

Abstract
Many types of cancer, including glioma, melanoma, non-small cell lung cancer (NSCLC), among others, are resistant to proapoptotic stimuli and thus poorly responsive to current therapies based on the induction of apoptosis in cancer cells. The current investigation describes the synthesis and anticancer evaluation of unique C12-Wittig derivatives of polygodial, a terpenenoid dialdehyde isolated from Persicaria hydropiper (L.) Delabre. These compounds were found to undergo an unprecedented pyrrole formation with primary amines in a chemical model system, a reaction that could be relevant in the biological environment and lead to the pyrrolation of lysine residues in the target proteins. The anticancer evaluation of these compounds revealed their promising activity against cancer cells displaying various forms of drug resistance, including resistance to proapoptotic agents. Mechanistic studies indicated that compared to the parent polygodial, which displays fixative general cytotoxic action against human cells, the C12-Wittig derivatives exerted their antiproliferative action mainly through cytostatic effects explaining their activity against apoptosis-resistant cancer cells. The possibility for an intriguing covalent modification of proteins through a novel pyrrole formation reaction, as well as useful activities against drug resistant cancer cells, make the described polygodial-derived chemical scaffold an interesting new chemotype warranting thorough investigation.
Keywords: glioblastoma, ion channel, capsaicin, vanilloid, resiniferatoxin, cannabidiol
1. Introduction
Apoptosis resistance is a hallmark of cancer, because defects in apoptosis regulators invariably accompany tumorigenesis and sustain malignant progression. Therefore, because most standard chemotherapeutic agents work by the induction of apoptosis in cancer cells, its disruption during tumor evolution can promote drug resistance and result in therapy failure [1–4]. Indeed, many types of cancer, such as the tumors of the lung, liver, stomach, esophagus, pancreas as well as melanomas and gliomas, are intrinsically resistant to the induction of apoptosis and thus refractory to the most of the currently available chemotherapeutic agents [5]. For example, patients afflicted by glioblastoma multiforme (GBM) [6], which responds poorly to conventional chemotherapy with proapoptotic agents [7–9], have a median survival expectancy of less than 14 months when treated with the best available protocol [10]. GBM is characterized by a deregulated tumor genome containing opportunistic deletions of tumor suppressor genes as well as amplification or mutational hyperactivation of receptor tyrosine kinase receptors. These genetic changes result in enhanced survival pathways and systematic defects in the apoptotic machinery. One solution to apoptosis resistance entails the complementation of cytotoxic therapeutic regimens with cytostatic agents and thus a search for novel cytostatic anticancer drugs that can overcome cancer cell resistance to apoptosis is an important pursuit [11–19].
Furthermore, tumors that initially respond to chemotherapy can still become refractory to the continuing treatment by developing a multi-drug resistant phenotype (MDR) affecting a broad spectrum of structurally and mechanistically diverse antitumor agents [20,21]. The development of MDR is common with many conventional chemotherapies, including the well-known vinca alkaloids [22] and taxanes [23]. Thus, the search for agents capable of circumventing MDR mechanisms is another important area of research aiming to combat drug-resistant cancers [24].
In the pursuit of agents active against drug-resistant cancers, our labs have been investigating compounds targeting ion channels [25,26], whose alterations often represent important mechanisms in the impairment of apoptosis and development of drug resistance [27]. Our attention was recently brought to the transient receptor potential vanilloid 1 receptor (TRPV1), which is a non-selective cation channel with preference for Ca2+ and identified as molecular target of a pungent component of hot chili pepper capsaicin, used as a spice in the culinary traditions of many countries and as an antinociceptive in traditional medicine [28]. Research has shown that in addition to its expression in sensory neurons and involvement in different modalities of pain, TRPV1 is also upregulated in various human cancer cells [29–31]. It has thus been proposed to be a an attractive target for the treatment of brain tumors [32], as it was shown that its activation with endogenous agonists leads to endoplasmic reticulum (ER) stress in human glioma cells, followed by cell death [32]. Many reports investigating TRPV1-targeting agents such as capsaicin [33–36], resiniferatoxin [33,37], capsazepine [34,35], and SB366791 [34], as potential anticancer agents, have appeared in the literature. Curiously, however, the group of α,β-unsaturated 1,4-dialdehyde terpenoids (Figure 1), studied for many diverse biological properties [38] and known for their TRPV1 agonistic activities [38–43] have not been studied as anticancer agents. Polygodial (1, Figure 1) is the most well known representative of these 1,4-dialdehydes and it was first isolated as a pungent component of the sprout of Persicaria hydropiper (L.) Delabre (Polygonaceae), a plant used as a popular condiment for sashimi in Japan [44]. Compound 1 tastes hot to the human tongue and possesses antifeedant activities [45–47], both of which are evidently mediated through TRPV1-targeting [38–43] possibly through the formation of a covalent complex [38]. Encouraged by several earlier reports of cytotoxic activity associated with 1 [49–54], we prepared a series of its chemical derivatives and studied their TRPV1 agonistic and anticancer activities in detail. A related publication resulting from this study describes the discovery of useful anticancer activities associated with 9-epipolygodial [55]. Herein, we present a series of C12-Wittig derivatives of 1 that exert their antiproliferative action mainly through cytostatic effects and possess promising activities against cancer cells resistant to apoptosis as well as those with an MDR phenotype. Furthermore, these compounds undergo an unprecedented pyrrole formation with primary amines, a reaction that could be relevant in a biological environment and lead to the pyrrolylation of lysine residues in the target proteins through this previously unknown chemical mechanism.
Figure 1.
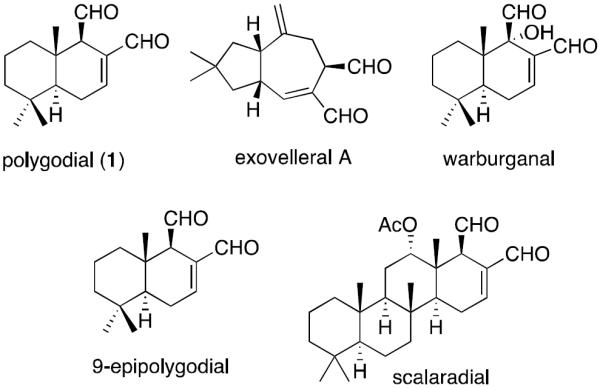
Structures of selected α,β-unsaturated 1,4-dialdehyde terpenoids.
2. Chemistry
During earlier studies of reactivity of 1 toward various nucleophiles, the possibility of pyrrole formation with an active site lysine was mentioned in the literature [46], although no pyrrole adduct was isolated in these experiments. In our own attempts [55] to demonstrate the feasibility of what could be referred to as the modified Paal-Knorr [56] pyrrolylation of proteins by 1, it was found that although N-alkyl pyrrole 2 (R = Bn, Figure 2A) formed, it was extremely unstable and readily oxidized in air. This stability problem was solved by the preparation and successful isolation of electron-deficient N-aryl pyrroles 2 (R = Ph and p-NO2-C6H4); however, these adducts are obviously not relevant biologically as the resultant covalent complex with a lysine residue would be an alkyl pyrrole and thus also readily oxidizable. Although examples of lysine pyrrolylation with small molecules are extremely rare [57], its involvement in n-hexane-induced axonal atrophy in the central nervous system is a well studied phenomenon. It has been demonstrated both in vitro and in vivo that 2,5-hexanedione, a neurotoxic n-hexane metabolite, pyrrolylates the lysine residues of axonal cytoskeleton proteins within specific regions of neurofilaments (Figure 2B) [58]. Importantly, subsequent oxidation of the 2,5-dimethylpyrrole adducts has been shown to be an obligatory event in the induction of neuropathy [59]. This line of reasoning would suggest that lysine pyrrolylation with a non-selective 1,4-dicarbonyl-containing small molecule could be generally toxic to cells, especially if the formed pyrrole adducts are unstable and may lead to further non-specific protein cross-linking events. Whether it is for this reason or another, as reported herein, our investigation of 1 as a potential anticancer agent led us to conclude that at the concentrations necessary to induce cancer cell death, 1 behaves as a toxic fixative compound with little promise as a potential drug.
Figure 2.

(A) Demonstration of the feasibility of the modified Paal-Knorr condensation of 1 to form pyrrole 2, (B) Paal-Knorr pyrrole formation implicated in the neurotoxicity of hexane and (C) novel pyrrolylation of primary amines with C12-Wittig derivatives reported herein.
It is likely that the susceptibility of N-alkyl pyrroles 2 toward oxidation is due to the presence of the conjugated C7,C8-alkene, which in addition to the electronic effects, possibly imparts significant strain by forcing the trans-fused ring B into planarity. Our chemical studies of 1 indicated that the C12-aldehyde is considerably more reactive than its C11-counterpart and thus it seemed possible to prepare C12-Wittig derivatives of the type 3 in Figure 2C, where X would be an electron-withdrawing group. Based on mechanistic considerations (intermediates A and B in Figure 2C), the formation of pyrroles 4 appeared feasible in the reaction of 3 with primary amines and adducts 4 were predicted to be more stable due to the absence of the destabilizing C7,C8-alkene. It was indeed discovered that polygodial (1) reacts selectively at the C12-aldehyde functionality with a variety of stabilized phosphorous ylides (Figure 2) producing a,b-unsaturated esters 5–10, nitrile 11 and ketone 12 with high regioselectivity and in excellent yields. The products arising from alkenylation of the C11-aldehyde were not detected in these reaction mixtures. In addition, a terminal alkyne-containing ester 13 was prepared for a possible future conjugate synthesis through click chemistry. Finally, when 5 was reacted with BnNH2 in THF in the presence of catalytic amount of AcOH, pyrrole 14 was cleanly formed in 85% yield and turned out to be a stable isolable compound.
3. Anticancer Evaluation
The synthesized C12-Wittig derivatives were evaluated for antiproliferative activities in a panel of cancer cell lines that included apoptosis-resistant human U373 glioblastoma (GBM) [60], human A549 non-small cell lung cancer (NSCLC) [61] and human SKMEL-28 melanoma [62] as well as apoptosis-sensitive human Hs683 anaplastic oligodendroglioma [60] and human MCF-7 breast cancer [63]. While 1 was ineffective in this cancer cell panel, the C12-Wittig derivatives showed moderate double-digit micromolar potencies. Of note, although the potencies in this series of compounds differed among the individual cell lines, the average GI50 values over the 72-hour treatment period were quite similar, indicating either similar intracellular target binding requirements unaffected by the nature of the type of the group X (Figure 3) or a covalent complex formation leading to an identical mode of target inhibition. Further analysis of these data revealed that the C12-Wittig derivatives did not discriminate between apoptosis resistant and apoptosis-sensitive cells and displayed comparable potencies in both cell types, indicating that compounds of this class are capable of overcoming apoptosis resistance (Table 1).
Figure 3.
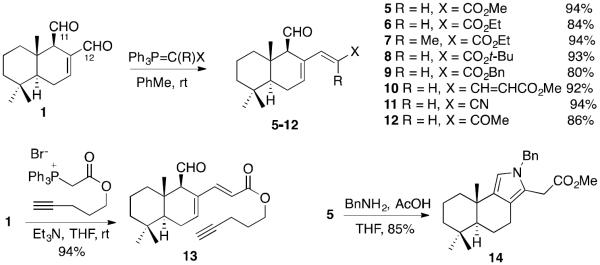
Synthesis of C12-Wittig derivatives 5–13 and formation of pyrrole 14 from 5 and BnNH2.
Table 1.
In Vitro growth inhibitory effects of the C12-Wittig derivatives of 1
| compound | GI50
in vitro values (μM)a |
|||||
|---|---|---|---|---|---|---|
| A549 | SKMEL-28 | MCF-7 | U373 | Hs683 | Mean + SEM | |
| 1 | 90 | 65 | 75 | >99 | 95 | > 85 ± 6 |
| 5 | 21 | 27 | 7 | 22 | 24 | 20 ± 3 |
| 6 | 25 | 29 | 30 | 37 | 29 | 30 ± 2 |
| 7 | 26 | 37 | 27 | 37 | 28 | 31 ± 2 |
| 8 | 29 | 41 | 32 | 40 | 29 | 34 ± 3 |
| 9 | 28 | 39 | 27 | 39 | 31 | 33 ± 3 |
| 10 | 29 | 25 | 31 | 27 | 12 | 25 ± 3 |
| 11 | 27 | 42 | 31 | 36 | 28 | 33 ± 3 |
| 12 | 24 | 28 | 20 | 25 | 22 | 24 ± 1 |
| 13 | 30 | 36 | 30 | 35 | 33 | 33 ± 1 |
Average concentration required to reduce the viability of cells by 50% after a 72 h treatment relative to a control, each experiment performed in sextuplicates, as determined by MTT assay.
Although the moderate double-digit potencies of the synthesized derivatives are somewhat unremarkable, the analysis of the actual experimental growth curves indicated that these compounds eliminate all cells in the cultures and generate no resistant populations (close to 0% cell viability) at concentrations just slightly exceeding their GI50 values, as shown for compound 5 in Figure 4A. The contrasting effects between derivative 5 and common pro-apoptotic agents paclitaxel and podophyllotoxin on apoptosis-resistant A549 NSCLC and U87 GBM cells are shown in Figures 4B and 4C. Indeed, the normally low nanomolar antiproliferative agents paclitaxel and podophyllotoxin have no effect on proliferation of ca. 50% of cells at concentrations up to 100 μM [15], whereas 5 exhibited growth inhibitory properties against most of the cells in these cultures and, with increasing concentration, reached the antiproliferative levels of a non-discriminate cytotoxic agent phenyl arsine oxide (PAO). Compound 5 demonstrated a similar behaviour in docetaxel-resistant SCC4 and cisplatin-resistant SCC25 human oral cancer cell lines, as well as the docetaxel-resistant PC-3 human prostate cells (data not shown).
Figure 4.

(A) The absense of resistant populations in all 5 cultures tested with analogue 5 and contrasting effects on viability of all cells between 5 and standard chemotherapeutic agents paclitaxel and podophyllotoxin in (B) A549 NSCLC and (C) U87 glioblastoma cell cultures. PODO = podophyllotoxin, PAO = phenyl arsine oxide.
Often, tumors initially respond to chemotherapy but eventually become refractory to the continuing treatment. Such acquired resistance commonly occurs through the development of a multi-drug resistant phenotype (MDR) [64,65] affecting many common chemotherapeutic agents, including the vinca alkaloids [66] and taxanes [67]. In addition, the development of apoptosis resistance and MDR mechanisms is often related and concurrent [68,69], and thus it was of interest to evaluate 5 against MDR cells. Towards this end, the MDR uterine sarcoma cell line MES-SA/Dx5, established by growing the parent uterine sarcoma MES-SA in the presence of increasing concentrations of doxorubicin and resistant to multiple functionally and structurally unrelated molecules [70], was utilized. It was found that paclitaxel and vinblastine lost their potency by a factor of a thousand when tested for antiproliferative activity against the MES-SA/Dx5 MDR cell line as compared with the parent MES-SA cells. In contrast, there was little variation in the sensitivities of the two cell lines towards 5 (Table 2).
Table 2.
Antiproliferative effects of 5 against MDR cells
| GI50
in vitro values (μM)a |
||
|---|---|---|
| MES-SA | MES-SA/Dx5 | |
|
|
||
| Paclitaxel | 0.007 | 10 |
| Vinblastine | 0.006 | 5 |
| 5 | 40 | 45 |
Concentration required to reduce the viability of cells by 50% after a 48 h treatment with the indicated compounds relative to a DMSO control from two independent experiments, each performed in 4 replicates, as determined by the MTT assay.
Due to the ability of 5 to overcome drug resistance, it was further challenged with a panel of GBM cell cultures maintained under neurosphere conditions. Neurospheres, known to promote the growth of stem-like cells from human glioma tissue, are generally resistant to radiation and chemotherapy [71–74]. Furthermore, compared with serum cultured glioma cell lines they have been shown on both histological and genetic levels to serve as a better model of human gliomas when injected into the brains of mice [75–78]. Figure 5 shows the results of cytotoxicity evaluation for compound 5 against GBM neurosphere cultures carrying a tumor suppressor cdkn2a deletion [79] as well as PDGFB [80] and EGFRvIII [81] and amplifications, representing frequent mutations in high-grade astrocytic tumors. The data indicate that compound 5 used at 20 μM (average GI50 in Table 1) shows effectiveness similar to that of cannabidiol (CBD) at 10 μM, an orphan drug advanced to phase II clinical trials for the treatment of GBM. From the mechanistic perspective, it is interesting that CBD loses its activity against cells with the cdkn2a deletion and PDGFB amplification that additionally lack the TRPV1 gene (Figure 5D), whereas 5 appears to be even more effective against these cells (Figure 5B) than against their counterparts containing the intact TRPV1 (Figure 5B). These data indicate that while the CBD-induced anticancer effects could indeed be mediated at least in part through TRPV1, which is consistent with the literature data suggesting activation of this receptor by CBD [82], 5 appears to work through a TRPV1-independent mechanism.
Figure 5.
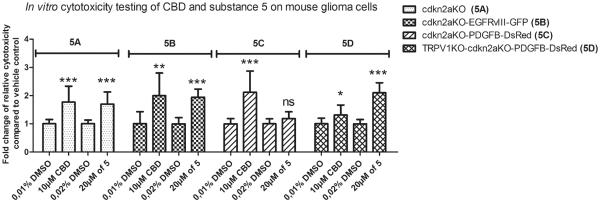
Activity of 5 against neurosphere glioma cell cultures with clinically relevant mutations. Transgenic mouse glioma cells of defined molecular subtypes were generated by forced expression of EGFRvIII (classical GBM subtype), PDGFB (proneural GBM subtype) in cdkn2a-deficient and cdkn2a/TRPV1-doubly deficient subventricular neural precursors (NPC). These primary cell cultures were treated for 24 hours either with 10 μM CBD versus a corresponding vehicle-control (containing 0,01% DMSO) or 20 μM of 5 versus vehicle control (0,02% DMSO). Cytotoxicity was measured 24 h after incubation and base-line cytotoxicity levels in the controls were arbitrarily defined as 1. Read-outs from treated cells were normalized to their respective vehicle controls and fold change of relative cytotoxicity was calculated. Each bar represents the mean ± SD from two independent experiments; satistical significance, as determined by unpaired t-tests, is indicated: *** represents p < 0,001; ** p < 0,005; * p=0,0257; ns not significant).
4. Evaluation of Vanilloid Activities
To confirm that the synthesized derivatives of 1 do not have an effect on TRPV1, compound 5 was assessed for the inhibition of specific binding of [3H]-resiniferatoxin (RTX) in rat spinal cord membranes [41]. The results (Figure 6A) demonstrate that in contrast to 1, which displayed 76% inhibition at the concentration of 10 μM, compound 5 failed to show any activity in this assay. In a complementary assessment of the TRPV1-mediated effects, measurements of Ca2+ entry into MDA-MB-231 breast cancer cells abundantly expressing TRPV1 receptors [83] were performed (Figure 6B and C). In agreement with the [3H]-RTX TRPV1 assay, 1 at its average GI50 concentration of 80 μM (from Table 1) caused a transient and synchronous [Ca2+]i increase in all tested cells. Moreover this Ca2+ response can be prevented by the TRPV1 blocker capsazepine indicating the activation of TRPV1 in the plasma membrane of MDA-MB-231 cells (data not shown). On the other hand, compound 5 at its average GI50 of 20 μM (from Table 1) displayed a different pattern with sustained and asynchronous [Ca2+] increases displaying a different Ca2+-signature induced by compound 5 compared to 1.
Figure 6.
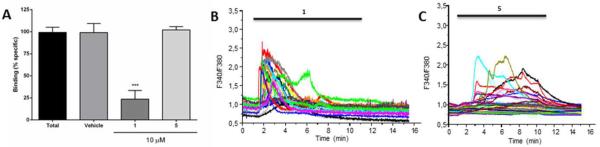
(A) Evaluation of compound 5 in a [3H]-RTX TRPV1 displacement assay. Effects of 1 and 5 at the concentration of 10 μM on the specific binding of [3H]-RTX to the vanilloid site of TRPV1 receptor from rats spinal cord membranes. Results are expressed as mean ± S.E.M from 3 independent experiments, analyzed by one way analysis of variance (ANOVA), followed by Dunnett's multiple comparison test (***p<0.05 and ****p<0.001). (B and C) Evaluation of 5 for TRPV1 activity in MDA-MB-231 breast cancer cells. (B) Effect of 1 (80 μM) on MDA-MB-231 [Ca2+]i. (C) Effect of 5 (20 μM) on MDA-MB-231 [Ca2+]i.
5. Computer modeling
In an attempt to understand why the Wittig derivatization at C12 of 1 leads to compounds devoid of TRPV1 activity, capsaicin, 1 and Wittig derivative 5 were docked to the capsaicin/RTX binding site on TRPV1 (Figure 7). The receptor for these studies was obtained by the refinement of the cryo-EM structure of TRPV1 (protein data bank (PDB) ID 3J5R) [84], as described in our related manuscript [55]. The docking of capsaicin reveals that it is well accommodated in this pocket, with the polar phenolic moiety orientated toward the polar “southern” region of the binding pocket and the apolar alkyl chain extending up into the apolar “northern” region of the pocket (Figure 7, left). The phenolic proton of 1 is well positioned to form a hydrogen bonding interaction to the carboxylate of Glu570 and the amide proton linking the alkyl chain is well suited to form another hydrogen bonding interaction to Thr550 (as was observed for the reduced polygodiol derivative described in our related manuscript) [55]. Compound 1 is similarly accommodated in the pocket, although being somewhat smaller, hydrogen bonding interactions occur between the aldehydes and the residues Thr550 and Tyr511, thereby allowing the hydrophobic region of the molecule to be well placed in the “northern” hydrophobic region of the binding pocket. Interestingly, docking of the Wittig derivative 5 suggests a similar pose as the best possible orientation; however, this would require that the apolar ester chain protrudes deeply into the polar “southern” region of the pocket, with the methyl group in close proximity to the negatively charged carboxylate of Glu570. This undesirable mismatched interaction may well account for the loss of TRPV1 activity for 5.
Figure 7.
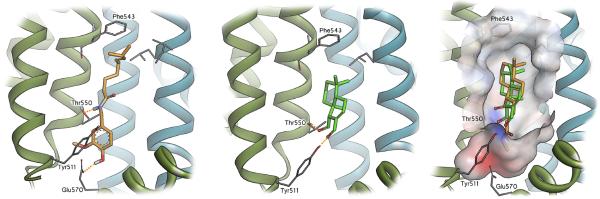
Molecular modeling showing the capsaicin binding region of TRPV1, with the likely binding pose of capsaicin (left). Compound 1 is also well accommodated in this pocket (middle), but the Wittig derivative 5 (right - displayed in orange and overlayed with 1 in green) is required to bind with its apolar ester chain embedded in the polar “southern” region of the pocket (as highlighted by the solvent interpolated charge surface).
6. Studies of morphological changes in affected cancer cells
Although many reports investigating vanilloid agonists as potential anticancer agents have appeared in the literature, the involvement of TRPV1 in mediating their antiproliferative effects has been questioned on many occasions as vanilloid antagonists have consistently failed to prevent capsaicin or RTX-induced cell death [33–37]. In the related publication [55], we show that these observations also apply to α,β-unsaturated 1,4-dialdehyde terpenoids, such as 1 and 9-epipolygodial, and that cancer cell death induced by these compounds cannot be explained by TRPV1-targeting. In an effort to gain insight into possible mechanisms associated with antiproliferative properties of 1 and the C12-Wittig derivatives, the morphological changes in cells treated with these compounds were studied with computer-assisted phase-contrast microscopy [14] (quantitative videomicroscopy). Figure 8 shows that 1, at a concentration equalling its GI50 value against the U373 cell line (Table 1), exhibited strong fixative effects on these GBM cells. In contrast, the C12-Wittig derivatives 5 and 13 inhibited cancer cell proliferation without inducing cell death when assayed at their GI50 concentrations (Table 1).
Figure 8.

In vitro videomicroscopic analysis of the anticancer effects of 1 and its C12-Wittig derivatives 5 and 13. The U373 human glioma cell line was treated with polygodial, 5 and 13 at their mean GI50 concentrations (Table 1) or left untreated. Videomicroscopy enabled taking pictures of the culture field every 4 minutes. The experiment was conducted once in triplicate. While the morphology of cells treated with 1 was fixed over time, 5 and 13 exerted cytostatic effects on U373 cells.
To confirm the results obtained in the videomicroscopy experiments, trypan blue assay was employed to detect necrotic and late apoptotic cells that had lost their plasma membrane integrity (Figure 9). Indeed, while cells treated with 1 were all blue-stained before methanol fixation, the 13-treated cells were still alive after 72 h of treatment. These observations support the results of the MTT assay and provide an explanation for the effectiveness of the C12-Wittig derivatives against cells, which display resistance to apoptosis induction. In contrast, at the concentrations necessary to induce cancer cell death 1 behaves as a toxic fixative compound of little promise as a potential drug.
Figure 9.
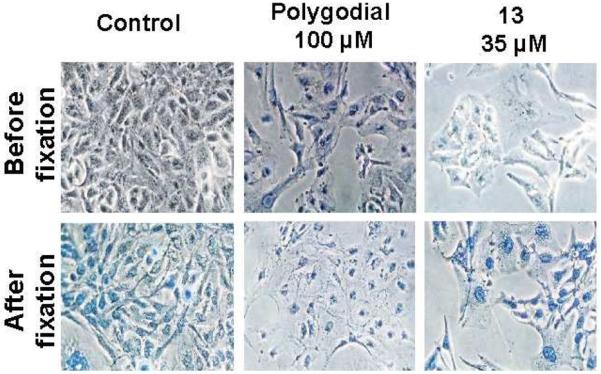
Viability of U373 cells by trypan blue staining. U373 cells were treated for 72 h with 1 or 13 and stained with trypan blue. The experiment was conducted once in triplicate. After having taken pictures, cells were fixed with ice-cold methanol and again stained with trypan blue as internal positive control. While cells treated with 1 were all blue-stained before methanol fixation, the 13-treated cells were still alive after 72 h of treatment.
7. Conclusion
Cancers characterized by an intrinsic resistance to the induction of apoptosis are refractory to the most of the currently available chemotherapeutic agents and present a formidable clinical challenge [1–5]. For example, GBM is one of the most feared of all human diseases both due to the certain fatal outcome and a rapid debilitating loss of cognitive function. The therapy-related improvement of overall survival has been counted in months, not years, for the last 40 years and the GBM clinic is in dire need of conceptually new treatment strategies [6–8]. A great deal of recent research has been aimed at overcoming the apoptosis resistance of GBM cells by rendering them more susceptible to therapy-induced apoptosis [9]. Thus, in GBM cells, several key regulatory elements of cell homeostasis and apoptosis are altered through inactivating mutations, methylation, or altered expression. These alterations affect the p53 protein, the BCL-2 protein family, the inhibitor of apoptosis proteins (IAPs) or receptor tyrosine kinases (e.g. the epidermal growth factor receptor (EGFR)) and their downstream signaling cascades. All of these represent attractive targets for therapeutic interventions and have been pursued by various researchers [9,85]. However, to date, the results from the clinical trials of a number of such agents, mostly targeting growth factor pathways, have been disappointing. For example, a randomized, controlled, phase II trial conducted with erlotinib, a small molecule targeting the EGFR signaling, unfortunately showed no therapeutic benefit [86].
The identification of agents whose mode of action is not based on cell death represents an alternative approach. Indeed, delaying proliferation in cancer cells over certain periods of time will induce cell death that usually occurs via apoptosis. In this case, apoptosis is a consequence and not a cause of the drug-induced effects. The present investigation led to the discovery of such agents, which represent novel derivatives of polygodial, a sesquiterpenoid widely studied as a TRPV1 agonist. Indeed, the biological effects of these compounds are not TRPV1-mediated and compared to the parent polygodial, which displays a fixative general cytotoxic action against human cells, the C12-Wittig derivatives exert their antiproliferative action mainly through cytostatic effects explaining their activity against apoptosis-resistant cancer cells. Furthermore, these novel derivatives maintain activity against MDR cells as well as GBM neurosphere cultures carrying tumor suppressor and growth factor receptor mutations representing an import challenge in the clinical management of high-grade astrocytic tumors. These compounds are produced in an efficient one-step synthesis from polygodial using a selective Wittig derivatization of the C12-aldehyde group. Lastly, the possibility for an intriguing covalent modification of proteins through a novel pyrrole formation reaction make the described polygodial-derived chemical scaffold an interesting chemotype for the investigation of novel ways of covalent modification of proteins with small molecules of biological and therapeutic relevance.
8. Experimental Section
8.1 General Experimental
All reagents, solvents and catalysts were purchased from commercial sources (Acros Organics and Sigma-Aldrich) and used without purification. All reactions were performed in oven-dried flasks open to the atmosphere or under nitrogen or argon and monitored by thin layer chromatography (TLC) on TLC precoated (250 μm) silica gel 60 F254 glass-backed plates (EMD Chemicals Inc.). Visualization was accomplished with UV light, iodine and p-anisaldehyde stains. 1H and 13C NMR spectra were recorded on a Bruker 400 spectrometer. Chemical shifts (δ) are reported in ppm relative to the TMS internal standard. Abbreviations are as follows: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet). Polygodial (1) was purchased from VWR.
8.2 General procedure for the Wittig reaction
To a solution of 1 (3 mg, 0.0128 mmol, 1 eq) in dichloromethane (3 mL) was added a selected Wittig reagent (5 eq). The resultant mixture was stirred at room temperature for 20 h. After completion of the reaction, as monitored by TLC, the reaction mixture was filtered and the filtrate was concentrated under reduced pressure. The crude product was purified by preparative TLC (EtOAc/Hexane, 1:10) to obtain Wittig products (5–12).
8.3 Compound 5
94%; 1H NMR (400 MHz, CDCl3) δ 9.47 (d, J = 4.8 Hz, 1H), 7.33 (d, J = 16.3 Hz, 1H), 6.53 – 6.49 (m, 1H), 5.50 (d, J = 16.3 Hz, 1H), 3.71 (s, 3H), 2.83 (s, 1H), 2.37 – 2.16 (m, 2H), 1.87 – 1.80 (m, 1H), 1.53 – 1.44 (m, 3H), 1.38 – 1.30 (m, 1H), 1.23 – 1.16 (m, 2H), 1.00 (s, 3H), 0.94 (s, 3H), 0.90 (s, 3H); 13C NMR (100 MHz, C6D6): δ 203.8, 167.2, 146.9, 141.1, 130.7, 116.8, 62.9, 51.2, 48.3, 41.9, 40.2, 37.2, 33.2, 32.9, 24.7, 22.2, 18.3, 15.3; HRMS (ESI) calcd for C18H26NaO3 (M+Na) 313.1780, found 313.1779.
8.4 Compound 6
84%; 1H NMR (400 MHz, C6D6) δ 9.36 (d, J = 4.7 Hz, 1H), 7.53 (d, J = 16.4 Hz, 1H), 5.89 – 5.86 (m, 1H), 5.87 (d, J = 16.4 Hz, 1H), 4.13 – 4.00 (m, 2H), 2.63 – 2.58 (m, 1H), 1.79 – 1.63 (m, 3H), 1.26 – 1.18 (m, 3H), 1.07 (td, J = 13.0, 5.8 Hz, 1H), 0.98 (t, J = 8.0 Hz, 3H), 0.95 – 0.88 (m, 1H), 0.75 – 0.69 (m, 1H), 0.66 (s, 3H), 0.65 (s, 3H), 0.64 (s, 3H); 13C NMR (100 MHz, C6D6): δ 203.9, 166.8, 146.7, 140.9, 130.7, 117.3, 62.9, 60.3, 48.4, 41.9, 40.2, 37.2, 33.2, 32.9, 24.7, 22.2, 18.3, 15.3, 14.3; HRMS m/z (ESI) calcd for C19H28NaO3 (M+Na) 327.1936, found 327.1933.
8.5 Compound 7
94%; 1H NMR (400 MHz, C6D6) δ 9.46 (d, J = 4.4 Hz, 1H), 7.29 – 7.25 (m, 1H), 5.73 – 5.69 (m, 1H), 4.11 – 3.97 (m, 2H), 2.68 – 2.63 (m, 1H), 2.07 (d, J = 1.5 Hz, 3H), 1.85 – 1.71 (m, 2H), 1.66 – 1.60 (m, 1H), 1.28 – 1.21 (m, 3H), 1.14 – 1.05 (m, 1H), 0.99 (t, J = 7.1 Hz, 3H), 0.93 – 0.87 (m, 2H), 0.78 (s, 3H), 0.72 (s, 3H), 0.70 (s, 3H); 13C NMR (100 MHz, C6D6) δ 202.8, 167.9, 139.1, 134.0, 132.9, 130.1, 66.0, 60.7, 48.6, 42.0, 40.1, 37.2, 33.2, 32.9, 24.2, 22.0, 18.5, 15.4, 14.9, 14.3; HRMS (ESI) calcd for C20H30NaO3 (M+Na) 341.2093, found 341.2091.
8.6 Compound 8
93%; 1H NMR (400 MHz, C6D6) δ 9.37 (d, J = 4.7 Hz, 1H), 7.49 (d, J = 16.2 Hz, 1H), 5.91 – 5.81 (m, 2H), 2.66 – 2.58 (m, 1H), 1.80 – 1.63 (m, 3H), 1.42 (s, 9H), 1.26 – 1.17 (m, 3H), 1.13 – 1.03 (m, 1H), 0.98 – 0.93 (m, 1H), 0.73 (dd, J = 11.3, 5.2 Hz, 1H), 0.66 (s, 3H), 0.65 (s, 3H), 0.63 (s, 3H); 13C NMR (100 MHz, C6D6) δ 204.0, 166.3, 146.0, 140.4, 130.8, 119.0, 80.0, 62.9, 48.4, 41.9, 40.2, 37.2, 33.2, 32.9, 28.2 (3C), 24.7, 22.2, 18.3, 15.3; HRMS (ESI) calcd for C21H32NaO3 (M+Na) 355.2249, found 355.2250.
8.7 Compound 9
80%; 1H NMR (400 MHz, C6D6) δ 9.32 (d, J = 4.7 Hz, 1H), 7.50 (d, J = 16.3 Hz, 1H), 7.24 – 7.20 (m, 2H), 7.11 – 7.03 (m, 3H), 5.86 (d, J = 16.3 Hz, 1H), 5.84 – 5.80 (m, 1H), 5.18 (d, J = 12.4 Hz, 1H), 5.06 (d, J = 12.4 Hz, 1H), 2.60 – 2.55 (m, 1H), 1.78 – 1.63 (m, 3H), 1.26 – 1.21 (m, 3H), 1.12 – 1.03 (m, 1H), 0.99 – 0.94 (m, 1H), 0.72 (dd, J = 11.3, 5.1 Hz, 1H), 0.65 (s, 6H), 0.64 (s, 3H); 13C NMR (100 MHz, C6D6) δ 203.8, 166.6, 147.3, 141.2, 136.9, 130.8, 128.7 (2C), 128.6 (2), 128.2, 116.8, 66.3, 62.8, 48.4, 41.9, 40.2, 37.2, 33.2, 32.9, 24.8, 22.2, 18.3, 15.3; HRMS (ESI) calcd for C24H30NaO3 (M+Na) 389.2093, found 389.2090.
8.8 Compound 10
92%; 1H NMR (400 MHz, CDCl3) δ 9.46 (d, J = 5.0 Hz, 1H), 7.23 (ddd, J = 15.9, 10.9, 0.8 Hz, 1H), 6.54 (d, J = 15.9 Hz, 1H), 6.35 – 6.30 (m, 1H), 5.93 (dd, J = 15.9, 10.9 Hz, 1H), 5.81 (d, J = 15.9 Hz, 1H), 3.73 (s, 3H), 2.83 (s, 1H), 2.35 – 2.13 (m, 2H), 1.87 – 1.79 (m, 1H), 1.53 – 1.43 (m, 3H), 1.38 – 1.28 (m, 1H), 1.23 – 1.15 (m, 2H), 1.01 (s, 3H), 0.94 (s, 3H), 0.90 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 206.0, 167.5, 144.9, 142.5, 137.7, 131.1, 124.7, 120.3, 62.9, 51.5, 48.7, 41.8, 40.2, 37.4, 33.2, 33.1, 24.6, 22.3, 18.1, 15.6; HRMS m/z (ESI) calcd for C20H28NaO3 (M+Na) 339.1936, found 339.1936.
8.9 Compound 11
94%; 1H NMR (400 MHz, C6D6) δ 9.07 (d, J = 4.5 Hz, 1H), 6.37 (d, J = 16.9 Hz, 1H), 5.56 – 5.49 (m, 1H), 4.60 (d, J = 16.9 Hz, 1H), 2.29 (s, 1H), 1.72 – 1.51 (m, 3H), 1.26 – 1.15 (m, 4H), 1.09 – 0.87 (m, 2H), 0.63 (s, 6H), 0.57 (s, 3H); 13C NMR (100 MHz, C6D6) δ 203.0, 151.5, 141.3, 130.4, 118.0, 95.4, 62.2, 48.2, 41.8, 40.2, 37.1, 33.1, 32.8, 24.6, 22.1, 18.2, 15.3; HRMS (ESI) calcd for C17H23NNaO (M+Na) 280.1677, found 280.1676.
8.10 Compound 12
86%; 1H NMR (400 MHz, C6D6) δ 9.34 (d, J = 4.7 Hz, 1H), 7.13 (d, J = 16.5 Hz, 1H), 5.94 (d, J = 16.5 Hz, 1H), 5.91 – 5.87 (m, 1H), 2.65 – 2.60 (m, 1H), 1.85 (s, 3H), 1.82 – 1.68 (m, 3H), 1.29 – 1.20 (m, 3H), 1.13 (td, J = 12.8, 5.1 Hz, 1H), 1.02 – 0.93 (m, 1H), 0.80 – 0.75 (m, 1H), 0.70 (s, 3H), 0.68 (s, 3H), 0.67 (s, 3H); 13C NMR (100 MHz, C6D6) δ 203.9, 195.8, 144.2, 141.5, 130.9, 125.2, 63.0, 48.5, 41.9, 40.3, 37.3, 33.2, 32.9, 28.4, 24.9, 22.2, 18.3, 15.4; HRMS (ESI) calcd for C18H26NaO2 (M+Na) 297.1830, found 297.1830.
8.11 Compound 13
To a solution of 4-pentynyl (triphenylphosphoranylidene)acetate (11.9 mg, 0.026 mmol) in THF (2 mL) was added triethylamine (3.5 μL, 0.026 mmol) and stirred at rt for 10 min. This was followed by the addition of 1 (4.0 mg, 0.0170 mmol) in THF (1 mL) and the resultant mixture was stirred at room temperature for 20 h. After completion of the reaction, as monitored by TLC, the reaction mixture was filtered and the filtrate was concentrated under reduced pressure. The crude product was purified by preparative TLC (9/91 EtOAc/Hexane) to obtain 5.5 mg of 13 (94% yield); 1H NMR (400 MHz, C6D6) δ 9.35 (d, J = 4.7 Hz, 1H), 7.50 (d, J = 16.3 Hz, 1H), 5.89 – 5.85 (m, 1H), 5.82 (d, J = 16.3 Hz, 1H), 4.19 – 4.07 (m, 2H), 2.60 (s, 1H), 1.93 (td, J = 7.1, 2.6 Hz, 2H), 1.79 – 1.73 (m, 1H), 1.72 (t, J = 2.6 Hz, 1H), 1.71 – 1.60 (m, 2H), 1.58 – 1.49 (m, 2H), 1.27 – 1.18 (m, 3H), 1.14 – 1.04 (m, 1H), 0.99 – 0.89 (m, 1H), 0.73 (dd, J = 11.3, 5.2 Hz, 1H), 0.67 (s, 3H), 0.66 (s, 3H), 0.65 (s, 3H); 13C NMR (100 MHz, C6D6) δ 203.9, 166.7, 147.0, 141.1, 130.7, 116.9, 83.1, 69.4, 63.1, 62.9, 48.4, 41.9, 40.3, 37.2, 33.2, 32.9, 27.9, 24.8, 22.2, 18.3, 15.4, 15.4; HRMS m/z (ESI) calcd for C22H30NaO3 (M+H) 365.2093, found 365.2092.
8.12 Compound 14
To a solution of 5 (3.4 mg, 0.012 mmol) and benzylamine (1.4 μL, 0.013 mmol) in THF (2 mL) was added AcOH (4.1 μL, 0.07 mmol). The mixture was stirred at rt for 40 h. After completion of the reaction, as monitored by TLC, the reaction mixture was concentrated under reduced pressure and co-distilled with toluene. The crude product was purified by preparative TLC (EtOAc/Hexane, 1:10) to obtain 3.8 mg of 14 (85% yield); 1H NMR (400 MHz, C6D6) δ 7.09 – 7.03 (m, 2H), 7.02 – 6.98 (m, 1H), 6.91 – 6.86 (m, 2H), 6.21 (s, 1H), 4.86 (d, J = 16.4 Hz, 1H), 4.80 (d, J = 16.4 Hz, 1H), 3.31 (d, J = 15.6 Hz, 1H), 3.27 (d, J = 15.6 Hz, 1H), 3.15 (s, 3H), 2.80 – 2.72 (m, 1H), 2.60 (ddd, J = 15.6, 11.8, 7.1 Hz, 1H), 2.00 – 1.92 (m, 1H), 1.87 – 1.65 (m, 3H), 1.55 – 1.45 (m, 2H), 1.45 – 1.38 (m, 2H), 1.33 (s, 3H), 1.22 (td, J = 13.5, 3.7 Hz, 1H), 0.92 (s, 6H); 13C NMR (100 MHz, C6D6) δ 170.7, 139.6, 135.5, 133.0, 128.8 (2C), 128.4 (2C), 127.3, 126.7, 114.4, 52.2, 51.3, 50.7, 42.6, 40.2, 34.9, 33.8, 33.3, 30.9, 26.1, 23.2, 21.8, 20.2, 19.8; HRMS m/z (ESI) calcd for C25H33NNaO2 (M+Na) 402.2409, found 402.2410.
8.13 Cell culture
Human cancer cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA), the European Collection of Cell Culture (ECACC, Salisbury, UK) and the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Braunschweig, Germany). Human mammary carcinoma MCF-7 (ATCC HTB22) cells were cultured in RPMI supplemented with 10% FBS. The U87 cells (ATCC HTB-14) were cultured in DMEM culture medium, while the A549 cells (DSMZ ACC107) were cultured in RPMI culture medium supplemented with 10% heat-inactivated FBS. The GBM Hs683 (ATCC HTB-138) cells were cultivated in DMEM supplemented with 10% FBS. The human uterine sarcoma MES-SA (ATCC CRL1966) and MES-SA/Dx5 cells were cultured in RPMI-1640 medium supplemented with 10% FBS with MES SA/Dx5 maintained in the presence of 500 nM Doxorubicin (Sigma). SKMEL-28 cells (ATCC HTB72) and U373 GBM cells (ECACC 08061901) were cultured in RPMI culture medium supplemented with 10% heat-inactivated FBS. Cell culture media were supplemented with 4 mM glutamine (Lonza code BE17-605E), 100 μg/mL gentamicin (Lonza code 17-5182), and penicillin-streptomycin (200 units/ml and 200 μg/ml) (Lonza code 17-602E). The MDA-MB-231 (ATCC HTB-26) epithelial mammary adenocarcinoma cells were cultured in Eagle's minimum essential medium (EMEM; Invitrogen) containing 5% fetal calf serum (FCS, Cambrex), 2 mM L-glutamine (Invitrogen), 0.06% HEPES (Invitrogen) and penicillin (50 IU/ml)/ streptomycin (50 lg/ml; Invitrogen) at 37 °C in a humidified atmosphere of 5% CO2 in air. Transformed mouse NPCs were cultured in suspension under neurosphere conditions at 37 °C in a humidified atmosphere of 95% O2 and 5% CO2 in DMEM F12 (Invitrogen 11320-074) supplemented with 1x B27 supplement (Invitrogen 17504-044), 5% penicillin-streptomycin (Biochrom 10378-017), 10 ng/ml EGF (R&D systems 236-EG), 10 ng/ml FGF (PeproTech 100-18B).
8.14 Antiproliferative Properties
Antiproliferative properties of the synthesized compounds were evaluated by the MTT assay. All compounds were dissolved in DMSO at a concentration of either 100 mM or 50 mM prior to cell treatment. The cells were trypsinized and seeded at various cell concentrations depending on the cell type. The cells were grown for 24 h to 72 h, treated with compounds at concentrations ranging from 0.001 to 100 M and incubated for 48 or 72 h in 100 or 200 μL media depending on the cell line used. The number of experiments and replicates varied depending on the cell line. Cells treated with 0.1% DMSO were used as a negative control; 1 μM PAO was used as a positive control.
8.15 Selection of Doxorubicin Resistant Cells
Selection of the MES-SA/Dx5 cell line was done according to Harker et al [70]. The cells were split and allowed to adhere overnight. The next day cells were initially exposed to doxorubicin (DOX) at the concentration of 100 nM, which represented the GI50 concentration. The cells were maintained at this DOX concentration until their growth rate reached that of the untreated cells. The DOX concentration was then increased in two-fold increments following the same growth criteria at each concentration to a final DOX concentration of 500 nM. Each new DOX concentration required approximately 2 passages to reach the growth rate of the untreated cells.
8.16 CytoTox-Fluor™ Cytotoxicity Assay
The CytoTox-Fluor cytotoxicity assay from Promega has been used according to manufacturer′s instructions. In brief, 0,015 ×106 cells/well were plated in 24 well-plates in 450 μl (5 replicates per condition) then they received 50 μl of culture medium (DMEM-F12 without phenol red) supplemented with the drugs or respective vehicle control. After 24 hours of incubation at 37 °C, 20 μl of cell suspension was transferred to a black 384 well-plate and mixed with 20 μl of bis-AAF-R110 substrate dilution. After 2 hours of incubation at 37 °C, the fluorescence intensity was measured using the Tecan InfiniteF200 fluorescence plate reader (485 nm Ex/520 nm Em). Blank was subtracted from all wells and the fluorescence read-out for untreated cells (vehicle control) was normalized to 1. Read-outs from cells receiving different treatment conditions were normalized to those of untreated cells and fold change of relative cytotoxicity compared to untreated cells was calculated for each well. Graphs were generated using the GraphPad Prism software.
8.17 [3H]-Resiniferatoxin Binding Assay
To evaluate the possible affinity of different analogues to the vanilloid site of TRPV1, a [3H]-resiniferatoxin ([3H]-RTX) binding assay was performed as previously described [77,78]. Briefly, rats spinal cord were homogenized in buffer A (pH 7.4, 5 mM KCl, 5.8 mM NaCl, 2 mM MgCl2, 0.75 mM CaCl2, 137 mM sucrose, and 10 mM HEPES) and centrifuged for 10 minutes at 1000g at 4 °C and the supernatant was further centrifuged for 30 min at 35,000g at 4 °C. The resulting pellets were than resuspended in buffer A and frozen until assayed. The binding reaction was performed in a final volume of 500 μL, containing buffer A (plus 0.25 mg/mL bovine serum albumin, BSA), membranes (0.5 mg/mL), and 2 nM [3H]-RTX in the presence or absence of analogues of 1 (10 μM). For the measurement of the nonspecific binding, 100 μM nonradioactive RTX were included used. The reaction was started by incubating tubes at 37 °C during 60 minutes, and stopped by transferring the tubes to ice bath and adding 100 μg of bovine α1-acid glycoprotein (to reduce nonspecific binding). Finally, the bound and free membranes [3H]-RTX were separated by centrifuging for 30 min at 35,000g at 4 °C. The pellet was used to quantify the scintillation counting. The specific binding was calculated as the difference of the total and nonspecific binding and the results were measured as % of specific binding.
8.18 Intracellular Ca2+ measurements
Cells were grown on glass coverslips for fluorescence imaging. The cytosolic calcium was measured using Fura-2-loaded cells. Cells were loaded for 45 min at 37 °C in a humidified atmosphere of 5% CO2 in air with 3.3 μM Fura-2/AM prepared in saline solution. Fluorescence was excited at 350 and 380 nm alternately using a monochromator (Polychrome IV; TILL Photonics, Planegg, Germany), and captured by a Cool SNAP HQ camera (Princeton Instruments, France) after filtration through a long-pass filter (510 nm). Metafluor software 7.0 (Molecular Devices) was used for acquisition and analysis. All recordings were carried out at room temperature. The cells were perfused with the saline solutions comprising of (in mM): NaCl 140, KCl 5, CaCl2 2, MgCl2 2, HEPES 10 and Glucose 5 (pH adjusted to 7.4 with NaOH).
8.19 Computer modeling
Molecular modelling was performed using Discovery Studio 4.5 (DS). The receptor template was obtained from the PDB (ID 3J5R) and chains B and D were retained for the simulations. Protein preparation was carried out using the Prepare Protein protocol launched from within DS. All docking simulations were carried out using a modified CDocker protocol with pregeneration of ligand conformations to adequately sample conformational space. Minimizations were carried out within DS employing the CHARMm forcefield (version 39.1).
Supplementary Material
A number of unique C12-Wittig derivatives of the vanilloid polygodial were prepared.
The C12-Wittig derivatives were found to be more potent than polygodial.
The C12-Wittig derivatives maintained potency against drug-resistant cancer cells.
The C12-Wittig derivatives form pyrroles with amines in an unprecedented manner.
The C12-Wittig derivatives exert their anticancer action through cytostatic effects.
Acknowledgments
This project was supported by grants from the National Institute of General Medical Sciences (P20GM103451), National Cancer Institute (CA186046-01A1), Welch Foundation (AI-0045), and National Science Foundation (NSF award 0946998). Part of the experiments in this work were made possible by the Belgian Brain Tumor Support (BBTS; Belgium). SR and LF acknowledge their NMT Presidential Research Support. LF acknowledges Samantha Saville and National Science Foundation (NSF award IIA-1301346). RK is a director of research with the Fonds National de la Recherche Scientifique (FRS-FNRS; Belgium). SCP and WvO gratefully acknowledge support from the National Research Foundation (NRF)-South Africa, as well as Stellenbosch University. The authors are grateful to Anntherese Kornienko for her help with creating Figure 4.
Abbreviations
- ATCC
American Type Culture Collection
- BSA
bovine serum albumin
- CBD
cannabidiol
- cdkn2a
cyclin-dependent kinase inhibitor 2A
- DMEM
Dulbecco's modified Eagle's medium
- DSMZ
Deutsche Sammlung von Mikroorganismen and Zellkulturen
- ECACC
European Collection of Cell Culture
- EMEM
Eagle's minimum essential medium
- EGFRvIII
epidermal growth factor receptor variant III
- FBS
fetal bovine serum
- GBM
glioblastoma multiforme
- HEPES
4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid
- MDR
multidrug resistance
- MS
mass spectrometry
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NSCLC
non-small-cell lung cancer
- NPC
neural precursor
- PAO
phenyl arsine oxide
- P-gp
P-glycoprotein
- PDGFB
platelet-derived growth factor subunit B
- RPMI
Roswell Park Memorial Institute
- RTX
resiniferatoxin
- TRPV1
transient receptor potential vanilloid 1 receptor.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Kaufmann SH, Earnshaw WC. Induction of apoptosis by cancer chemotherapy. Exp. Cell Res. 2000;256:42–49. doi: 10.1006/excr.2000.4838. [DOI] [PubMed] [Google Scholar]
- [2].Kornienko A, Mathieu V, Rastogi S, Lefranc F, Kiss R. Therapeutic agents triggering nonapoptotic cancer cell death. J. Med. Chem. 2013;56:4823–4839. doi: 10.1021/jm400136m. [DOI] [PubMed] [Google Scholar]
- [3].Savage P, Stebbing J, Bower M, Crook T. Why does cytotoxic chemotherapy cure only some cancers? Nat. Clin. Pract. Oncol. 2009;6:43–52. doi: 10.1038/ncponc1260. [DOI] [PubMed] [Google Scholar]
- [4].Wilson TR, Johnston PG, Longley DB. Anti-apoptotic mechanisms of drug resistance in cancer. Curr. Cancer Drug Targets. 2009;9:307–319. doi: 10.2174/156800909788166547. [DOI] [PubMed] [Google Scholar]
- [5].Brenner H. Long-term survival rates of cancer patients achieved by the end of the 20th century: a period analysis. Lancet. 2002;360:1131–1135. doi: 10.1016/S0140-6736(02)11199-8. [DOI] [PubMed] [Google Scholar]
- [6].Agnihotri S, Burrell KE, Wolf A, Jalali S, Hawkins C, Rutka JT, Zadeh G. Glioblastoma, a Brief Review of History, Molecular Genetics, Animal Models and Novel Therapeutic Strategies. Arch. Immunol. Ther. Exp. 2013;61:25–41. doi: 10.1007/s00005-012-0203-0. [DOI] [PubMed] [Google Scholar]
- [7].Adamson C, Kanu OO, Mehta AI, Di C, Lin N, Mattox AK. Glioblastoma multiforme: a review of where we have been and where we are going. Expert Opin. Invest. Drugs. 2009;18:1061–1083. doi: 10.1517/13543780903052764. [DOI] [PubMed] [Google Scholar]
- [8].Stupp R, Hottinger AF, van den Bent MJ, Dietrich PY, Brandes AA. Frequently asked questions in the medical management of high-grade glioma: a short guide with practical answers. Ann. Oncol. 2008;19:209–216. doi: 10.1093/annonc/mdn474. [DOI] [PubMed] [Google Scholar]
- [9].Krakstad C, Chekenya M. Survival signalling and apoptosis resistance in glioblastomas: opportunities for targeted therapeutics. Mol. Cancer. 2010;9:135. doi: 10.1186/1476-4598-9-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- [11].Van Goietsenoven G, Andolfi A, Lallemand B, Cimmino A, Lamoral-Theys D, Gras T, et al. Amaryllidaceae alkaloids belonging to different structural subgroups display activity against apoptosis-resistant cancer cells. J. Nat. Prod. 2010;73:1223–1227. doi: 10.1021/np9008255. [DOI] [PubMed] [Google Scholar]
- [12].Lamoral-Theys D, Andolfi A, Van Goietsenoven G, Cimmino A, Le Calvé B, Wauthoz N, et al. Lycorine, the main phenanthridine Amaryllidaceae alkaloid, exhibits significant antitumor activity in cancer cells that display resistance to proapoptotic stimuli: an investigation of structure-activity relationship and mechanistic insight. J. Med. Chem. 2009;52:6244–6256. doi: 10.1021/jm901031h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Evdokimov NM, Lamoral-Theys D, Mathieu V, Andolfi A, Pelly S, van Otterlo WAL, et al. In search of a cytostatic agent derived from the alkaloid lycorine: synthesis and growth inhibitory properties of lycorine derivatives. Bioorg. Med. Chem. 2011;19:7252–7261. doi: 10.1016/j.bmc.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Luchetti G, Johnston R, Mathieu V, Lefranc F, Hayden K, Andolfi A, et al. Bulbispermine: a crinine-type Amaryllidaceae alkaloid exhibiting cytostatic activity toward apoptosis-resistant glioma cells. Chem. Med. Chem. 2012;7:815–822. doi: 10.1002/cmdc.201100608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Aksenov AV, Smirnov AN, Magedov IV, Reisenaur MR, Aksenov NA, Aksenova IV, Nguyen G, Johnston RK, Rubin M, Kiss R, Mathieu V, Lefranc F, Correa J, Cavazos DA, Brenner AJ, Rogelj S, Kornienko A, Frolova LV. Activity of 2-Aryl-2-(3-indolyl)acetohydroxamates Against Drug-Resistant Cancer Cells. J. Med. Chem. 2015;58:2206–2220. doi: 10.1021/jm501518y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Masi M, Frolova LV, Yu X, Mathieu V, Cimmino A, De Carvalho A, Kiss R, Rogelj S, Pertsemlidis A, Kornienko A, Evidente A. Jonquailine, a new pretazettine-type alkaloid isolated from Narcissus jonquilla quail, with activity against drug-resistant cancer. Fitoterapia. 2015;102:41–48. doi: 10.1016/j.fitote.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dasari R, Banuls LMY, Masi M, Pelly SC, Mathieu V, Green IR, van Otterlo WAL, Evidente A, Kiss R, Kornienko A. C1,C2-Ether Derivatives of the Amaryllidaceae Alkaloid Lycorine: Retention of Activity of Highly Lipophilic Analogues Against Apoptosis-Resistant Cancer Cells. Bioorg. Med. Chem. Lett. 2014;24:923–927. doi: 10.1016/j.bmcl.2013.12.073. [DOI] [PubMed] [Google Scholar]
- [18].Magedov IV, Lefranc F, Frolova LV, Banuls LMY, Peretti AS, Rogelj S, Mathieu V, Kiss R, Kornienko A. Antiproliferative Activity of 2,3-Disubstituted Indoles Toward Apoptosis-Resistant Cancers Cells. Bioorg. Med. Chem. Lett. 2013;23:3277–3282. doi: 10.1016/j.bmcl.2013.03.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lamoral-Theys D, Pottier L, Kerff F, Dufrasne F, Proutiere F, Wauthoz N, Neven P, Ingrassia L, Van Antwerpen P, Lefranc F, Gelbcke M, Pirotte B, Kraus JL, Neve J, Kornienko A, Kiss R, Dubois J. Simple di- and trivanillates exhibit cytostatic properties toward cancer cells resistant to pro-apoptotic stimuli. Bioorg. Med. Chem. 2010;18:3823–3833. doi: 10.1016/j.bmc.2010.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- [21].Saraswathy M, Gong SQ. Different strategies to overcome multidrug resistance in cancer. Biotechnol. Adv. 2013;31:1397–1407. doi: 10.1016/j.biotechadv.2013.06.004. [DOI] [PubMed] [Google Scholar]
- [22].Chen GK, Duran GE, Mangili A, Beketic-Oreskovic L, Sikic BI. MDR 1 activation is the predominant resistance mechanism selected by vinblastine in MES-SA cells. Br. J. Cancer. 2000;83:892–898. doi: 10.1054/bjoc.2000.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Geney R, Ungureanu M, Li D, Ojima I. Overcoming multidrug resistance in taxane chemotherapy. Clin. Chem. Lab. Med. 2002;40:918–925. doi: 10.1515/CCLM.2002.161. [DOI] [PubMed] [Google Scholar]
- [24].Frolova LV, Magedov IV, Romero AE, Karki M, Otero I, Hayden K, et al. Exploring natural product chemistry and biology with multicomponent reactions. 5. Discovery of a novel tubulin-targeting scaffold derived from the rigidin family of marine alkaloids. J. Med. Chem. 2013;56:6886–6900. doi: 10.1021/jm400711t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mathieu V, Chantome A, Lefranc F, Cimmino A, Miklos W, Paulitschke V, Mohr T, Maddau L, Kornienko A, Berger W, Vandier C, Evidente A, Delpire E, Kiss R. Sphaeropsidin A shows promising activity against drug-resistant cancer cells by targeting regulatory volume increase. Cell. Mol. Life Sci. 2015 doi: 10.1007/s00018-015-1902-6. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bury M, Girault A, Mégalizzi V, Spiegl-Kreinecker S, Mathieu V, Berger W, Evidente A, Kornienko A, Gailly P, Vandier C, Kiss R. Ophiobolin A induces paraptosis-like cell death in human glioblastoma cells by decreasing BKCa channel activity. Cell Death Dis. 2013;4:48–57. doi: 10.1038/cddis.2013.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hoffmann EK, Lambert IH. Ion channels and transporters in the development of drug resistance in cancer cells. Phil. Trans. R. Soc. B. 2014;369:1–16. doi: 10.1098/rstb.2013.0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- [29].Premkumar LS, Bishnoi M. Disease-Related Changes in TRPV1 Expression and Its Implications for Drug Development. Curr. Top. Med. Chem. 2011;11:2192–2209. doi: 10.2174/156802611796904834. [DOI] [PubMed] [Google Scholar]
- [30].Hartel M, di Mola FF, Selvaggi F, Mascetta G, Wente MN, Felix K, Giese NA, Hinz U, Di Sebastiano P, Buchler MW, Friess H. Vanilloids in pancreatic cancer: potential for chemotherapy and pain management. Gut. 2006;55:519–528. doi: 10.1136/gut.2005.073205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ziglioli F, Frattini A, Maestroni U, Dinale F, Ciufifeda M, Cortellini P. Vanilloid-mediated apoptosis in prostate cancer cells through a TRPV-1 dependent and a TRPV-1-independent mechanism. Acta. Biomed. 2009;80:13–20. [PubMed] [Google Scholar]
- [32].Stock K, Kumar J, Synowitz M, Petrosino S, Imperatore R, Smith ES, Wend P, Purfürst B, Nuber UA, Gurok U, Matyash V, Wälzlein JH, Chirasani SR, Dittmar G, Cravatt BF, Momma S, Lewin GR, Ligresti A, De Petrocellis L, Cristino L, Di Marzo V, Kettenmann H, Glass R. Neural precursor cells induce cell death of high-grade astrocytomas through stimulation of TRPV1. Nat Med. 2012;18:1232–1238. doi: 10.1038/nm.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hartel M, di Mola FF, Selvaggi F, Mascetta G, Wente MN, Felix K, Giese NA, Hinz U, Di Sebastiano P, Buchler MW, Friess H. Vanilloids in pancreatic cancer: potential for chemotherapy and pain management. Gut. 2006;55:519–528. doi: 10.1136/gut.2005.073205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Athanasiou A, Smith PA, Vakilpour S, Kumaran NM, Turner AE, Bagiokou D, Layfield R, Ray DE, Westwell AD, Alexander SPH, Kendall DE, Lobo DN, Watson SA, Lophatanon A, Muir KA, Guo D, Bates TE. Vanilloid receptor agonists and antagonists are mitochondrial inhibitors: How vanilloids cause nonvanilloid receptor mediated cell death. Biochem. Biophys. Res. Commun. 2007;354:50–55. doi: 10.1016/j.bbrc.2006.12.179. [DOI] [PubMed] [Google Scholar]
- [35].Gonzales CB, Kirma NB, De La Chapa JJ, Chen R, Henry MA, Luo S, Hargreaves KM. Vanilloids induce oral cancer apoptosis independent of TRPV1. Oral Oncol. 2014;50:437–447. doi: 10.1016/j.oraloncology.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Skrzypski M, Sassek M, Abdelmessih S, Mergler S, Grotzinger C, Metzke D, Wojciechowicz T, Nowak KW, Strowski MZ. Capsaicin induces cytotoxicity in pancreatic neuroendocrine tumor cells via mitochondrial action. Cell. Signal. 2014;26:41–48. doi: 10.1016/j.cellsig.2013.09.014. [DOI] [PubMed] [Google Scholar]
- [37].Farfariello V, Liberati S, Morelli MB, Tomassoni D, Santoni M, Nabissi M, Giannantoni A, Santoni G, Amantini C. Resiniferatoxin induces death of bladder cancer cells associated with mitochondrial dysfunction and reduces tumor growth in a xenograft mouse model. Chem. Biolog. Interact. 2014;224:128–135. doi: 10.1016/j.cbi.2014.10.020. [DOI] [PubMed] [Google Scholar]
- [38].Sterner O, Szallasi A. Novel natural vanilloid receptor agonists: new therapeutic targets for drug development. TiPS. 1999;20:459–465. doi: 10.1016/s0165-6147(99)01393-0. [DOI] [PubMed] [Google Scholar]
- [39].Andre AE, Ferreira J, Malheiros A, Yunes RA, Calixto JB. Evidence for the involvement of vanilloid receptor in the antinociception produced by the dialdeydes unsaturated sesquiterpenes polygodial and drimanial in rats. Neuropharmacology. 2004;46:590–597. doi: 10.1016/j.neuropharm.2003.10.008. [DOI] [PubMed] [Google Scholar]
- [40].Monica CD, De Petrocellis L, Di Marzo V, Landi R, Izzo I, Spinella A. Enantioselective synthesis and vanilloid activity evaluation of 1-b-(pmethoxycinnamoyl)polygodial, an antinociceptive compound from Drymis winteri barks. Bioorg. Med. Chem. Lett. 2007;17:6444–6447. doi: 10.1016/j.bmcl.2007.10.001. [DOI] [PubMed] [Google Scholar]
- [41].Andre E, Campi B, Trevisani M, Ferreira J, Malheiros A, Yunes RA, Calixto JB, Geppetti P. Pharmacological characterisation of the plant sesquiterpenes polygodial and drimanial as vanilloid receptor agonists. Biochem. Pharmacol. 2006;71:1248–1254. doi: 10.1016/j.bcp.2005.12.030. [DOI] [PubMed] [Google Scholar]
- [42].D'Acunto M, Monica CD, Izzo I, De Petrocellis L, di Marzo V, Spinella A. Enantioselective synthesis of 3(S)-hydroxy polygodial derivatives and evaluation of their vanilloid activity. Tetrahedron. 2010;66:9785–9789. [Google Scholar]
- [43].Iwasaki Y, Tanabe M, Kayama Y, Abe M, Kashio M, Koizumi K, Okumura Y, Morimitsu Y, Tominaga M, Ozawa Y, Watanabe T. Miogadial and miogatrial with α,β-unsaturated 1,4-dialdehyde moieties—Novel and potent TRPA1 agonists. Life Sci. 2009;85:60–69. doi: 10.1016/j.lfs.2009.04.017. [DOI] [PubMed] [Google Scholar]
- [44].Ohsuka A. The structure of tadeonal and isotadeonal, components of Polygonum Hydropiper L. Nippon Kagaku Zasshi. 1963;84:748–752. [Google Scholar]
- [45].Nakanishi K, Kubo I. Studies on warburganal, muzigadial and related compounds. Israel J. Chem. 1977;16:28–31. [Google Scholar]
- [46].Cimino G, De Rosa S, De Stefano S, Sodano G. The chemical defense of four Mediterranean nudibranchs. Comp. Eiorhem. Pbysiol. 1982;73B:471–474. [Google Scholar]
- [47].Kubo I, Ganjian I. Insect antifeedant terpenes, hot-tasting to humans. Experientia. 1981;37:1063–1064. doi: 10.1007/BF02085009. [DOI] [PubMed] [Google Scholar]
- [48].Cimino G, Spinella A, Sodano G. Identification of an intermediate in the reaction between polygodial and methylamine in biomimetic conditions. Tetrahedron Lett. 1984;25:4151–4152. [Google Scholar]
- [49].Allouche N, Cécile A, Martin MT, Dumontet V, Guéritte F, Litaudon M. Cytotoxic sesquiterpenoids from Winteraceae of Caledonian rainforest. Phytochemistry. 2009;70:546–553. doi: 10.1016/j.phytochem.2009.01.012. [DOI] [PubMed] [Google Scholar]
- [50].Tozyo T, Yasuda F, Nakai H, Tada H. Cytotoxic hydroxypolygodials. X-Ray molecular structure of (1R,3S,5aS,-9aS,9bR)-1,3,5,5a,6,7,8,9,9a,9b-decahydro-1,3-dimethoxy-6,6,9a-trimethylnaphtho[1,2-c]furan. J. Chem. Soc. Perkin Trans. 1. 1992;46:1859–1866. [Google Scholar]
- [51].Barrero AF, Corte′s M, Manzaneda EA, Cabrera E, Chahboun R, Lara M, Rivas AR. Synthesis of 11,12-Epoxydrim-8,12-en-11-ol, 11,12-Diacetoxydrimane, and Warburganal from (-)-Sclareol. J. Nat. Prod. 1999;62:1488–1491. doi: 10.1021/np990140q. [DOI] [PubMed] [Google Scholar]
- [52].Anke H, Sterner O. Comparison of the antimicrobial and cytotoxic activities of twenty unsaturated sesquiterpene dialdehydes from plants and mushrooms. Planta Med. 1991;57:344–346. doi: 10.1055/s-2006-960114. [DOI] [PubMed] [Google Scholar]
- [53].Forsby A, Andersson M, Lewan L, Sterner O. The cytotoxicity of 22 sesquiterpenoid unsaturated dialdehydes, as determined by the neutral red absorption assay and by protein determination. Toxicol In Vitro. 1991;5:9–14. doi: 10.1016/0887-2333(91)90043-d. [DOI] [PubMed] [Google Scholar]
- [54].Andersson M, Bocchio F, Sterner O, Forsby A, Lewan L. Structure-activity relationships for unsaturated dialdehydes 7. The membrane toxicity of 15 sesquiterpenoids measured as the induction of ATP leakage in ELD cells. The correlation of the activity with structural descriptors by the multivariate PLS method. Toxicol In Vitro. 1993;7:1–6. doi: 10.1016/0887-2333(93)90106-f. [DOI] [PubMed] [Google Scholar]
- [55].Dasari R, De Carvalho A, Medellin DC, Middleton KN, Hague F, Volmar MNM, Frolova LV, Rossato MF, De La Chapa JJ, Dybdal-Hargreaves NF, Pillai A, Mathieu V, Rogelj S, Gonzales CB, Calixto JB, Evidente A, Gautier M, Munirathinam G, Glass R, Burth P, Pelly SC, van Otterlo WAL, Kiss R, Kornienko A. Synthetic and Biological Studies of sesquiterpene polygodial: activity of 9-epipolygodial against drug resistant cancer cells. Submitted. [DOI] [PMC free article] [PubMed]
- [56].Amarnath V, Anthony DC, Amarnath K, Valentine WM, Wetterau LA, Graham DG. Intermediates in the Paal-Knorr Synthesis of Pyrroles. J. Org. Chem. 1991;56:6924–6931. [Google Scholar]
- [57].Schneimann MJ, Beaudry CM, Genung NE, Canham SM, Untiedt NL, Karanikolas BDW, Sutterlin C, Overman LE. Divergent Synthesis and Chemical Reactivity of Bicyclic Lactone Fragments of Complex Rearranged Spongian Diterpenes. J. Am. Chem. Soc. 2011;133:17494–17503. doi: 10.1021/ja207727h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhang L, Gavin T, DeCaprio AP, LoPachin RM. gamma-Diketone Axonopathy: Analyses of Cytoskeletal Motors and Highways in CNS Myelinated Axons. Toxicol. Sci. 2010;117:180–189. doi: 10.1093/toxsci/kfq176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Graham DG, Anthony DC, Boekelheide K, Maschmann NA, Richards RG, Wolfram JW, Shaw BR. Studies of the molecular pathogenesis of hexane neuropathy. II. Evidence that pyrrole derivatization of lysyl residues leads to protein crosslinking. Toxicol. Appl. Pharmacol. 1982;64:415–422. doi: 10.1016/0041-008x(82)90237-x. [DOI] [PubMed] [Google Scholar]
- [60].Lefranc F, Nuzzo G, Hamdy NA, Fakhr I, Moreno Y Banuls L, Van Goietsenoven G, Villani G, Mathieu V, van Soest R, Kiss R, Ciavatta ML. In vitro pharmacological and toxicological effects of norterpene peroxides isolated from the Red Sea sponge Diacarnus erythraeanus on normal and cancer cells. J. Nat. Prod. 2013;76:1541–1547. doi: 10.1021/np400107t. [DOI] [PubMed] [Google Scholar]
- [61].Mathieu A, Remmelink M, D'Haene N, Penant S, Gaussin JF, Van Ginckel R, Darro F, Kiss R, Salmon I. Development of a chemoresistant orthotopic human nonsmall cell lung carcinoma model in nude mice: Analyses of tumor heterogeneity in relation to the immunohistochemical levels of expression of cyclooxygenase-2, ornithine decarboxylase, lung-related resistance protein, prostaglandin E synthetase, and glutathione-S-transferase-alpha (GST)-alpha-, GST-mu, and GST-pi. Cancer. 2004;101:1908–1918. doi: 10.1002/cncr.20571. [DOI] [PubMed] [Google Scholar]
- [62].Mathieu V, Pirker C, Martin de Lasalle E, Vernier M, Mijatovic T, De Neve N, Gaussin JF, Dehoux M, Lefranc F, Berger W, Kiss R. J. Cell. Mol. Med. 2009;13:3960–3972. doi: 10.1111/j.1582-4934.2009.00708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Frolova LV, Magedov IV, Romero AE, Karki M, Otero I, Hayden K, Evdokimov NM, Banuls LMY, Rastogi SK, Smith WR, Lu SL, Kiss R, Shuster CB, Hamel E, Betancourt T, Rogelj S, Kornienko A. Exploring Natural Product Chemistry and Biology with Multicomponent Reactions. 5. Discovery of a Novel Tubulin-Targeting Scaffold Derived from the Rigidin Family of Marine Alkaloids. J. Med. Chem. 2013;56:6886–6900. doi: 10.1021/jm400711t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- [65].Saraswathy M, Gong SQ. Different strategies to overcome multidrug resistance in cancer. Biotechnol. Adv. 2013;31:1397–1407. doi: 10.1016/j.biotechadv.2013.06.004. [DOI] [PubMed] [Google Scholar]
- [66].Chen GK, Duran GE, Mangili A, Beketic-Oreskovic L, Sikic BI. MDR1 activation is the predominant resistance mechanism selected by vinblastine in MES-SA cells. Br. J. Cancer. 2000;83:892–898. doi: 10.1054/bjoc.2000.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Geney R, Ungureanu M, Li D, Ojima I. Overcoming multidrug resistance in taxane chemotherapy. Clinical Chem. Lab. Med. 2002;40:918–925. doi: 10.1515/CCLM.2002.161. [DOI] [PubMed] [Google Scholar]
- [68].Ruefli AA, Tainton KM, Darcy PK, Smyth MJ, Johnstone RW. Pglycoprotein inhibits caspase-8 activation but not formation of the death inducing signal complex (disc) following Fas ligation. Cell Death Differ. 2002;9:1266–1272. doi: 10.1038/sj.cdd.4401081. [DOI] [PubMed] [Google Scholar]
- [69].Johnstone RW, Cretney E, Smyth MJ. P-glycoprotein protects leukemia cells against caspase-dependent, but not caspase-independent, cell death. Blood. 1999;93:1075–1085. [PubMed] [Google Scholar]
- [70].Harker WG, Sikic BI. Multidrug (pleiotropic) resistance in doxorubicin-selected variants of the human sarcoma cell line MES-SA. Cancer Research. 1985;45:4091–4096. [PubMed] [Google Scholar]
- [71].Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JM. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- [72].Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Johannessen TC, Bjerkvig R, Tysnes BB. DNA repair and cancer stemlike cells - Potential partners in glioma drug resistance? Cancer Treat Rev. 2008;34:558–567. doi: 10.1016/j.ctrv.2008.03.125. [DOI] [PubMed] [Google Scholar]
- [74].Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27:1749–1758. doi: 10.1038/sj.onc.1210811. [DOI] [PubMed] [Google Scholar]
- [75].Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks BP. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- [76].Yuan X, Curtin J, Xiong Y, Liu G, Waschsmann-Hogiu S, Black KL, Yu JS. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene. 2004;23:9392–9400. doi: 10.1038/sj.onc.1208311. [DOI] [PubMed] [Google Scholar]
- [77].Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- [78].Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, Park JK, Fine HA. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- [79].Purkait S, Jha P, Sharma MC, Suri V, Sharma M, Kale SS, Sarkar C. CDKN2A deletion in pediatric versus adult glioblastomas and predictive value of p16 immunohistochemistry. Neuropathol. 2013;33:405–412. doi: 10.1111/neup.12014. [DOI] [PubMed] [Google Scholar]
- [80].Guo P, Hu B, Gu W, Xu L, Wang D, Huang HJS, Cavenee WK, Cheng SC. Platelet-Derived Growth Factor-B Enhances Glioma Angiogenesis by Stimulating Vascular Endothelial Growth Factor Expression in Tumor Endothelia and by Promoting Pericyte Recruitment. Amer. J. Pathol. 2003;162:1083–1093. doi: 10.1016/S0002-9440(10)63905-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Gan HK, Kaye AH, Luwor RB. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 2009;16:748–754. doi: 10.1016/j.jocn.2008.12.005. [DOI] [PubMed] [Google Scholar]
- [82].Iannotti FA, Hill CL, Leo A, Alhusaini A, Soubrane C, Mazzarella E, Russo E, Whalley BJ, Di Marzo V, Stephens GJ. Nonpsychotropic Plant Cannabinoids, Cannabidivarin (CBDV) and Cannabidiol (CBD), Activate and Desensitize Transient Receptor Potential Vanilloid 1 (TRPV1) Channels in Vitro: Potential for the Treatment of Neuronal Hyperexcitability. ACS Chem. Neurosci. 2014;5:1131–1141. doi: 10.1021/cn5000524. [DOI] [PubMed] [Google Scholar]
- [83].Ligresti A, Moriello AS, Starowicz K, Matias I, Pisanti S, De Petrocellis L, Laezza C, Portella G, Bifulco M, Di Marzo V. Antitumor Activity of Plant Cannabinoids with Emphasis on the Effect of Cannabidiol on Human Breast Carcinoma. J. Pharmacol. Exp. Ther. 2006;318:1375–1387. doi: 10.1124/jpet.106.105247. [DOI] [PubMed] [Google Scholar]
- [84].Cao E, Liao M, Cheng D, Julius D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature. 2013;504:113–118. doi: 10.1038/nature12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Eisele G, Weller M. Targeting apoptosis pathways in glioblastoma. Cancer Lett. 2013;332:335–345. doi: 10.1016/j.canlet.2010.12.012. [DOI] [PubMed] [Google Scholar]
- [86].van den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MC, Kros JM, Carpentier AF, Clement PM, Frenay M, Campone M, Baurain JF. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J. Clin. Oncol. 2009;27:1268–1274. doi: 10.1200/JCO.2008.17.5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.