F cells for the indicated periods of time, fixed and processed for immunochemical analysis. In experiments in which amylin and CTX were concurrently incubated with cells, immunoconfocal microscopy revealed a punctuated staining pattern of CTX and amylin oligomers on the cell PM, exhibiting high spectral overlap (yellow) and a high co-localization coefficient (R = 0.74 ± 0.09) in discrete membrane regions (Trikha and Jeremic 2011). Changes in the cell morphology characterized by the appearances of elongated protrusions were frequently observed in cultures treated with human amylin (Trikha and Jeremic 2011). Amylin oligomer binding and uptake were particularly noticeable in protrusions. However, amylin oligomers also co-patched with the lipid raft marker CTX in other PM regions (Trikha and Jeremic 2011, 2013). This suggests that amylin oligomers accumulate at specific microdomains, possibly lipid rafts, on the cell PM prior to their uptake.
To exclude a possible modulatory effect of CTX on amylin binding, sequential incubations of rat insulinoma cells with amylin and CTX were performed. Following the incubation with amylin, cells were further incubated with CTX for 30 min at 4 ° C. Amylin oligomers and CTX once more co-localized on the cell PM (Fig. 4.6a, 30 min). Prolonging the incubation period from 30 min to 24 h allowed amylin oligomers in the microdomains to internalize, as demonstrated by a ~50 % drop in amylin oligomer/CTX colocalization values (Fig. 4.6a) (Trikha and Jeremic 2011). Upon depletion of PM cholesterol with BCD/Lov, a significant decrease in colocalization of amylin oligomers with cholera toxin on the cell PM was observed indicating amylin/CTX particle de-clustering and their dispersions across the cell surface (Fig. 4.6a). In line with this finding, image and single-particle analysis revealed that the mean particle area of PM-bound amylin oligomers (Fig. 4.6a, right boxed panel) in cholesterol-depleted cells (BCD/Lov) decreased significantly as compared to control cells (Fig. 4.6a, particle analysis). Conversely, the number of amylin oligomer clusters, or puncta, on the PM of cholesterol-depleted cells increased by threefold relative to control cells (Fig. 4.6a, particle analysis). Consequently, cell surface coverage by amylin oligomers increased by ~twofold in cells with reduced PM cholesterol content as compared to control cells (Fig. 4.6a, right boxed panel). This inhibitory effect of BCD/Lov on clustering of amylin oligomers on the PM was also observed at earlier time points, 30 min and 3 h (data not shown). These results demonstrate that the seeding (nucleation) capacity of amylin oligomers and their ability to form a dense network of amyloid aggregates on the PM were augmented in cells with impaired cholesterol homeostasis.
Clustering of amylin oligomers on the PM was fully restored following replenishment of PM cholesterol (Fig. 4.6a), indicating that amylin oligomer deposition on the PM is modulated by cholesterol and is reversible. These findings are in full compliance with the inhibitory and clustering effect of cholesterol on amylin aggregation on synthetic membranes (Figs. 4.4 and 4.5) (Cho et al. 2008, 2009).
In contrast to oligomers (Fig. 4.6a), fewer amylin monomers co-localized with the lipid raft marker CTX on the PM during the first 30 min of incubation with cells (Fig. 4.6b). However, analogous to the binding of oligomers (control 30 min, Fig. 4.6a right boxed panel), amylin monomers exhibited a discrete, punctuated staining pattern on the cell PM during that period (control 30 min, Fig. 4.6b right boxed panel). Interestingly, the degree of colocalization between amylin monomers and CTX also decreased significantly upon cholesterol depletion with BCD/Lov (Fig. 4.6b), albeit to much lesser extent than for amylin oligomers (Fig. 4.6a). In contrast to oligomers, the number and the mean particle area of amylin monomer puncta at the cell PM remained almost the same in control and BCD/Lov-treated cells (Fig. 4.6b image and graphs) (Trikha and Jeremic 2011), indicating that clustering of amylin monomers into microdomains on the cell PM is not affected by PM cholesterol. Specificity of amylin and CTX binding is further demonstrated in experiments in which transferrin, a marker of clathrin-dependent endocytosis, was used (Trikha and Jeremic 2011, 2013). Colocalization analysis revealed that neither amylin monomers nor oligomers colocalize with transferrin on the cell PM (Trikha and Jeremic 2011). Similarly, CTX and Trf were found to bind to distinct regions on the cell PM. Collectively, our studies depicted in Fig. 4.6 (Trikha and Jeremic 2011) imply that PM cholesterol reversibly and specifically determines binding and distribution of amylin oligomers, but not monomers, on the cell PM.
We also investigated whether, and to what extent, variations in PM cholesterol levels affect amylin toxicity in rat and human pancreatic islet cells. Cells were treated or not with BCD and/or Lov and then exposed to human amylin. Following 24 h incubation, cells were analyzed by confocal microscopy for the presence or absence of apoptotic markers, cleaved caspase-3 and PS externalization (Fig. 4.7). Amylin was toxic to cultured human islet cells, albeit with notably higher potency (LD50 = 2.5 μM) as compared to its toxic effect in rat insulinoma cells (LD50 = 15 μM). Human amylin (hA, 2 μM) evoked apoptosis in 43 ± 5 % of cultured human islet cells. The combination of BCD (5 mM, 10 min) and lovostatin (0.5 μM, 24 h), reduces PM cholesterol to 67 ± 4 % relative to controls (Trikha and Jeremic 2011), which in turn increases amylin toxicity by a 34 ± 5 % as compared to cells with normal membrane cholesterol content (Fig. 4.7, image and histogram). Addition of soluble cholesterol together with BCD/Lov replenishes cholesterol levels (94 ± 5 % in BCD/Lov/Chol relative to controls), which reverses the stimulatory effect of BCD/Lov on amylin toxicity. As demonstrated in rat insulinoma cells (Trikha and Jeremic 2011), Lov alone had a small insignificant stimulatory effect on amylin toxicity in human islet cells (<10 %, data not shown). Finally, incubation of human islet cells with soluble cholesterol (50 μg/ml) increases PM cholesterol levels by 14 ± 6 % relative to control cells (Trikha and Jeremic 2011) and further attenuates amylin toxicity (Fig. 4.7). A strong inverse relationship between PM cholesterol levels and amylin toxicity in human islets is obtained (Fig. 4.7, graph). The inhibitory effect of PM cholesterol on amylin toxicity in human islets was confirmed with the LDH-release assay (Trikha and Jeremic 2011). Similarly, Western blot analysis demonstrates extracellular accumulation of low-molecular-weight amylin monomers and dimers and intermediate-sized oligomers upon PM-cholesterol depletion with BCD/Lov, and their efficient clearance by islet cells upon PM cholesterol reloading with soluble cholesterol (BCD/Lov/Chol). In line with these findings, confocal microscopy shows that cholesterol supplementation stimulates, while PM cholesterol depletion decreases amylin oligomer internalization in human islet cells (Trikha and Jeremic 2011). These results demonstrate a major role of PM cholesterol in amylin turnover and toxicity in human islets.
4.8 Role of Cholesterol and Phospholipids in Aggregation and Toxicity of Other Amyloid Proteins
Amyloid formation, or amyloidogenesis, an intrinsic property of all polypeptides (Chiti and Dobson 2006) is a process by which soluble proteins aggregate into insoluble, structurally conserved unbranched fibers that are characterized by resistance to proteinase K digestion, dye binding specificity, and ordered β-sheet-rich structure (Sipe et al. 2010). Amyloids can be broadly categorized into detrimental and functional. Detrimental amyloids, which cause protein misfolding in amyloid diseases, include huntingtin implicated in Huntington’s disease, α-synuclein implicated in Parkinson’s disease, prion protein implicated in Creutzfield-Jacob’s disease, superoxide dismutase implicated in amyotrophic lateral sclerosis, and amyloid-β (Aβ) peptide implicated in Alzheimer’s disease, transthyretin implicated in transthyretin familial amyloidosis, Tau implicated in frontotemporal lobar degeneration etc. Some of these proteins, including Aβ, α-synuclein, serum amyloid A (implicated in inflammation-linked amyloidosis) and other apolipoproteins (implicated in systemic amyloidosis and atherosclerosis) are described in other chapters in this volume. Functional amyloids are an integral part of the normal physiology of the cell and include curli, chaplin, URE2p and PmeL17. Curli found in E. coli plays a role in biofilm formation and mediates infection. Chaplin found in Streptomyces plays a role in protection against water surface tension. URE2p found in S. Cerevisiae plays a role in nitrogen catabolism, and Pmel17 found in humans play a role in melanin synthesis (Granzotto et al. 2011; Lau et al. 2007; Rymer and Good 2000).
High local concentrations of proteins accumulating on biological surfaces, such as the plasma or organelle membranes, and the physicochemical properties of membrane lipids that interact with these proteins may accelerate aggregation and fibril formation (Burke et al. 2013). Interestingly, while there have been reports of cholesterol and phospholipids stimulating amyloid aggregation and enhancing amyloid-mediated toxicity, there have also been contrasting reports describing that cholesterol and phospholipids reduce aggregation and toxicity of a variety of amyloid proteins. Moreover, distinct mechanisms are reported for the effects of cholesterol and phospholipids on amyloid protein aggregation and toxicity. It is widely reported that high levels of cholesterol aggravate Alzheimer’s disease (Cossec et al. 2010) by promoting the generation of Aβ peptide (Barrett et al. 2012) and stimulating its aggregation. Barrett et al. have shown that the carboxyl terminal transmembrane domain of amyloid precursor protein binds cholesterol and is cleaved by γ-secretase to generate Aβ peptides. This promotes the possible use of cholesterol lowering drugs, like statins, in the control of Alzheimer’s pathology (McGuinness and Passmore 2010). Another elegant study that supports the enhancing role of cholesterol in the aggregation of amyloid precursor proteins (APP) (Hayashi et al. 2000) provides evidence that APP is not present in caveolae or caveoli-like domains of the cell membrane but in cholesterol rich microdomains which may be linked to the maturation of APP in a cell type specific manner. Exogenously applied amyloid beta peptides and tau protein have also been reported to accumulate in lipid rafts, in turn affecting their mobility (Williamson et al. 2008).
There are multiple reports that anionic phospholipids like phosphatidylserine provide ‘docking sites’ where amyloid aggregates can be nucleated, resulting in toxicity through disruption of the membrane (Zhao et al. 2004, 2005; Lee et al. 2002). For example, native-like aggregates of the toxic prion protein Ure2p dock preferentially on PS, speeding up aggregation and dampening toxicity (Pieri et al. 2009). The conversion of another normal cellular predominantly α-helical prion protein isoform, PRPC, to the protease-resistant, β-sheet-rich PRPSC isoform is enhanced by the presence of cholesterol-rich phospholipid membranes at ambient conditions (Rymer and Good 2000). In addition to cholesterol and phospholipids, fatty acids are abundantly found in membranes. For example, docosahexaenoic acid (DHA) is abundant in the vicinity of α-synuclein in neuronal membranes of patients with Parkinson’s disease, and has been reported to enhance α-synuclein aggregation (De Franceschi et al. 2011).
At the physiological concentration (5 μM) at which cholesterol is found in the cerebrospinal fluid, it is reported to slow down, but not block, aggregation of β-amyloid complexes, in the presence or absence of metal ions. On the other hand, cholesterol compensates for membrane damage induced by Aβ peptides, preventing their toxic effects (Granzotto et al. 2011). Lipid rafts formed by cholesterol-rich microdomains in membranes are an attachment target for Aβ peptides. Antagonists of platelet-derived growth factor and inhibitors of phospholipase A2 inhibit the release of cholesterol from cholesterol esters, thereby reducing lipid rafts that traffic Aβ peptides, which in turn lead to the increase of cytosolic degradation of Aβ (Simmons et al. 2014). The apoptotic accumulation and redistribution of Aβ peptides on the neuronal cell membrane and in lipid rafts is reported to depend on a non-receptor tyrosine kinase that increases phosphorylation of membrane associated proteins (Williamson et al. 2008). Apolipoprotein E4, an established risk factor for Alzheimer’s disease and cerebral amyloid angiopathy, affects clearance of Aβ peptides. ApoE4 preferentially forms plaques in the cerebral vessels rather than cerebral parenchyma by altering the ratio of Aβ (1–40) and Aβ (1–42) (Fryer et al. 2005).
In summary, during the last three decades investigators working in the amyloid field have made major progress by illuminating important regulatory mechanisms and cellular factors implicated in turnover and function of amyloid proteins such as human amylin. Studies reveal exactly how membrane-associated lipids and cholesterol modulate aggregation of human amylin and other amyloid proteins and how islet amyloid is formed and processed in cells, interacts with membranes and affects cellular functions. The knowledge gained in these studies offers exciting opportunity for development of novel approaches and drugs that may eradicate or slow down progression of amyloid-associated diseases.
Acknowledgments
This work was supported by the NIH grant RO1DK091845 and the ICR Basic Science Islet Distribution Program (to A.J.).
Abbreviations
- AFM
Atomic force microscopy
- BCD
Methylbetacyclodextrin
- CD
Circular dichroism
- CTX
Cholera toxin
- DOPC
1,2-dioleoyl-phosphatidylcholine
- DOPS
1,2-dioleoylphosphatidylserine
- EM
Electron microscopy
- HFIP
Hexafluoride isopropanol
- hIAPP
Human islet amyloid peptide
- Lov
Lovostatin
- PM
Plasma membranes
- T2DM
Type 2 diabetes mellitus
- ThT
Thioflavin-T
- Trf
Transferrin
Contributor Information
Sanghamitra Singh, Department of Biological Sciences, The George Washington University, 2023 G Street NW, Washington, DC 20052, USA.
Saurabh Trikha, Department of Biological Sciences, The George Washington University, 2023 G Street NW, Washington, DC 20052, USA.
Diti Chatterjee Bhowmick, Department of Biological Sciences, The George Washington University, 2023 G Street NW, Washington, DC 20052, USA.
Anjali A. Sarkar, Department of Biological Sciences, The George Washington University, 2023 G Street NW, Washington, DC 20052, USA
Aleksandar M. Jeremic, Email: jerema@gwu.edu, Department of Biological Sciences, The George Washington University, 2023 G Street NW, Washington, DC 20052, USA
References
- Abedini A, Raleigh DP. The role of His-18 in amyloid formation by human islet amyloid polypeptide. Biochemistry. 2005;44(49):16284–16291. doi: 10.1021/bi051432v. [DOI] [PubMed] [Google Scholar]
- Abedini A, Schmidt AM. Mechanisms of islet amyloidosis toxicity in type 2 diabetes. FEBS Lett. 2013;587(8):1119–1127. doi: 10.1016/j.febslet.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancsin JB. Amyloidogenesis: historical and modern observations point to heparan sulfate proteoglycans as a major culprit. Amyloid. 2003;10(2):67–79. doi: 10.3109/13506120309041728. [DOI] [PubMed] [Google Scholar]
- Barrett PJ, Song Y, Van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A, et al. The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science. 2012;336(6085):1168–1171. doi: 10.1126/science.1219988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brender JR, Hartman K, Reid KR, Kennedy RT, Ramamoorthy A. A single mutation in the nonamyloidogenic region of islet amyloid polypeptide greatly reduces toxicity. Biochemistry. 2008a;47(48):12680–12688. doi: 10.1021/bi801427c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brender JR, Lee EL, Cavitt MA, Gafni A, Steel DG, Ramamoorthy A. Amyloid fiber formation and membrane disruption are separate processes localized in two distinct regions of IAPP, the type-2-diabetes-related peptide. J Am Chem Soc. 2008b;130(20):6424–6429. doi: 10.1021/ja710484d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke KA, Yates EA, Legleiter J. Biophysical insights into how surfaces, including lipid membranes, modulate protein aggregation related to neurodegeneration. Front Neurol. 2013;4:17. doi: 10.3389/fneur.2013.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caminos JE, Bravo SB, Garces MF, Gonzalez CR, Cepeda LA, Gonzalez AC, et al. Vaspin and amylin are expressed in human and rat placenta and regulated by nutritional status. Histol Histopathol. 2009;24(8):979–990. doi: 10.14670/HH-24.979. [DOI] [PubMed] [Google Scholar]
- Cao P, Tu LH, Abedini A, Levsh O, Akter R, Patsalo V, et al. Sensitivity of amyloid formation by human islet amyloid polypeptide to mutations at residue 20. J Mol Biol. 2012;421(2–3):282–295. doi: 10.1016/j.jmb.2011.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P, Abedini A, Wang H, Tu LH, Zhang X, Schmidt AM, et al. Islet amyloid polypeptide toxicity and membrane interactions. Proc Natl Acad Sci U S A. 2013a;110(48):19279–19284. doi: 10.1073/pnas.1305517110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P, Marek P, Noor H, Patsalo V, Tu LH, Wang H, et al. Islet amyloid: from fundamental biophysics to mechanisms of cytotoxicity. FEBS Lett. 2013b;587(8):1106–1118. doi: 10.1016/j.febslet.2013.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Rand RP. The influence of cholesterol on phospholipid membrane curvature and bending elasticity. Biophys J. 1997;73(1):267–276. doi: 10.1016/S0006-3495(97)78067-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Chiu CC, Singh S, de Pablo JJ. Effect of proline mutations on the monomer conformations of amylin. Biophys J. 2013;105(5):1227–1235. doi: 10.1016/j.bpj.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho WJ, Jena BP, Jeremic AM. Nanoscale imaging and dynamics of amylin-membrane interactions and its implication in type II diabetes mellitus. Methods Cell Biol. 2008;90:267–286. doi: 10.1016/S0091-679X(08)00813-3. [DOI] [PubMed] [Google Scholar]
- Cho WJ, Trikha S, Jeremic AM. Cholesterol regulates assembly of human islet amyloid polypeptide on model membranes. J Mol Biol. 2009;393(3):765–775. doi: 10.1016/j.jmb.2009.08.055. [DOI] [PubMed] [Google Scholar]
- Clark A, Nilsson MR. Islet amyloid: a complication of islet dysfunction or an aetiological factor in type 2 diabetes? Diabetologia. 2004;47(2):157–169. doi: 10.1007/s00125-003-1304-4. [DOI] [PubMed] [Google Scholar]
- Cooper GJ, Willis AC, Clark A, Turner RC, Sim RB, Reid KB. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc Natl Acad Sci U S A. 1987;84(23):8628–8632. doi: 10.1073/pnas.84.23.8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossec JC, Marquer C, Panchal M, Lazar AN, Duyckaerts C, Potier MC. Cholesterol changes in Alzheimer’s disease: methods of analysis and impact on the formation of enlarged endosomes. Biochim Biophys Acta. 2010;1801(8):839–845. doi: 10.1016/j.bbalip.2010.03.010. [DOI] [PubMed] [Google Scholar]
- Costes S, Gurlo T, Rivera JF, Butler PC. UCHL1 deficiency exacerbates human islet amyloid polypeptide toxicity in beta-cells: evidence of interplay between the ubiquitin/proteasome system and autophagy. Autophagy. 2014;10(6):1004–1014. doi: 10.4161/auto.28478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Franceschi G, Frare E, Pivato M, Relini A, Penco A, Greggio E, et al. Structural and morphological characterization of aggregated species of alpha-synuclein induced by docosahexaenoic acid. J Biol Chem. 2011;286(25):22262–22274. doi: 10.1074/jbc.M110.202937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel MF, Yigittop H, Elgersma RC, Rijkers DT, Liskamp RM, de Kruijff B, et al. Islet amyloid polypeptide inserts into phospholipid monolayers as monomer. J Mol Biol. 2006;356(3):783–789. doi: 10.1016/j.jmb.2005.12.020. [DOI] [PubMed] [Google Scholar]
- Engel MF, Khemtemourian L, Kleijer CC, Meeldijk HJ, Jacobs J, Verkleij AJ, et al. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc Natl Acad Sci U S A. 2008;105(16):6033–6038. doi: 10.1073/pnas.0708354105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, Westermark G, Chan SJ, Steiner DF. Altered gene structure and tissue expression of islet amyloid polypeptide in the chicken. Mol Endocrinol. 1994;8(6):713–721. doi: 10.1210/mend.8.6.7935487. [DOI] [PubMed] [Google Scholar]
- Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM, et al. Human apolipoprotein E4 alters the amyloid-beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci. 2005;25(11):2803–2810. doi: 10.1523/JNEUROSCI.5170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazit E. A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002;16(1):77–83. doi: 10.1096/fj.01-0442hyp. [DOI] [PubMed] [Google Scholar]
- German MS, Moss LG, Wang J, Rutter WJ. The insulin and islet amyloid polypeptide genes contain similar cell-specific promoter elements that bind identical beta-cell nuclear complexes. Mol Cell Biol. 1992;12(4):1777–1788. doi: 10.1128/mcb.12.4.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsbury C, Kistler J, Aebi U, Arvinte T, Cooper GJ. Watching amyloid fibrils grow by time-lapse atomic force microscopy. J Mol Biol. 1999;285(1):33–39. doi: 10.1006/jmbi.1998.2299. [DOI] [PubMed] [Google Scholar]
- Granzotto A, Suwalsky M, Zatta P. Physiological cholesterol concentration is a neuroprotective factor against beta-amyloid and beta-amyloid-metal complexes toxicity. J Inorg Biochem. 2011;105(8):1066–1072. doi: 10.1016/j.jinorgbio.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Green JD, Goldsbury C, Kistler J, Cooper GJ, Aebi U. Human amylin oligomer growth and fibril elongation define two distinct phases in amyloid formation. J Biol Chem. 2004;279(13):12206–12212. doi: 10.1074/jbc.M312452200. [DOI] [PubMed] [Google Scholar]
- Haataja L, Gurlo T, Huang CJ, Butler PC. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr Rev. 2008;29(3):303–316. doi: 10.1210/er.2007-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Mizuno T, Michikawa M, Haass C, Yanagisawa K. Amyloid precursor protein in unique cholesterol-rich microdomains different from caveolae-like domains. Biochim Biophys Acta. 2000;1483(1):81–90. doi: 10.1016/s1388-1981(99)00174-2. [DOI] [PubMed] [Google Scholar]
- Hoppener JW, Lips CJ. Role of islet amyloid in type 2 diabetes mellitus. Int J Biochem Cell Biol. 2006;38(5–6):726–736. doi: 10.1016/j.biocel.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Hoppener JW, Ahren B, Lips CJ. Islet amyloid and type 2 diabetes mellitus. N Engl J Med. 2000;343(6):411–419. doi: 10.1056/NEJM200008103430607. [DOI] [PubMed] [Google Scholar]
- Huang CJ, Gurlo T, Haataja L, Costes S, Daval M, Ryazantsev S, et al. Calcium-activated calpain-2 is a mediator of beta cell dysfunction and apoptosis in type 2 diabetes. J Biol Chem. 2011;285(1):339–348. doi: 10.1074/jbc.M109.024190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaikaran ET, Clark A. Islet amyloid and type 2 diabetes: from molecular misfolding to islet pathophysiology. Biochim Biophys Acta. 2001;1537(3):179–203. doi: 10.1016/s0925-4439(01)00078-3. [DOI] [PubMed] [Google Scholar]
- Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes. 1999;48(3):491–498. doi: 10.2337/diabetes.48.3.491. [DOI] [PubMed] [Google Scholar]
- Jayasinghe SA, Langen R. Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry. 2005;44(36):12113–12119. doi: 10.1021/bi050840w. [DOI] [PubMed] [Google Scholar]
- Jayasinghe SA, Langen R. Membrane interaction of islet amyloid polypeptide. Biochim Biophys Acta. 2007;1768(8):2002–2009. doi: 10.1016/j.bbamem.2007.01.022. [DOI] [PubMed] [Google Scholar]
- Jha S, Sellin D, Seidel R, Winter R. Amyloidogenic propensities and conformational properties of ProIAPP and IAPP in the presence of lipid bilayer membranes. J Mol Biol. 2009;389(5):907–920. doi: 10.1016/j.jmb.2009.04.077. [DOI] [PubMed] [Google Scholar]
- Kayed R, Bernhagen J, Greenfield N, Sweimeh K, Brunner H, Voelter W, et al. Conformational transitions of islet amyloid polypeptide (IAPP) in amyloid formation in vitro. J Mol Biol. 1999;287(4):781–796. doi: 10.1006/jmbi.1999.2646. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300(5618):486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Khemtemourian L, Killian JA, Hoppener JW, Engel MF. Recent insights in islet amyloid polypeptide-induced membrane disruption and its role in beta-cell death in type 2 diabetes mellitus. Exp Diabetes Res. 2008;2008:421287. doi: 10.1155/2008/421287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight JD, Miranker AD. Phospholipid catalysis of diabetic amyloid assembly. J Mol Biol. 2004;341(5):1175–1187. doi: 10.1016/j.jmb.2004.06.086. [DOI] [PubMed] [Google Scholar]
- Knight JD, Hebda JA, Miranker AD. Conserved and cooperative assembly of membrane-bound alpha-helical states of islet amyloid polypeptide. Biochemistry. 2006;45(31):9496–9508. doi: 10.1021/bi060579z. [DOI] [PubMed] [Google Scholar]
- Konarkowska B, Aitken JF, Kistler J, Zhang S, Cooper GJ. The aggregation potential of human amylin determines its cytotoxicity towards islet beta-cells. FEBS J. 2006;273(15):3614–3624. doi: 10.1111/j.1742-4658.2006.05367.x. [DOI] [PubMed] [Google Scholar]
- Koo BW, Miranker AD. Contribution of the intrinsic disulfide to the assembly mechanism of islet amyloid. Protein Sci. 2005;14(1):231–239. doi: 10.1110/ps.041051205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krampert M, Bernhagen J, Schmucker J, Horn A, Schmauder A, Brunner H, et al. Amyloidogenicity of recombinant human pro-islet amyloid polypeptide (ProIAPP) Chem Biol. 2000;7(11):855–871. doi: 10.1016/s1074-5521(00)00034-x. [DOI] [PubMed] [Google Scholar]
- Lau TL, Gehman JD, Wade JD, Perez K, Masters CL, Barnham KJ, et al. Membrane interactions and the effect of metal ions of the amyloidogenic fragment Abeta(25–35) in comparison to Abeta(1–42) Biochim Biophys Acta. 2007;1768(10):2400–2408. doi: 10.1016/j.bbamem.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Lee G, Pollard HB, Arispe N. Annexin 5 and apolipoprotein E2 protect against Alzheimer’s amyloid-beta-peptide cytotoxicity by competitive inhibition at a common phosphatidylserine interaction site. Peptides. 2002;23(7):1249–1263. doi: 10.1016/s0196-9781(02)00060-8. [DOI] [PubMed] [Google Scholar]
- Lutz TA. Amylinergic control of food intake. Physiol Behav. 2006;89(4):465–471. doi: 10.1016/j.physbeh.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Lutz TA. The role of amylin in the control of energy homeostasis. Am J Physiol Regul Integr Comp Physiol. 2010;298(6):R1475–R1484. doi: 10.1152/ajpregu.00703.2009. [DOI] [PubMed] [Google Scholar]
- MacArthur DL, de Koning EJ, Verbeek JS, Morris JF, Clark A. Amyloid fibril formation is progressive and correlates with beta-cell secretion in transgenic mouse isolated islets. Diabetologia. 1999;42(10):1219–1227. doi: 10.1007/s001250051295. [DOI] [PubMed] [Google Scholar]
- Martin C. The physiology of amylin and insulin: maintaining the balance between glucose secretion and glucose uptake. Diabetes Educ. 2006;32(Suppl 3):101S–104S. doi: 10.1177/0145721706288S237. [DOI] [PubMed] [Google Scholar]
- Martinez A, Kapas S, Miller MJ, Ward Y, Cuttitta F. Coexpression of receptors for adrenomedullin, calcitonin gene-related peptide, and amylin in pancreatic beta-cells. Endocrinology. 2000;141(1):406–411. doi: 10.1210/endo.141.1.7261. [DOI] [PubMed] [Google Scholar]
- Marzban L, Trigo-Gonzalez G, Zhu X, Rhodes CJ, Halban PA, Steiner DF, et al. Role of beta-cell prohormone convertase (PC)1/3 in processing of pro-islet amyloid polypeptide. Diabetes. 2004;53(1):141–148. doi: 10.2337/diabetes.53.1.141. [DOI] [PubMed] [Google Scholar]
- McGuinness B, Passmore P. Can statins prevent or help treat Alzheimer’s disease? J Alzheimers Dis. 2010;20(3):925–933. doi: 10.3233/JAD-2010-091570. [DOI] [PubMed] [Google Scholar]
- McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, et al. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature. 1998;393(6683):333–339. doi: 10.1038/30666. [DOI] [PubMed] [Google Scholar]
- Mirzabekov TA, Lin MC, Kagan BL. Pore formation by the cytotoxic islet amyloid peptide amylin. J Biol Chem. 1996;271(4):1988–1992. doi: 10.1074/jbc.271.4.1988. [DOI] [PubMed] [Google Scholar]
- Miyazato M, Nakazato M, Shiomi K, Aburaya J, Toshimori H, Kangawa K, et al. Identification and characterization of islet amyloid polypeptide in mammalian gastrointestinal tract. Biochem Biophys Res Commun. 1991;181(1):293–300. doi: 10.1016/s0006-291x(05)81416-0. [DOI] [PubMed] [Google Scholar]
- Morfis M, Tilakaratne N, Furness SG, Christopoulos G, Werry TD, Christopoulos A, et al. Receptor activity-modifying proteins differentially modulate the G protein-coupling efficiency of amylin receptors. Endocrinology. 2008;149(11):5423–5431. doi: 10.1210/en.2007-1735. [DOI] [PubMed] [Google Scholar]
- Moriarty DF, Raleigh DP. Effects of sequential proline substitutions on amyloid formation by human amylin20–29. Biochemistry. 1999;38(6):1811–1818. doi: 10.1021/bi981658g. [DOI] [PubMed] [Google Scholar]
- Mulder H, Ahren B, Sundler F. Islet amyloid polypeptide and insulin gene expression are regulated in parallel by glucose in vivo in rats. Am J Physiol. 1996;271(6 Pt 1):E1008–E1014. doi: 10.1152/ajpendo.1996.271.6.E1008. [DOI] [PubMed] [Google Scholar]
- Munishkina LA, Fink AL. Fluorescence as a method to reveal structures and membrane-interactions of amyloidogenic proteins. Biochim Biophys Acta. 2007;1768(8):1862–1885. doi: 10.1016/j.bbamem.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Nakazato M, Asai J, Miyazato M, Matsukura S, Kangawa K, Matsuo H. Isolation and identification of islet amyloid polypeptide in normal human pancreas. Regul Pept. 1990;31(3):179–186. doi: 10.1016/0167-0115(90)90004-g. [DOI] [PubMed] [Google Scholar]
- Nishi M, Sanke T, Seino S, Eddy RL, Fan YS, Byers MG, et al. Human islet amyloid polypeptide gene: complete nucleotide sequence, chromosomal localization, and evolutionary history. Mol Endocrinol. 1989;3(11):1775–1781. doi: 10.1210/mend-3-11-1775. [DOI] [PubMed] [Google Scholar]
- Opie EL. On the relation of chronic interstitial pancreatitis to the Islands of Langerhans and to diabetes melutus. J Exp Med. 1901;5(4):397–428. doi: 10.1084/jem.5.4.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padrick SB, Miranker AD. Islet amyloid: phase partitioning and secondary nucleation are central to the mechanism of fibrillogenesis. Biochemistry. 2002;41(14):4694–4703. doi: 10.1021/bi0160462. [DOI] [PubMed] [Google Scholar]
- Pieri L, Bucciantini M, Guasti P, Savistchenko J, Melki R, Stefani M. Synthetic lipid vesicles recruit native-like aggregates and affect the aggregation process of the prion Ure2p: insights on vesicle permeabilization and charge selectivity. Biophys J. 2009;96(8):3319–3330. doi: 10.1016/j.bpj.2008.12.3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper K, Brickwood S, Turnpenny LW, Cameron IT, Ball SG, Wilson DI, et al. Beta cell differentiation during early human pancreas development. J Endocrinol. 2004;181(1):11–23. doi: 10.1677/joe.0.1810011. [DOI] [PubMed] [Google Scholar]
- Poyner DR, Sexton PM, Marshall I, Smith DM, Quirion R, Born W, et al. International Union of Pharmacology. XXXII. The mammalian calcitonin gene-related peptides, adrenomedullin, amylin, and calcitonin receptors. Pharmacol Rev. 2002;54(2):233–246. doi: 10.1124/pr.54.2.233. [DOI] [PubMed] [Google Scholar]
- Quist A, Doudevski I, Lin H, Azimova R, Ng D, Frangione B, et al. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc Natl Acad Sci U S A. 2005;102(30):10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riediger T, Zuend D, Becskei C, Lutz TA. The anorectic hormone amylin contributes to feeding-related changes of neuronal activity in key structures of the gut-brain axis. Am J Physiol Regul Integr Comp Physiol. 2004;286(1):R114–R122. doi: 10.1152/ajpregu.00333.2003. [DOI] [PubMed] [Google Scholar]
- Ritzel RA, Meier JJ, Lin CY, Veldhuis JD, Butler PC. Human islet amyloid polypeptide oligomers disrupt cell coupling, induce apoptosis, and impair insulin secretion in isolated human islets. Diabetes. 2007;56(1):65–71. doi: 10.2337/db06-0734. [DOI] [PubMed] [Google Scholar]
- Rivera JF, Costes S, Gurlo T, Glabe CG, Butler PC. Autophagy defends pancreatic beta cells from human islet amyloid polypeptide-induced toxicity. J Clin Invest. 2014;124(8):3489–3500. doi: 10.1172/JCI71981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rymer DL, Good TA. The role of prion peptide structure and aggregation in toxicity and membrane binding. J Neurochem. 2000;75(6):2536–2545. doi: 10.1046/j.1471-4159.2000.0752536.x. [DOI] [PubMed] [Google Scholar]
- Salamekh S, Brender JR, Hyung SJ, Nanga RP, Vivekanandan S, Ruotolo BT, et al. A two-site mechanism for the inhibition of IAPP amyloidogenesis by zinc. J Mol Biol. 2011;410(2):294–306. doi: 10.1016/j.jmb.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Schulz-Schaeffer W, Hartmann T, Schulz JB, Simons M. Cholesterol depletion reduces aggregation of amyloid-beta peptide in hippocampal neurons. Neurobiol Dis. 2006;23(3):573–577. doi: 10.1016/j.nbd.2006.04.015. [DOI] [PubMed] [Google Scholar]
- Simmons C, Ingham V, Williams A, Bate C. Platelet-activating factor antagonists enhance intracellular degradation of amyloid-beta42 in neurons via regulation of cholesterol ester hydrolases. Alzheimers Res Ther. 2014;6(2):15. doi: 10.1186/alzrt245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1(1):31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJ, et al. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid. 2010;17(3–4):101–104. doi: 10.3109/13506129.2010.526812. [DOI] [PubMed] [Google Scholar]
- Smith PE, Brender JR, Ramamoorthy A. Induction of negative curvature as a mechanism of cell toxicity by amyloidogenic peptides: the case of islet amyloid polypeptide. J Am Chem Soc. 2009;131(12):4470–4478. doi: 10.1021/ja809002a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susa AC, Wu C, Bernstein SL, Dupuis NF, Wang H, Raleigh DP, et al. Defining the molecular basis of amyloid inhibitors: human islet amyloid polypeptide-insulin interactions. J Am Chem Soc. 2014;136(37):12912–12919. doi: 10.1021/ja504031d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trikha S, Jeremic AM. Clustering and internalization of toxic amylin oligomers in pancreatic cells require plasma membrane cholesterol. J Biol Chem. 2011;286(41):36086–36097. doi: 10.1074/jbc.M111.240762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trikha S, Jeremic AM. Distinct internalization pathways of human amylin monomers and its cytotoxic oligomers in pancreatic cells. PLoS One. 2013;8(9):e73080. doi: 10.1371/journal.pone.0073080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu LH, Raleigh DP. Role of aromatic interactions in amyloid formation by islet amyloid polypeptide. Biochemistry. 2013;52(2):333–342. doi: 10.1021/bi3014278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagoner PK, Chen C, Worley JF, Dukes ID, Oxford GS. Amylin modulates beta-cell glucose sensing via effects on stimulus-secretion coupling. Proc Natl Acad Sci U S A. 1993;90(19):9145–9149. doi: 10.1073/pnas.90.19.9145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Xu J, Finnerty J, Furuta M, Steiner DF, Verchere CB. The prohormone convertase enzyme 2 (PC2) is essential for processing pro-islet amyloid polypeptide at the NH2-terminal cleavage site. Diabetes. 2001;50(3):534–539. doi: 10.2337/diabetes.50.3.534. [DOI] [PubMed] [Google Scholar]
- Westermark P, Wernstedt C, Wilander E, Sletten K. A novel peptide in the calcitonin gene related peptide family as an amyloid fibril protein in the endocrine pancreas. Biochem Biophys Res Commun. 1986;140(3):827–831. doi: 10.1016/0006-291x(86)90708-4. [DOI] [PubMed] [Google Scholar]
- Westermark P, Engstrom U, Johnson KH, Westermark GT, Betsholtz C. Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc Natl Acad Sci U S A. 1990;87(13):5036–5040. doi: 10.1073/pnas.87.13.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark G, Westermark P, Eizirik DL, Hellerstrom C, Fox N, Steiner DF, et al. Differences in amyloid deposition in islets of transgenic mice expressing human islet amyloid polypeptide versus human islets implanted into nude mice. Metabolism. 1999;48(4):448–454. doi: 10.1016/s0026-0495(99)90102-6. [DOI] [PubMed] [Google Scholar]
- Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev. 2011;91(3):795–826. doi: 10.1152/physrev.00042.2009. [DOI] [PubMed] [Google Scholar]
- Williamson R, Usardi A, Hanger DP, Anderton BH. Membrane-bound beta-amyloid oligomers are recruited into lipid rafts by a fyn-dependent mechanism. FASEB J. 2008;22(5):1552–1559. doi: 10.1096/fj.07-9766com. [DOI] [PubMed] [Google Scholar]
- Wiltzius JJ, Sievers SA, Sawaya MR, Cascio D, Popov D, Riekel C, et al. Atomic structure of the cross-beta spine of islet amyloid polypeptide (amylin) Protein Sci. 2008;17(9):1467–1474. doi: 10.1110/ps.036509.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wookey PJ, Lutz TA, Andrikopoulos S. Amylin in the periphery II: an updated mini-review. Sci World J. 2006;6:1642–1655. doi: 10.1100/tsw.2006.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young A, Denaro M. Roles of amylin in diabetes and in regulation of nutrient load. Nutrition. 1998;14(6):524–527. doi: 10.1016/s0899-9007(98)00044-6. [DOI] [PubMed] [Google Scholar]
- Zhang S, Liu J, Dragunow M, Cooper GJ. Fibrillogenic amylin evokes islet beta-cell apoptosis through linked activation of a caspase cascade and JNK1. J Biol Chem. 2003;278(52):52810–52819. doi: 10.1074/jbc.M308244200. [DOI] [PubMed] [Google Scholar]
- Zhang S, Liu H, Chuang CL, Li X, Au M, Zhang L, et al. The pathogenic mechanism of diabetes varies with the degree of overexpression and oligomerization of human amylin in the pancreatic islet beta cells. FASEB J. 2014;28(12):5083–5096. doi: 10.1096/fj.14-251744. [DOI] [PubMed] [Google Scholar]
- Zhao H, Tuominen EK, Kinnunen PK. Formation of amyloid fibers triggered by phosphatidylserine-containing membranes. Biochemistry. 2004;43(32):10302–10307. doi: 10.1021/bi049002c. [DOI] [PubMed] [Google Scholar]
- Zhao H, Jutila A, Nurminen T, Wickstrom SA, Keski-Oja J, Kinnunen PK. Binding of endostatin to phosphatidylserine-containing membranes and formation of amyloid-like fibers. Biochemistry. 2005;44(8):2857–2863. doi: 10.1021/bi048510j. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Jayawardhana D, Guan X. Stochastic study of the effect of ionic strength on non covalent interactions in protein pores. Biophys J. 2008;94(4):1267–1275. doi: 10.1529/biophysj.107.117598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Hu R, Sciacca MF, Brender JR, Chen H, Ramamoorthy A, et al. Non-selective ion channel activity of polymorphic human islet amyloid polypeptide (amylin) double channels. Phys Chem Chem Phys. 2014;16(6):2368–2377. doi: 10.1039/c3cp53345j. [DOI] [PMC free article] [PubMed] [Google Scholar]