

Abstract
Describing the process of spontaneous mutation is fundamental for understanding the genetic basis of disease, the threat posed by declining population size in conservation biology, and much of evolutionary biology. Directly studying spontaneous mutation has been difficult, however, because new mutations are rare. Mutation accumulation (MA) experiments overcome this by allowing mutations to build up over many generations in the near absence of natural selection. Here, we sequenced the genomes of 85 MA lines derived from six genetically diverse strains of the green alga Chlamydomonas reinhardtii. We identified 6843 new mutations, more than any other study of spontaneous mutation. We observed sevenfold variation in the mutation rate among strains and that mutator genotypes arose, increasing the mutation rate approximately eightfold in some replicates. We also found evidence for fine-scale heterogeneity in the mutation rate, with certain sequence motifs mutating at much higher rates, and clusters of multiple mutations occurring at closely linked sites. There was little evidence, however, for mutation rate heterogeneity between chromosomes or over large genomic regions of 200 kbp. We generated a predictive model of the mutability of sites based on their genomic properties, including local GC content, gene expression level, and local sequence context. Our model accurately predicted the average mutation rate and natural levels of genetic diversity of sites across the genome. Notably, trinucleotides vary 17-fold in rate between the most and least mutable sites. Our results uncover a rich heterogeneity in the process of spontaneous mutation both among individuals and across the genome.
Understanding the processes that generate new genetic variation from mutation is a key goal of genetics research. In humans, for example, new mutations cause Mendelian genetic disorders, play a direct role in polygenic disease (e.g., Veltman and Brunner 2012), and are a major factor in cancers (e.g., Alexandrov et al. 2013a). New mutations also play a central role in evolutionary biology, since the variation that fuels adaptive evolution is ultimately derived from advantageous mutations. It is widely believed that the majority of new mutations that affect functional elements of the genome are deleterious, and the input of these mutations is pivotal in explaining the evolution of recombination and sex (for review, see Otto 2009).
When new mutations are harmful, theory predicts that the mutation rate should evolve toward zero, because individuals with higher mutations rates will suffer a greater mutational load. The mutation rate is always greater than zero in nature, however, ranging over seven orders of magnitude (for review, see Drake 2006), and two main explanations have been proposed for this. One explanation is that there is a limit to the accuracy of DNA repair, due to a trade-off between the benefit of further reducing the mutation rate and the costs of increased fidelity (Kimura 1967). Alternatively, a “selection-drift” barrier may constrain progress toward a lower mutation rate when the selective advantage of further improvement becomes so small that new mutations decreasing the mutation rate are effectively neutral (Lynch 2010). Evidence for a selection-drift barrier comes from the negative correlation between the mutation rate per generation and effective population size (Ne) (Sung et al. 2012). However, when mutation rate is expressed per cell division, there is much less variation between species and little relationship with Ne, consistent with the constraint on the fidelity of replication hypothesis. It is currently difficult to fully evaluate the support for these hypotheses, however, because studies of mutation are restricted to a small number of taxa, few genotypes per species, and a limited number of mutation events.
Although there is clear evidence for variation between species, we know relatively little about the extent of mutation rate variation within species. Individuals with an unusually high mutation rate have been isolated from natural populations of prokaryotes (Matic et al. 1997; Sundin and Weigand 2007), but to our knowledge no natural mutators have been found in eukaryotes. This discrepancy likely stems from the fact that prokaryotes are asexual whereas eukaryotes are predominantly sexual. Theory predicts that in an asexual population, a mutator allele can hitchhike to high frequency if it generates a beneficial allele on the same genetic background (Johnson 1999). In contrast, recombination in sexual populations uncouples a mutator from a linked beneficial allele, so the mutator allele is then expected to be selected against because of its association with linked deleterious mutations (for review, see Drake et al. 1998). Although a smaller amount of mutation rate variation is expected in sexual than asexual species, mutations that alter the mutation rate are, nevertheless, expected to occur and potentially provide the basis for mutation rate evolution. Mutation rate variation within a species may also reflect mutation-selection balance, whereby new deleterious alleles that alter the mutation rate continually arise and are purged by selection. In this scenario, intra-specific mutation rate variation will reflect the distribution of phenotypic effects of mutations that alter DNA repair and stability and the effectiveness of selection against them. In the largest study of spontaneous mutation in humans, there was little evidence for mutation rate variation among individuals after accounting for parental age (Kong et al. 2012). Father's age was also an important factor explaining mutation rate variation in chimpanzees (Venn et al. 2014). Similarly, there was no evidence of mutation rate variation between two strains in both Caenorhabditis elegans and Caenorhabditis briggsae (Denver et al. 2012). There is evidence from Drosophila that individuals in poor condition have elevated mutation rates (Sharp and Agrawal 2012), and a separate study comparing two inbred lines revealed a 2.4-fold difference in the rate of mutation (Schrider et al. 2013). Moreover, two independent experiments in Chlamydomonas reinhardtii suggested that there is a fivefold difference in the mutation rate between two natural strains (Ness et al. 2012; Sung et al. 2012).
In addition to mutation rate variation within and between species, there is also evidence that the mutation rate varies across the genome. Such heterogeneity is expected to alter the rate of evolution across the genome and to create variation in the susceptibility of genes or sites to deleterious or beneficial mutations. There is clear evidence for the existence of fine-scale variation in the rate of mutation. At the scale of individual sites, G:C positions tend to mutate at higher rates than A:T positions, and transitions from G:C→A:T are the most common change in a broad range of species, including bacteria (Hershberg and Petrov 2010), animals (Kong et al. 2012; Schrider et al. 2013), fungi (Zhu et al. 2014), and plants (Ness et al. 2012). Similarly, the bases surrounding a mutated site have a strong effect on mutability. The high frequency of G:C→A:T transitions in mammals, for example, is driven by the deamination of methylated CpG sites (Ehrlich and Wang 1981). In general, the bases flanking a particular site, referred to as the “sequence context,” are one of the best predictors of mutation rate (Michaelson et al. 2012; Neale et al. 2012; Samocha et al. 2014; Zhu et al. 2014). However, investigations concerning the underlying mechanisms and the consistency of sequence context effects on mutability are only now emerging.
At a broader genomic scale, evidence for mutation rate heterogeneity is weaker. Sequencing of mutation accumulation (MA) lines in Saccharomyces cerevisiae (Zhu et al. 2014) and Drosophila melanogaster (Schrider et al. 2013) found no evidence of mutation rate variation between chromosomes. Although there is evidence that mutation rate increases as a function of replication timing (Stamatoyannopoulos et al. 2009; Lang and Murray 2011), this finding has not been supported by direct estimates of the mutation rate (Samocha et al. 2014; Zhu et al. 2014). A variety of other genomic properties have been linked to increased susceptibility to mutation, including transcription level, nucleosome occupancy, DNase hypersensitivity, and recombination rate (e.g., Michaelson et al. 2012). If these factors strongly influence mutation and generate variation between sites or large scale patterns of mutation rate variation, it is important to quantify their effects in order to facilitate better predictive models of DNA sequence evolution.
Detailed investigations of the process of spontaneous mutation and the extent of mutation rate variation have been limited by the rarity of spontaneous mutations, which has constrained direct observation of sufficient numbers of mutations to infer the underlying biology. Sequencing of parents and their offspring is an increasingly common method for directly identifying de novo mutations (e.g., Keightley et al. 2014a,b). Although this approach has advantages, it is currently very expensive to sequence sufficient offspring to observe large numbers of mutations and has therefore only been applied on a large scale in humans (Kong et al. 2012). Another approach is to maintain experimental populations for many generations under minimal natural selection to allow mutations to accumulate regardless of their fitness consequences. Increasing the strength of genetic drift by bottlenecking the population in each generation allows random, unbiased accumulation of all but the strongest deleterious mutations. These mutation accumulation experiments have been used in a variety of species to investigate the phenotypic effects of new mutations (for review, see Halligan and Keightley 2009) and are now being paired with whole genome sequencing to identify individual mutations. MA studies have generally been limited to sequencing a small number of genomes, and only two studies have tested for heterogeneity in mutation rate across the genome (Schrider et al. 2013; Zhu et al. 2014), and no study has included more than two ancestral genotypes from a single species. In this study, we sequenced the genomes of 85 MA lines derived from six genetically diverse strains of the model green alga C. reinhardtii. We identified 6843 mutations, sevenfold more than any previous MA study, and integrate these data with detailed annotation of genomic properties to investigate the process of spontaneous mutation with unprecedented detail. Specifically, we address the following questions: (1) What is the relative frequency of different kinds of mutation, including the base spectrum and rate of insertion and deletion mutations? (2) What is the extent of mutation rate variation between individuals within a species? (3) Is there evidence of mutation rate heterogeneity across the genome and what genomic properties predict the rate of mutation at individual sites?
Results
We conducted a mutation accumulation experiment in six genetically diverse strains of C. reinhardtii that were chosen to broadly cover the geographic range of known C. reinhardtii samples in North America (Table 1). Fifteen replicate MA lines from each of the six ancestral strains were initiated for a total of 90 MA lines. Eighty-five of the initial 90 MA lines survived to the end of the experiment. The mean number of generations undergone by each MA line was estimated to be 940 (range 403–1130). We used Illumina whole genome sequencing to identify de novo mutations in an average of 75.4 Mbp per line (72.5% of genome, range 58.5–84.9 Mbp) (see Methods for details on mutation calling). In total, we identified 6843 mutations, including 5716 single nucleotide mutations (SNMs) and 1127 short indels. To confirm our mutation calls, we Sanger-sequenced a random sample of 138 mutations. One hundred fifteen of 117 SNMs and 19 of 21 indels were confirmed, implying a detection rate of 98.3% and 90.5% for SNMs and indels, respectively. A complete list of all mutations and their annotations can be found in Supplemental Table S1.
Table 1.
Ancestral strains of Chlamydomonas reinhardtii used for mutation accumulation

Mutation rate variation among genotypes
The mutation rate across all MA lines was µ = 11.5 × 10−10 per site per generation. The SNM and indel mutation rates were µSNM = 9.63 × 10−10 and µINDEL = 1.90 × 10−10, respectively. Mutation rate varied considerably among the MA replicates and between ancestral strains. Mutation rates of the individual MA lines ranged over nearly two orders of magnitude from MA replicate 4 from CC-1952 (µCC-1952-MA4 = 0.57 × 10−10) to MA replicate 1 from CC-2344 (µCC-2344-MA1 = 49.4 × 10−10). There was significant variation in the mean mutation rate among the ancestral strains (F(1,5) = 30.96, P < 0.0001) (see Fig. 1). Post-hoc Tukey tests showed that strain CC-1373 had an average mutation rate significantly higher than all of the other strains (µ = 28.1 × 10−10, P = 0.01 to <0.001). Its rate was nearly sevenfold higher than strain CC-1952 (µ = 4.05 × 10−10), which had the lowest mutation rate, and was significantly lower than strain CC-1373 (P < 0.001), CC-2931 (µ = 15.6 × 10−10, P < 0.001), and CC-2342 (µ = 11.1 × 10−10, P < 0.01). Within strains CC-2344 and CC-2931, there were individual MA lines with unusually high mutation rates, 3.5× and 8.0× above their respective strain means, i.e., µ estimates for µCC-2344-MA1 = 56.9 × 10−10, CC-2344 (confidence interval [CI] = 2.6–12.0 × 10−10), and µCC-2931-MA5 = 36.2 × 10−10 (CC-2931 CI = 7.2–20.0 × 10−10) are outside the 99.99% CI of their ancestral strain mutation rates. We also found that one MA line, CC-1952-MA4, had an unusually low mutation rate of µCC-2931-MA5 = 36.2 × 10−10 compared to its strain mean and may be a hypomutator, although further investigation would be needed to distinguish this possibility from the alternate, which is that with 85 MA lines, one accumulated an unusually low number of mutations by chance.
Figure 1.
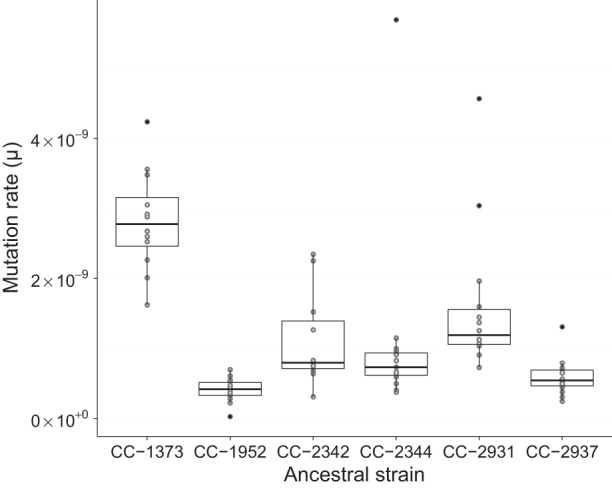
Variation in mutation rate between strains. Mutation rate (µ = mutations/[sites × generations]) for each of the MA lines, categorized based on their ancestral strain. The boxes outline the first to third quartile of the mutation rate in lines from a given ancestral strain, the horizontal lines within the boxes indicate the median mutation rate, and the whiskers extend to the last data point that is within 1.5× the inter-quartile range; points outside the whiskers are filled black.
Indel mutations
Short deletions (613) were significantly more common than insertions (514) (χ2 = 8.7, P < 0.005), and these deletions also tended to be larger (mean lengths = −7.9 and +5.9, respectively, Mann–Whitney U test, W = 112,604.5, P < 2.2 × 10−16). MA lines of strain CC-2931 had an unusually high number of indels (408) due to an abundance of 9-bp deletions, i.e., 120 of 408 indels in CC-2931 were 9-bp deletions compared to a mean of five 9-bp deletions in each of the other strains. These deletions did not appear to have any shared sequence motif nor were they associated with coding exons, repetitive sequence, or any genomic property that we could identify. After adjusting for the excess of 9-bp deletions in CC-2931 by setting it to the mean number of 9-bp deletions found in the other strains, there were similar numbers of insertions and deletions, but deletions were still significantly longer (W = 100,759.5, P = 3.3 × 10−9).
Spatial heterogeneity
Mutation rate measured in 200-kbp sliding windows ranged from 0.0 to 23.5 × 10−10 (variance, σ2 = 1.3 × 10−19). By comparing the distribution of mutation rates for each window with a simulated distribution, much of this variation could be accounted for as noise around the genome average (simulated variance, σ2 = 1.2 × 10−19, Kolmogorov-Smirnov [KS] test D = 0.038, P = 0.43). In 1000 simulations where mutation positions were randomized, the 95% confidence interval of simulated mutation rates was µ = 5.3–18.3 × 10−10 compared to a 95% CI of µ = 4.8–19.4 × 10−10 in the observed data. Only 8% of 200-kbp windows were above the 95th percentile of simulated mutation rates, suggesting a slight excess of windows with a high mutation rate.
We detected a significant deviation in the distribution of minimum inter-mutation distance compared to that expected under simulation (KS test: D = 0.048, P = 4.5 × 10−14) (Fig. 2). This was caused by the presence of an excess of mutations clustered very near to one another (<100 bp apart), and most of the clusters were mutations at adjacent sites. By chance, we expected no adjacent mutations, but we observed 55 mutations where two adjacent sites were mutated. Each of these was visually inspected in the Integrative Genomics Viewer (IGV) (Thorvaldsdóttir et al. 2012) and appeared to be genuine and not the result of alignment or sequencing errors. Twenty-seven of these clustered mutations occurred at CpC sites, and 25 of 27 mutated to ApA/ApT/TpA/TpT. We also found a number of indels where a short sequence was replaced by an unrelated stretch of sequence. Clusters of mutations were only ever found together in the same MA line. When we limited our analysis to test for the presence of mutations occurring at nearby sites in different lines, there was no evidence for an excess of clustering (KS test: D = 0.02, P = 0.13).
Figure 2.
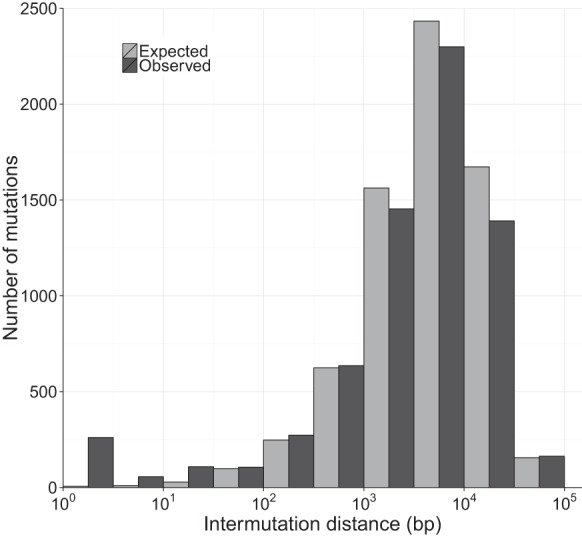
Expected and observed distributions of inter-mutation distance. Comparison of observed (dark gray) and expected (light gray) distributions of the distance between mutations. In this plot, inter-mutation distance was measured as the nearest mutation irrespective of the MA line or strain it occurred in. The expected distribution was generated by randomizing the location of mutations in each MA line and recalculating the inter-mutation distances. The simulation was repeated 1000 times, and the average of those iterations is shown here.
Base composition
Treating the strands symmetrically, we found a significantly nonrandom distribution of the six possible SNMs (χ2 = 1630.3, P < 0.0001) (Fig. 3). Mutations occurring at C:G sites were 4.2× more frequent than mutations at A:T sites, after correcting for genomic base composition, and this pattern was consistent across all MA lines and ancestral strains. Transitions from C:G→T:A were overrepresented nearly twofold compared to what would be expected if mutations occurred at all sites with equal probability. Although transitions from A:T→G:C were more common than the other mutations possible at A:T sites, they were still less common than any mutation type at C:G sites. Transversions from A:T→C:G or T:A were the least common type of mutation and were found 2.4× less frequently than expected.
Figure 3.
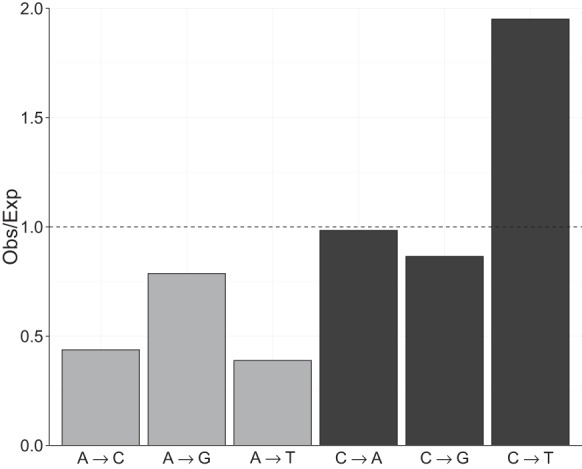
Mutation base spectrum of single nucleotide mutations. Base mutation spectrum of 5716 single nucleotide mutations (SNMs). The deviation of the mutation rate for each of the six possible SNMs relative to its expectation based on equal mutation rates was calculated as the observed number of mutations of each kind divided by the number of mutations expected if mutations occurred randomly with respect to base. Background base composition was calculated only from sites that have high-quality genotype calls (callable sites).
To assess the effect of the local sequence context on mutation rate, we measured the frequency of the bases surrounding random A:T and C:G sites in the genome and compared this to the base frequencies in the window surrounding SNMs (Fig. 4). We found nonrandom patterns surrounding all six kinds of mutation, but the extent of the deviation was strongest for mutations at C:G sites. The deviation was particularly strong in the 2–4 bp upstream of mutations at C:G sites and, to a lesser extent, 1 bp downstream from all mutation types. Specifically, the composition of the two nucleotides immediately upstream of mutated C:G sites was strongly biased. In the case of the CTC trinucleotide, for example, where the final C was mutated, that mutation rate was 4.5× the background rate.
Figure 4.
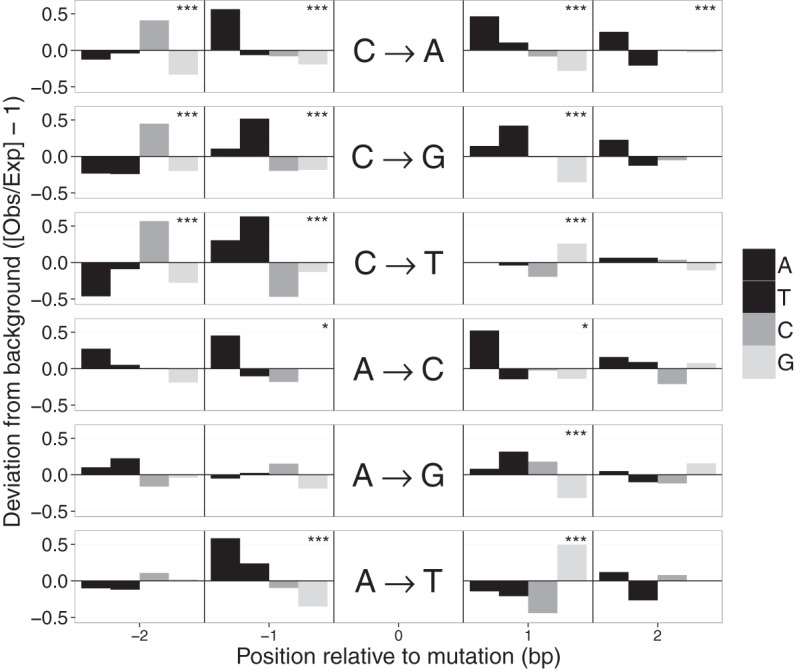
Sequence context of spontaneous mutations. Deviations in the local sequence context of the 2 bp flanking mutated sites. Deviations were calculated from the observed frequency of each base (A, T, C, G) in the flanks of mutated sites and the expected background composition based on flanking sequences of 106 random A:T or C:G sites. Each horizontal panel represents one of the six possible mutations indicated in the center. Significant deviations from the background base composition at each position were detected with tests and indicated as (*) P < 0.05 or (***) P < 0.001 (α-values were adjusted for multiple tests using a Bonferroni correction).
Mutability
We used logistic regression to identify the genomic properties that best differentiated mutated from nonmutated sites. We generated a training set of all 6843 mutations and a random set of 100,000 nonmutated sites. In the regression model, the genomic properties of these 106,843 sites (see Supplemental Table S2) were used as predictors and the presence or absence of mutations at a site as the response. With this model, we then calculated the probability of mutation, or “mutability,” for every site in the genome (see Methods for details). To assess the accuracy of the model, we binned sites in the genome based on their mutability (range 0.0–1.0) and calculated the observed mutation rate in each bin (bin width = 0.01). The predicted mutability of sites was strongly correlated with the observed mutation rate (R2 = 0.953, weighted by number of site-generations per bin) (Fig. 5). To ensure that the fit was not due to using the same mutations to generate the model and assess its fit, we also trained a model using a random subset of 1000 mutations and excluded these sites when assessing the fit. As with the full data set, predicted and observed mutability were highly correlated (R2 = 0.88). The fit was slightly poorer, presumably because using fewer mutations to calculate mutation rates led to more noise. Although mutability ranged from nearly 0 to 1.0, we found that 99.9% of the genome had mutability values between 0.01 and 0.30, corresponding to a range of mutation rates from 0.25 to 55.9 × 10−10. The top 25% of the genome by mutability accounts for 57% of all mutations. Mutability was highest for sites in 3′ and 5′ UTRs (predicted µ = 1.37 × 10−9) and lowest for zero- and fourfold degenerate sites (predicted µ = 7.92 × 10−10). If selection was acting in our MA experiment, despite recurrent bottlenecks, the lower mutability at zerofold sites might be caused by selective constraint. Assuming equal mutation rate across annotation categories, and controlling for the number of high-quality sites, we found only a slight deficit in zero- and fourfold degenerate mutated sites, consistent with the mutability model. We believe that the deficit is unlikely to be driven by selection, because the two categories are similarly underrepresented (zerofold Obs/Exp = 1292/1426 = 0.85, and fourfold Obs/Exp = 429/464 = 0.88), and selection on synonymous SNMs is not expected to overcome drift in our MA experiment. Furthermore, we found a slight overrepresentation of mutations at twofold degenerate sites (twofold Obs/Exp = 328/306 = 1.017), which are expected to be subject to stronger constraint than fourfold degenerate sites. These results suggest that the most likely explanation for lower mutability at zero- and fourfold degenerate sites is that other genomic properties are reducing their mutability relative to other sites in the genome.
Figure 5.
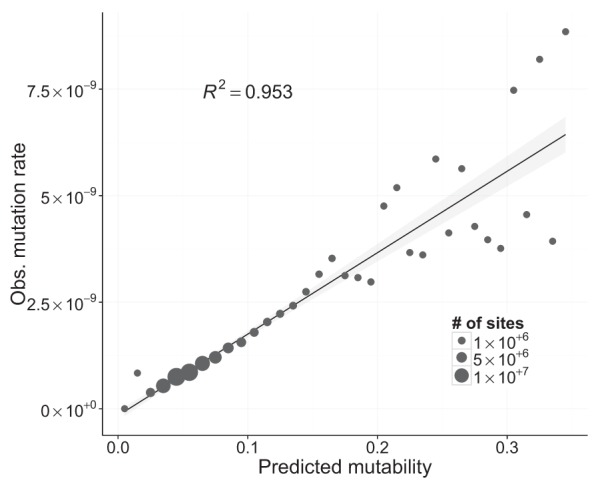
Linear fit between observed mutation rate and predicted mutability. Mutability was estimated using a logistic regression, where the presence or absence of a mutation was the response variable, and a variety of genomic properties were used as predictors (see Supplemental Table S2). Each point represents multiple genomic sites placed in discrete bins (width = 0.01) based on each site's mutability score. The size of each point is proportional to the number of sites in the genome with a given mutability. Observed mutation rates for each point were calculated as the number of observed mutations divided by the total number of callable sites-generations in that bin. The linear regression was weighted by the number of sites in each bin, and the shaded gray area around the line represents the 95% confidence region.
In neutrally evolving haploid DNA, the level of nucleotide diversity (θπ) is expected to be twice the product of mutation rate and the effective population size (2Neµ). We binned silent sites (intergenic, intronic, and fourfold degenerate sites) into 100 uniformly spaced mutability categories from 0.0 to 1.0 and calculated θπ for each bin using natural variation in the six ancestral strains used to initiate the MA lines. We found that, as predicted, sites with higher mutability have higher neutral genetic diversity (Fig. 6).
Figure 6.
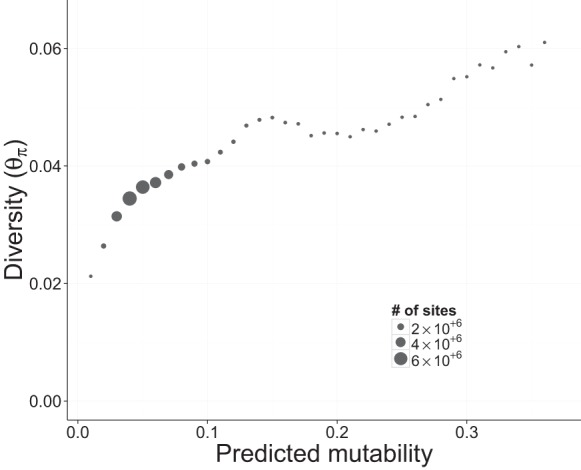
Relationship between natural genetic diversity and predicted mutability. Each point represents multiple genomic sites placed in discrete bins (width = 0.01) based on the predicted mutability of each site. Only putatively neutral sites (intronic, intergenic, and fourfold degenerate sites) were included in this figure. Nucleotide diversity (θπ) was calculated in each bin from the six ancestral strains used to start the mutation accumulation lines. The size of each point is proportional to the number of sites in the genome with a given mutability.
Factors influencing mutability
Based on the estimated parameters of the model of mutability, we extracted the relative contribution of different genomic properties to mutation. To allow comparison among the genomic properties, we scaled continuous predictors so that a change from 0 to 1 was a change of one standard deviation. We found that GC content of the surrounding genome strongly influenced the mutability at a site. Increasing the GC content of the 10 bp surrounding a site increased its mutability (GC% 10 bp, odds ratio = 1.38), but at larger scales GC content was negatively related to mutability (GC% 1000 bp, odds ratio = 0.12). The negative relationship between GC content and mutation rate was supported by a highly significant correlation between the observed mutation rate and GC content across the genome (R2 = 0.831, P < 0.001) (see Supplemental Fig. S1). Reflecting similar patterns of sequence context described above, the trinucleotide sequence in which a mutation occurred also had a strong effect on mutability. The most mutable trinucleotides were CTC and CAC, where the final C was the mutant position (odds ratio = 3.54 and 2.02, respectively), and the least mutable were GTT and AGA (odds ratio = 0.57 and 0.58, respectively). It was not possible to combine the triplets into a single predictor, but the maximum difference in mutability between triplets indicated a strong effect of sequence context on mutability. A number of other genomic properties increased mutability, such as gene density (odds ratio = 1.17) and whether a site was upstream of a transcription start site (odds ratio = 1.13). Interestingly, although a change of one standard deviation in transcription level had little effect on mutability (odds ratio = 1.02), the most highly transcribed sites in the genome were 3.7× more mutable than untranscribed sites.
Discussion
In total, we detected 6843 mutations, the largest set of characterized spontaneous mutations to date. The rate of mutation across all MA lines was µ = 11.5 × 10−10/site/generation, and the mutation rates for SNMs and small indels were 9.63 × 10−10 and 1.90 × 10−10, respectively. There are, therefore, five SNMs for each small indel, consistent with previous results in C. reinhardtii (Ness et al. 2012) and similar to Arabidopsis thaliana (5:1) (Ossowski et al. 2010) but substantially lower than the ratios recently reported from MA studies in S. cerevisiae (33:1) (Zhu et al. 2014) and D. melanogaster (12:1) (Schrider et al. 2013). Our large set of mutations, and the inclusion of multiple natural genotypes, allowed detailed examination of mutation rate variation between individuals within a species and mutation rate heterogeneity across the genome.
Within-species mutation rate variation
Our estimate of mutation rate in C. reinhardtii is 14.2- and 4.6-fold higher than estimates of Sung et al. (2012) and Ness et al. (2012), respectively. Our new estimate is partly increased by the higher rate in MA lines derived from ancestor CC-1373, but even after excluding this strain, the mutation rate is still substantially higher than the two previous estimates. The two MA lines (CC-2937-MA1, CC-2937-MA2) that were used in the study of Ness et al. (2012) continued to accumulate mutations in the present experiment for an average of ∼611 generations, and the final mutation rate estimates for each of these two lines are within the confidence intervals of their earlier estimates. Our experiment did not include strain CC-124 used in Sung et al. (2012), so we cannot directly compare the estimated mutation rate to this study. Only a single MA line (CC-1952-MA4) had a mutation rate as low as Sung et al. (2012), but the mean of all MA lines derived from that ancestor was nine times higher. Whether the low estimate of Sung et al. (2012) is the result of methodological differences or biological variation between strain CC-124 and the six strains included in our study remains to be determined.
We observed substantial within-species variation in the mutation rate (Fig. 1). MA lines derived from strain CC-1373 had an average rate more than three times higher than the mean of the other strains. MA experiments in diploid species generally start with inbred lines, and it is possible that the mutation rate could be affected by recessive mutation rate modifiers that are not expressed in nature. However, C. reinhardtii is haploid, so the elevated rate in CC-1373 is presumably caused by a mutation modifier that arose since collection or by natural variation expressed in nature. In bacteria, there is evidence that mutator lines can evolve during adaptation to a new environment (Sniegowski et al. 1997). However, CC-1373 is the slowest growing of the ancestral strains, indicating that it is not well adapted to laboratory conditions. A MA experiment in Drosophila provided evidence that individuals in poor condition have a higher mutation rate (Sharp and Agrawal 2012), so it is possible that the higher mutation rate in CC-1373 also reflects its poor condition. At the other end of the spectrum, CC-1952 had the lowest mutation rate, nearly sevenfold lower than that of CC-1373. The extent of intra-specific mutation rate variation we found implies that measuring the mutation rate for a species from a single genotype may not adequately reflect the species as a whole, and inter-specific differences in mutation rate may actually reflect limited sampling within species.
In general, theory predicts that selection is expected to drive the mutation rate toward zero, because alleles that increase the mutation rate will generate deleterious alleles and thereby reduce fitness (for review, see Sniegowski and Raynes 2013). However, mutation rates are always above zero in nature, which is usually explained by the cost of increased fidelity or by the “selection-drift barrier” imposed when selection for increasingly small improvements becomes too weak to counteract genetic drift. Under both hypotheses, the extent of intra-specific mutation rate variation may reflect mutation-selection balance in genes that affect DNA repair, replication fidelity, or the susceptibility to DNA damage. In our experiment, we detected at least two MA lines with mutation rates significantly higher than their strain means (i.e., CC-2344-MA1 and CC-2931-MA5 had mutation rates 8.0× and 3.5× above their respective strain means) (Fig. 1). It is likely that these two lines acquired mutations that damaged DNA repair or stability, concordant with the presence of two mutations in DNA repair proteins in CC-2344-MA1 (one nonsynonymous, one 5′ UTR) and four such mutations in CC-2931-MA5 (all nonsynonymous) (see Supplemental Table S3 for detailed annotations of these mutations). However, 26 of 85 MA lines also acquired one or more mutations that affect known DNA repair-associated proteins (seven nonsynonymous) but did not have elevated mutation rates. It is possible that many of these mutations did not substantially alter the mutation rate or that the mutations arose too late in the experiment to cause a detectable elevation of mutation rate. The increase in mutation rate in line CC-2344-MA1 was greater than the extent of natural variation among ancestral strains, suggesting that mutations that strongly alter mutation rate are common and may segregate in natural populations until purged by selection. Therefore, the high mutation rate of CC-1373 may be caused by a naturally occurring mutator allele. Alternatively, if C. reinhardtii is primarily asexual in nature, theory predicts that, if a mutator allele results in a linked beneficial allele, the mutator will hitchhike to high frequency. A key parameter determining whether selection will favor higher mutation rates is the rate of recombination, but the frequency of sex and recombination in natural populations of C. reinhardtii is currently unknown.
Spatial heterogeneity in mutation rate
By examining the spectrum of mutations and the local sequence context in which they occur, we found clear evidence for heterogeneity in mutation rate at fine scales. In particular, the rate of mutation at C:G sites (12.2 × 10−10) was 2.4× higher than at A:T sites (5.19 × 10−10), and transitions from C:G→T:A occurred at twice the rate expected if all mutations occurred at equal rates (Fig. 3). The transition-transversion ratio (Ts:Tv) is 1.03 in the SNMs detected in our experiment, in contrast to a Ts:Tv of 1.52 in the standing variation between ancestral strains. The difference between these two ratios indicates that selection or GC-biased gene conversion, rather than the underlying mutational process, is driving up the Ts:Tv ratio in nature. An AT-biased mutation spectrum is consistent with a growing body of evidence suggesting that it might be universal in both prokaryotes (Hershberg and Petrov 2010) and eukaryotes (e.g., Zhu et al. 2014). Additionally, we found that the sequence flanking a mutated site strongly influenced the mutation rate. In mammals, methylated CpG sites are frequently deaminated, causing C to T transitions, but in C. reinhardtii there is only weak evidence of CpG methylation, and our data reveal only a small excess of CpG motifs in C to T mutations (Fig. 4). The most mutable triplet (CTC) had a mutation rate more than 10× higher than the least mutable triplet (GCA), and after accounting for background triplet frequencies, a mutation from CTC to CTT was 17× more likely than a mutation from AAA to AAG. Interestingly, this CTC triplet appears to be highly mutable across a very wide diversity of organisms, including fungi (Zhu et al. 2014), plants, and animals (Alexandrov et al. 2013b). In human tumor genomes, there is a predominance of C to T and C to G mutations in the same CTCG sequence motif, which has been linked with the APOBEC family of cytidine deaminases (Alexandrov et al. 2013b). Given that this motif has been found repeatedly, it seems probable that the mutability of other sequence motifs may be shared across species; however, the mechanisms underlying this phenomenon are unknown. The fact that the mutation rate can vary to this extent over very short scales has consequences for the evolution of DNA and protein sequence. In the future, incorporation of direct measurements of mutability into models of sequence change will facilitate better predictions of disease susceptibility and molecular evolution (see Michaelson et al. 2012; Neale et al. 2012; Samocha et al. 2014).
By comparing the distribution of inter-mutation distances to a random expectation, we found that there is an excess of mutations clustered within 1–10 bp of one another (Fig. 2). The fact that these clusters all occur within MA lines suggests that each represents a single multinucleotide mutation (MNM) event. In total, there were 80 pairs and two trios of MNMs within 10 bp of one another, implying that 2.8% of SNMs arise through clustered mutations. The average proportion of MNMs was similar in MA studies of S. cerevisiae, D. melanogaster, C. elegans, and A. thaliana (3.4%) and genome sequencing studies of humans (1%–4%) (Schrider et al. 2011; Harris and Nielsen 2014). The generation of these clusters has been linked to error-prone polymerases such as Pol ζ in S. cerevisiae (Stone et al. 2012; Northam et al. 2013). In human and S. cerevisiae, the Pol ζ enzyme creates an excess of GpC to ApA or TpT MNMs (Northam et al. 2013; Harris and Nielsen 2014). Although we did not observe a similar excess of mutations at GpC sites, we found that 27 of 55 dinucleotide MNMs occur at CpC sites and that 25 of these resulted in ApA/ApT/TpA/TpT dinucleotides. The fact that MNMs have been observed in a broad array of taxa indicates that such mutations are a widespread phenomenon that potentially affects a significant proportion of variation. MNMs, therefore, violate the assumption of independence between SNP sites and could potentially lead to misinferences about the nature of selection in the genome. Additionally, by altering two or more nearby sites, MNMs have the potential to move between fitness peaks that would otherwise require maladaptive single mutations as intermediates.
At large genomic scales, we found little evidence for heterogeneity of the mutation rate. For example, the mutation rate variation among 200-kbp windows could be largely accounted for by random fluctuations. Although we found clear evidence of fine-scale variation in mutation rate, the variation appears to be evenly spread along the chromosome. This effect can be seen in our predictive model of mutation, where the mutability of sites in 200-kbp windows averages out, so that the standard deviation among windows equates to ∼7.5% of the mean (i.e., mean mutability = 0.069, SD = 0.005). Our findings are consistent with direct measurements of mutation rate in D. melanogaster (Schrider et al. 2013), S. cerevisiae (Zhu et al. 2014), and humans (Kong et al. 2012), where no evidence of large-scale variation in the mutation rate was detected. Although, comparative evidence suggests that substitution rate varies at the scale of megabases in mammals, this may be driven by selection or GC-biased gene conversion during recombination. From our observations and direct estimates of mutation rate variation in other species, we conclude that the causes of mutational heterogeneity do not appear to operate at the scale of tens of kilobases, and if heterogeneity exists at this scale, it will require even more precise measurements of the mutation rate.
Factors that predict mutability
Our model of mutability identified a number of other genomic properties that predict the rate of spontaneous mutation and create heterogeneity between sites. For example, the %GC of the 10 bp around a mutated site was positively correlated with mutability (odds ratio = 1.38, SD = 16.3%), probably because G:C bases and GC-rich triplets were more mutable. However, the GC content of the 1000 bp surrounding a site was negatively associated with its mutability (e.g., %GC of 1000-bp window, odds ratio = 0.12, SD = 5.4%). A negative correlation between mutability and GC content in humans has been attributed to higher melting temperatures of GC-rich DNA (Fryxell and Moon 2005). Because cytosine deamination is one of the most common sources of mutation and only occurs while DNA is single-stranded, mutation is less common in regions with high melting temperature (Frederico et al. 1993). An alternate explanation for our observations is that sites with a high mutation rate, for an unknown reason, evolve low GC content because mutation is AT-biased.
Our model of mutability also revealed an effect of gene expression when comparing untranscribed DNA to the most highly transcribed genes (odds ratio = 3.71). However, because most regions are untranscribed and the variance of transcription in expressed genes is relatively low, transcription level overall had little effect on mutability (odds ratio = 1.02, SD = 108.3 FPKM). It is commonly reported that highly expressed genes are the most evolutionarily conserved; therefore, an elevated mutation rate would predict that more deleterious mutations should occur in high expression genes, and therefore, more purifying selection would be required to conserve these sequences. The mean mutability score varied across sites with different annotations. The 5′ and 3′ UTRs had the highest mutability (predicted µ = 1.5 × 10−9), which is consistent with the observation in humans, where these regulatory regions are often found in accessible chromatin (DNase hypersensitive sites), to allow binding of transcription factors, and could lead to DNA damage (Michaelson et al. 2012). Consistent with an increased mutation rate and AT-biased mutation, UTRs have the lowest GC content of any broad category of sites (56.7%). Although the model predicted a higher rate in UTRs, we did not observe an elevation in observed mutation rate, possibly because, even with nearly 7000 mutations, there was still insufficient power to detect such subtle variation. Overall, the model accurately predicted the observed mutation rate, demonstrating that average mutation rate can be predicted from key genomic properties (Fig. 5). However, variation in mutability may not be fully captured with this approach (Eyre-Walker and Eyre-Walker 2014). For a close fit between observed and predicted mutability, only the average mutability of each bin needs to be accurately predicted. There may still be unexplained variation around the mean within each bin, and we should be cautious about predictions of mutability for very small numbers of sites. However, for large groups of sites, the model accurately predicts the average mutation rate, and we can be confident in the genomic properties that best predict mutation rate.
The mutability model also revealed that mutation rate variation affects patterns of neutral genetic variation. We found a clear positive relationship between mutability and nucleotide diversity at silent sites (Fig. 6). The model identifies the genomic properties of sites that mutated in our experiment, and we show that, using these genomic properties, we are able to predict natural levels of genetic diversity. Diversity is determined both by the mutation rate and the amount of genetic drift (Ne). The strength of drift across the genome depends on the effects of hitchhiking (selective sweeps and background selection), which is determined by the frequency and strength of selection and the rate of recombination. Therefore, our measure of mutability may not have correlated well with diversity. However, by binning sites by mutability, we have removed regional heterogeneity in the strength of drift and can see that mutation rate heterogeneity has a clear correlation with standing levels of genetic diversity in natural populations. It is worth noting that our findings are correlations, and we cannot with certainty assign a causal link between mutability and diversity. However, it seems reasonable that mutation rate heterogeneity between sites could affect diversity, which is theoretically determined by the product of µ and Ne. Given that mutability varies greatly between nearby sites, local heterogeneity in mutation is an important consideration when using diversity and related statistics to infer selection or demography from population genomic data.
This study characterized the largest set of spontaneous mutations to date and demonstrated the value of combining MA with whole-genome sequencing. We found sevenfold variation in mutation rate among natural strains of C. reinhardtii. Although the mutation rate did not vary across large genomic windows, the mutation rate of individual sites was strongly affected by their flanking sequence, resulting in fine-scale heterogeneity of mutation rate. Other genomic properties, such as GC content, gene density, and expression level, also influenced mutability. Similar results across a wide diversity of species suggests that general properties of mutation exist and that models of sequence evolution could be improved to reflect these properties and better detect selection in the genome or estimate phylogenetic relationships. In the near future, rapidly evolving sequencing technologies will facilitate even more detailed investigation into the process of mutation from both MA and parent-offspring sequencing. One important avenue of future research will be a synthesis of findings from studies like ours with the underlying DNA repair and damage mechanisms to provide explanations for patterns of mutational heterogeneity between individuals and across the genome.
Methods
Mutation accumulation experiment
We conducted a mutation accumulation experiment in six genetically diverse strains of C. reinhardtii obtained from the Chlamydomonas Resource Center (chlamycollection.org). The strains were isolated from the wild between 1945 and 1993 and have not been selected for unusual phenotypes and should represent a sample of the naturally occurring variation over the geographic range of C. reinhardtii samples in North America (Table 1). To initiate the MA lines, a single colony from each of the six ancestral strains was streaked out, and we randomly selected 15 individual colonies to start the replicated MA lines (for a total of 90 MA lines). We bottlenecked the MA lines at regular intervals by selecting a random colony which was streaked onto a fresh agar plate. We calculated Ne for a MA line as the harmonic mean population size at each cell division from one cell to 12 divisions, yielding Ne = 6.5. The timing of transfers was chosen to avoid selecting against slow growing colonies, and we periodically checked that no additional colonies became visible after the transfer time. We estimated the number of generations undergone by each MA line over the course of the experiment by measuring the number of cells in colonies grown on agar plates after a period of growth equivalent to the times between transfers in the experiment. A more detailed description of the MA line creation and generation time estimation can be found in Morgan et al. (2014).
Sequencing and alignment
To extract DNA, we grew cells on 1.5% Bold's agar for 4 d until there was a high density of cells, at which point the cells were collected and frozen at −80°C. We disrupted the frozen cells using glass beads and extracted DNA using a standard phenol-chloroform extraction. Whole-genome resequencing was conducted using the Illumina GAII platform at BGI HongKong. The sequencing protocol was modified to accommodate the unusually high GC content of the C. reinhardtii genome (mean GC = 63.9%). Variation in GC content is known to cause uneven representation of sequenced fragments, especially when GC > 55% (Aird et al. 2011). We therefore used a modified PCR step in sequencing library preparation, following Aird et al. (2011) (3 min at 98°C; 10× [80 sec at 98°C, 30 sec at 65°C, 30 sec at 72°C]; 10 min at 72°C, with 2M betaine and slow temperature ramping 2.2°C/sec). We obtained ∼30× coverage of the genome (3 Gbp of 100-bp paired-end sequence) for each of the MA lines.
We aligned reads to the C. reinhardtii reference genome (version 5.3) (Merchant et al. 2007) using BWA 0.7.4-r385 (Li and Durbin 2009). We included the plastid genome (NCBI accession NC_005353), the mitochondrial genome (NCBI accession NC_001638), and the MT- locus (NCBI accession GU814015) to avoid misalignment of reads derived from these loci onto other parts of the nuclear genome. We tested a variety of values for the fraction of mismatching bases allowed in alignments, but variation about the default (n = 0.04) did not improve the number of high-quality reads mapped or genome coverage (results not shown). After alignment, we removed duplicate reads with the Picard tool MarkDuplicates (v1.90). To avoid calling false variants due to alignment errors, we used the GATK (v2.8-1) tools RealignerTargetCreator and IndelRealigner (McKenna et al. 2010; Depristo et al. 2011) to realign reads flanking potential insertions and deletions. We realigned all replicate MA lines from each starting strain together to ensure that the same alignment solutions were chosen in all lines derived from that strain. The realigned BAM files included all MA lines from a given ancestral strain and were then used to jointly call genotypes using the UnifiedGenotyper from GATK. We used the “--output_mode EMIT_ALL_SITES” option to output all genomic positions so that we could identify both high-quality sites regardless of whether they had mutated. We used a “heterozygosity” parameter of 0.01, but previous testing in C. reinhardtii showed that our genotyping is not sensitive to this prior as long as read depth is high, as it is in the present experiment (Ness et al. 2012). To identify short insertions and deletions (indels), we used the GATK v(2.8-1) tool “HaplotypeCaller,” which performs local reassembly of reads (i.e., indels called with UnifiedGenotyper were ignored). The six resulting Variant Call Format files (VCFs) (one per ancestral strain) were converted to wormtable databases using the python package WormTable v0.1.0 (Kelleher et al. 2013) which enabled efficient exploration of quality filters for mutation identification.
Mutation identification
MA lines within an ancestral strain were genetically identical at the start of the experiment, so any unique allele carried by a replicate within a strain was a candidate mutation. We applied a number of filters to genotype calls to identify mutations while minimizing false positive and false negative calls. A site was called as a mutation if within that ancestral strain:
The mapping quality (MQ) ≥ 90 and the PHRED called site quality (QUAL) ≥ 100.
All MA lines were “homozygous”; C. reinhardtii is haploid; therefore, this filter avoided mapping errors due to paralogous loci.
The genotype of exactly one MA line differed from the rest of the lines.
All nonmutated lines shared the same genotype.
At least two sequences have confident genotype calls.
Our mutation-calling algorithm also allowed us to identify contamination that may have occurred between MA lines in our experiment. For example, cross-contamination between MA lines derived from different ancestors would manifest as millions of false-positive mutations that were actually naturally occurring SNPs. If contamination occurred between two lines from the same ancestor, it would mean that all mutations to that point in the experiment were not unique, which would cause unusually low mutation rates in both lines. No evidence for contamination of this kind was seen.
Callable sites
To calculate mutation rates and define null expectations, we needed to know the total number of sites with equivalent quality to the new mutations, hereafter referred to as “callable” sites. However, the definitions and distributions of quality scores are often different for variant and invariant sites. We therefore inferred a second measure of quality for invariant sites that was comparable to that used for mutant sites. For each mutant site, we extracted the QUAL and MQ for the mutation and the nearest invariant site, under the assumption that, because most reads are shared between adjacent sites, the quality characteristics of the sites will be similar. We then estimated the correlation and relationship between quality scores at neighboring mutant and invariant sites using a linear model (MQ: R2 = 0.9996, P < 0.001, QUAL: R2 = 0.38, P < 0.001). The linear relationships between invariant and variant quality scores were used to predict appropriate MQ and QUAL thresholds for invariant sites (invariant MQ threshold = 90, invariant QUAL threshold = 36.4). Analogous to the mutation calling, a site was callable within an ancestral strain if no line was called as a heterozygote, all lines with mapped reads had the same genotype call, and at least two MA lines had genotype calls.
Sanger confirmation
We estimated the accuracy of our mutation calls using Sanger-sequencing. We randomly selected 192 mutation calls (32 per ancestral strain) including both short indels and SNMs. We amplified each locus in the putative mutant MA line and a nonmutated MA line from the same ancestral strain. Sequences were then visually inspected in SeqTrace v0.9.0 to confirm the presence of the mutated site.
Mutation rate calculations
We calculated the mutation rate (µ) in each replicate as, µ = mutations/(callable sites × MA generations). Whenever multiple MA lines were combined for mutation rate calculations, the number of callable sites and MA generations (site-generations) for each MA line was included to accurately account for differences among replicate lines. Similarly, all null expectations and mutation rate estimates for particular classes of sites take into account the number of site-generations for the specific positions included. To compare the average mutation rate of the six ancestral strains, we used the GLS function in R to fit a linear model to the individual mutation rate estimates of the MA lines. The model included mutation rate as the response variable and ancestral strain as a fixed factor. We allowed the variance to differ among ancestral lines using the varIdent function (Zuur et al. 2009). We then used the ghlt function to generate linear contrasts, allowing us to further explore differences among the ancestors.
Base composition and sequence context
Throughout our analyses of the mutation spectrum, we treated complementary mutations (C:G and A:T) symmetrically, such that there were six distinct SNMs (A:T→C:G, A:T→G:C, A:T→T:A, C:G→A:T, C:G→G:C, C:G→T:A). To assess the base spectrum of mutations, we calculated the frequency of each of the six mutation types relative to the expected frequency if all mutations were equally likely, calculated from the base composition of the callable sites. To analyze the local sequence context in which mutations occurred, we measured base composition at each of the positions 5 bp upstream of and downstream from the mutated site. To calculate the null expectation for sequence context, we estimated base composition in analogous windows surrounding 106 randomly selected callable sites. Separate expectations were generated for sites centered on A:T and C:G.
Spatial heterogeneity of mutation
To assess whether there was spatial heterogeneity in mutation rate, we calculated the mutation rate across the genome in sliding windows. We conducted the analysis with windows of 100 kbp, 200 kbp, 500 kbp, and 1 mbp, but because the results were qualitatively similar, we report only the 200-kbp analysis. The mutation rate of each window was calculated as the number of mutations in that window divided by the total number of callable sites × generations. To assess how the mutation rate in these windows varied relative to null expectations, we simulated a random distribution of mutations. For each MA line, we generated a corresponding simulated line where the number of mutations carried by that line was distributed among the 200-kbp windows in proportion to the number of callable site-generations in each window. This procedure was repeated 1000 times to generate an expected distribution of mutation rates across the 200-kbp windows.
We also tested for the presence of a nonrandom spatial distribution of mutations by comparing the observed distribution of inter-mutation distances to a simulated distribution. This approach differs from the analysis above because it can detect fine-scale clusters of mutations. We simulated data under a model where mutations occur randomly across the genome, while retaining the same number of mutations per MA line and accounting for differences in the callable genome positions. For each MA line, we generated a corresponding simulated sample by randomly assigning the number of mutations that occurred in that MA line to individual callable positions. This allowed us to assess whether there was significantly more clustering within and between lines while accounting for line-specific differences in callable sites. The observed and simulated distributions of inter-mutation distances were compared using the Kolmogorov–Smirnov (KS) test in R.
Mutability
To determine which genomic properties influenced the mutability of individual sites, we used regularized logistic regression to differentiate between the identified mutations and randomly selected callable sites. Our analysis was loosely based on the approach of Michaelson et al. (2012). For all 6843 mutations and 105 nonmutated sites, we collated a table of genomic properties and annotations to use as predictors in the logistic regression. Genomic properties included %GC, gene density, transcription level, recombination rate, nucleosome occupancy, and the trinucleotide sequence in which the site occurs (see Supplemental Table S2 for details). A number of genomic properties were calculated for each site in windows of varying size from 10 bp up to 1 Mbp. Categorical predictors were converted to multiple binary predictors (0/1 for each category level) to be fitted in the same model with numeric predictors.
With these predictors, we used the R package GLMnet (v1.9-8) (Friedman et al. 2010) to fit a logistic regression, where mutation class, mutant (1) or background (0), was the binary response variable. GLMnet fits generalized linear models with penalized maximum likelihood using ridge or lasso regression which provides more precise model-fitting than other methods, such as least-squares, when the predictors are inter-correlated. In ridge/lasso regression, a penalty is imposed when the sum of the correlation coefficients of the predictors is large. Therefore, when predictors are inter-correlated the values of their combined correlation coefficients are reduced either by shrinking one of the coefficients toward zero (lasso regression) or by shrinking both toward some medium value (ridge regression). The strength of the penalty against large correlation coefficients is determined by the regularization parameter (λ), which therefore determines the complexity of the model (the number predictors with nonzero correlation coefficients). The value of λ was chosen using the in-built cross-validation function (we selected λ that minimizes mean cross-validated error, “lambda.min”). The elastic net mixing parameter (α) determines whether lasso (α = 1) or ridge regression (α = 0) is used. The fit of the model was unchanged by the selection of α, and all results presented here used α = 0.01, where the coefficients of correlated predictors are shrunk together. Using the “predict” function of GLMnet model objects, we estimated mutability at each site in the genome as its probability of belonging to class “mutation,” given the genomic predictors at a given site (see Supplemental Material for R code used to fit the model and predict mutability).
Only the relative values of the predicted mutability are important, because the exact probabilities returned by the model are influenced by the proportion of mutated and nonmutated sites in the training set. In our training set, we included 6843 mutations and 105 nonmutated sites; therefore, the mean predicted mutability is ∼0.06 (6843/[105+6843]). We assessed the accuracy of the predicted mutability by binning sites into 100 mutability categories from 0.0 to 1.0. Within each mutability category, we estimated mutation rate as the number of observed mutations divided by the total number of site-generations in that category. For example, there were 13,948,935 sites with mutability between 0.04 and 0.05, 820 of which mutated in one of our 85 MA lines; because these sites experienced 68,822 generations of MA, µ for this bin is 820/(68,822 × 13,948,935) = 8.6 × 10−10. The observed mutation rate was predicted to be positively correlated with the mid-point mutability of the category.
To test whether mutability predicted long-term effects of mutation rate variation, we also calculated the relationship between mutability and natural levels of nucleotide diversity in the six ancestral strains used to start the MA lines. In neutrally evolving haploid DNA, the level of nucleotide diversity (θπ) is expected to be twice the product of the mutation rate and the effective population size (2Neµ); we therefore predict that the mutation rate should correlate positively with mutability. For this analysis, whether a site was variant was omitted from the model to avoid circularity in the relationship between diversity and mutability. We binned silent sites (intergenic, intronic, and fourfold degenerate sites) into 100 uniformly spaced mutability categories from 0.0 to 1.0 and calculated θπ for all sites in each bin.
To assess the relative contributions of each genomic property to mutability, we extracted the coefficients of each predictor from the model. To compare the log(odds ratio) of each genomic property on mutability, we scaled each predictor so that a change from 0.0 to 1.0 was a change of one standard deviation. As alternate scaling, we also normalized the predictors such that each ranged from exactly zero to one.
Data access
All sequence data from this study have been submitted to the EBI European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena) under accession number PRJEB9934.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.191494.115.
References
- Aird D, Ross MG, Chen W-S, Danielsson M, Fennell T, Russ C, Jaffe DB, Nusbaum C, Gnirke A. 2011. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol 12: R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg Å, Børresen-Dale A-L, et al. 2013a. Signatures of mutational processes in human cancer. Nature 500: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. 2013b. Deciphering signatures of mutational processes operative in human cancer. Cell Rep 3: 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver DR, Wilhelm LJ, Howe DK, Gafner K, Dolan PC, Baer CF. 2012. Variation in base-substitution mutation in experimental and natural lineages of Caenorhabditis nematodes. Genome Biol Evol 4: 513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW. 2006. Chaos and order in spontaneous mutation. Genetics 173: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW, Charlesworth B, Charlesworth D, Crow JF. 1998. Rates of spontaneous mutation. Genetics 148: 1667–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M, Wang RY. 1981. 5-Methylcytosine in eukaryotic DNA. Science 212: 1350–1357. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A, Eyre-Walker YC. 2014. How much of the variation in the mutation rate along the human genome can be explained? G3 (Bethesda) 4: 1667–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederico LA, Kunkel TA, Shaw BR. 1993. Cytosine deamination in mismatched base pairs. Biochemistry 32: 6523–6530. [DOI] [PubMed] [Google Scholar]
- Friedman J, Hastie T, Tibshirani R. 2010. Regularization paths for generalized linear models via coordinate descent. J Stat Soft 33: 1–22. [PMC free article] [PubMed] [Google Scholar]
- Fryxell KJ, Moon W-J. 2005. CpG mutation rates in the human genome are highly dependent on local GC content. Mol Biol Evol 22: 650–658. [DOI] [PubMed] [Google Scholar]
- Halligan DL, Keightley PD. 2009. Spontaneous mutation accumulation studies in evolutionary genetics. Annu Rev Ecol Evol Syst 40: 151–172. [Google Scholar]
- Harris K, Nielsen R. 2014. Error-prone polymerase activity causes multinucleotide mutations in humans. Genome Res 24: 1445–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA. 2010. Evidence that mutation is universally biased towards AT in bacteria. PLoS Genet 6: e1001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T. 1999. Beneficial mutations, hitchhiking and the evolution of mutation rates in sexual populations. Genetics 151: 1621–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley PD, Ness RW, Halligan DL, Haddrill PR. 2014a. Estimation of the spontaneous mutation rate per nucleotide site in a Drosophila melanogaster full-sib family. Genetics 196: 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley PD, Pinharanda A, Ness RW, Simpson F, Dasmahapatra KK, Mallet J, Davey JW, Jiggins CD. 2014b. Estimation of the spontaneous mutation rate in Heliconius melpomene. Mol Biol Evol 32: 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher J, Ness RW, Halligan DL. 2013. Processing genome scale tabular data with wormtable. BMC Bioinformatics 14: 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. 1967. On evolutionary adjustment of spontaneous mutation rates. Genet Res 9: 23–34. [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, et al. 2012. Rate of de novo mutations and the importance of father's age to disease risk. Nature 488: 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GI, Murray AW. 2011. Mutation rates across budding yeast Chromosome VI are correlated with replication timing. Genome Biol Evol 3: 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2010. Evolution of the mutation rate. Trends Genet 26: 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matic I, Radman M, Taddei F, Picard B, Doit C, Bingen E, Denamur E, Elion J. 1997. Highly variable mutation rates in commensal and pathogenic Escherichia coli. Science 277: 1833–1834. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant SS, Prochnik SE, Vallon O, Harris EH, Karpowicz SJ, Witman GB, Terry A, Salamov A, Fritz-Laylin LK, Maréchal-Drouard L, et al. 2007. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318: 245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson JJ, Shi Y, Gujral M, Zheng H, Malhotra D, Jin X, Jian M, Liu G, Greer D, Bhandari A, et al. 2012. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 151: 1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AD, Ness RW, Keightley PD, Colegrave N. 2014. Spontaneous mutation accumulation in multiple strains of the green alga, Chlamydomonas reinhardtii. Evolution 68: 2589–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, Lin C-F, Stevens C, Wang L-S, Makarov V, et al. 2012. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485: 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness RW, Morgan AD, Colegrave N, Keightley PD. 2012. An estimate of the spontaneous mutation rate in Chlamydomonas reinhardtii. Genetics 192: 1447–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northam MR, Moore EA, Mertz TM, Binz SK, Stith CM, Stepchenkova EI, Wendt KL, Burgers PMJ, Shcherbakova PV. 2013. DNA polymerases ζ and Rev1 mediate error-prone bypass of non-B DNA structures. Nucleic Acids Res 42: 290–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski S, Schneeberger K, Lucas-Lledó JI, Warthmann N, Clark RM, Shaw RG, Weigel D, Lynch M. 2010. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327: 92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto SP. 2009. The evolutionary enigma of sex. Am Nat 174: S1–S14. [DOI] [PubMed] [Google Scholar]
- Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, Kosmicki JA, Rehnström K, Mallick S, Kirby A, et al. 2014. A framework for the interpretation of de novo mutation in human disease. Nat Genet 46: 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrider DR, Hourmozdi JN, Hahn MW. 2011. Pervasive multinucleotide mutational events in eukaryotes. Curr Biol 21: 1051–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrider DR, Houle D, Lynch M, Hahn MW. 2013. Rates and genomic consequences of spontaneous mutational events in Drosophila melanogaster. Genetics 194: 937–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp NP, Agrawal AF. 2012. Evidence for elevated mutation rates in low-quality genotypes. Proc Natl Acad Sci 109: 6142–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniegowski P, Raynes Y. 2013. Mutation rates: How low can you go? Curr Biol 23: R147–R149. [DOI] [PubMed] [Google Scholar]
- Sniegowski PD, Gerrish PJ, Lenski RE. 1997. Evolution of high mutation rates in experimental populations of E. coli. Nature 387: 703–705. [DOI] [PubMed] [Google Scholar]
- Stamatoyannopoulos JA, Adzhubei I, Thurman RE, Kryukov GV, Mirkin SM, Sunyaev SR. 2009. Human mutation rate associated with DNA replication timing. Nat Genet 41: 393–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JE, Lujan SA, Kunkel TA. 2012. DNA polymerase ζ generates clustered mutations during bypass of endogenous DNA lesions in Saccharomyces cerevisiae. Environ Mol Mutagen 53: 777–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundin GW, Weigand MR. 2007. The microbiology of mutability. FEMS Microbiol Lett 277: 11–20. [DOI] [PubMed] [Google Scholar]
- Sung W, Ackerman MS, Miller SF, Doak TG, Lynch M. 2012. Drift-barrier hypothesis and mutation-rate evolution. Proc Natl Acad Sci 109: 18488–18492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2012. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 17: 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veltman JA, Brunner HG. 2012. De novo mutations in human genetic disease. Nat Rev Genet 13: 565–575. [DOI] [PubMed] [Google Scholar]
- Venn O, Turner I, Mathieson I, de Groot N, Bontrop R, McVean G. 2014. Nonhuman genetics. Strong male bias drives germline mutation in chimpanzees. Science 344: 1272–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YO, Siegal ML, Hall DW, Petrov DA. 2014. Precise estimates of mutation rate and spectrum in yeast. Proc Natl Acad Sci 111: E2310–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuur A, Ieno EN, Walker N, Saveliev AA, Smith GM. 2009. Mixed effects models and extensions in ecology with R. Springer-Verlag, New York. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.