

In this study, Dai et al. show that concentration-dependent autoactivation of the proapoptotic BCL2 family member BAK plays an important role in the control of intrinsic apoptosis. These findings provides novel insight into BAK oligomerization and demonstrate how applying this new knowledge can result in sensitivity of lymphohematopoietic cells to various death stimuli.
Keywords: apoptosis, BAK, BH3 mimetic
Abstract
Mitochondrial outer membrane permeabilization (MOMP), a key step in the intrinsic apoptotic pathway, is incompletely understood. Current models emphasize the role of BH3-only BCL2 family members in BAX and BAK activation. Here we demonstrate concentration-dependent BAK autoactivation under cell-free conditions and provide evidence that this autoactivation plays a key role in regulating the intrinsic apoptotic pathway in intact cells. In particular, we show that up to 80% of BAK (but not BAX) in lymphohematopoietic cell lines is oligomerized and bound to anti-apoptotic BCL2 family members in the absence of exogenous death stimuli. The extent of this constitutive BAK oligomerization is diminished by BAK knockdown and unaffected by BIM or PUMA down-regulation. Further analysis indicates that sensitivity of cells to BH3 mimetics reflects the identity of the anti-apoptotic proteins to which BAK is constitutively bound, with extensive BCLXL•BAK complexes predicting navitoclax sensitivity, and extensive MCL1•BAK complexes predicting A1210477 sensitivity. Moreover, high BAK expression correlates with sensitivity of clinical acute myelogenous leukemia to chemotherapy, whereas low BAK levels correlate with resistance and relapse. Collectively, these results inform current understanding of MOMP and provide new insight into the ability of BH3 mimetics to induce apoptosis without directly activating BAX or BAK.
Previous studies have identified an important role for the intrinsic apoptotic pathway in the response to a variety of anti-cancer treatments (Kaufmann and Earnshaw 2000; Johnstone et al. 2002), including DNA-damaging drugs (Villunger et al. 2003), spindle poisons (Tan et al. 2005), kinase inhibitors (Kuroda et al. 2006; Cragg et al. 2008; Gupta et al. 2012), and BH3 mimetics (van Delft et al. 2006). This pathway is regulated by BCL2 family members, a group of structurally related proteins with distinct pro- and anti-apoptotic roles (Chipuk and Green 2008; Martinou and Youle 2011; Strasser et al. 2011; Czabotar et al. 2014). When the balance of pro- and anti-apoptotic signaling favors cell death, two members of this family, BAX and BAK, permeabilize the mitochondrial outer membrane (MOM), leading to cytochrome c release, caspase 9 activation, and cellular disassembly (Jiang and Wang 2004; Taylor et al. 2008).
How BAX and BAK are activated has been a matter of debate. According to one model, their activation is triggered when cellular stresses lead to increased expression or activity of BH3-only members of the BCL2 family, such as BIM, PUMA, or BID (Chipuk and Green 2008; Strasser et al. 2011; Czabotar et al. 2014). Sharing homology in a single 15-amino-acid α-helical domain (Czabotar et al. 2014; Westphal et al. 2014), these proapoptotic proteins bind transiently to the hydrophobic groove of BAX or BAK (Dai et al. 2011, 2014; Czabotar et al. 2013; Edwards et al. 2013; Brouwer et al. 2014) or a secondary site on BAX (Gavathiotis et al. 2008; Edwards et al. 2013) to facilitate BAX/BAK oligomerization (Antonsson et al. 2001; Cheng et al. 2003) and MOM permeabilization (MOMP). Evidence in support of this direct activation model includes direct binding of BH3-only proteins (Kim et al. 2009; Dai et al. 2011, 2014) or BH3 peptides (Walensky et al. 2006; Gavathiotis et al. 2008; Czabotar et al. 2013; Brouwer et al. 2014) to BAX or BAK in vitro as well as the ability of this binding to trigger BAX/ BAK oligomerization and membrane permeabilization in cell-free systems (Kuwana et al. 2002; Letai et al. 2002; Dewson et al. 2008, 2009; Lovell et al. 2008; Oh et al. 2010; Dai et al. 2011; Du et al. 2011). Importantly, BAX and BAK BH3 peptides also bind and activate BAX and BAK in some contexts in vitro (Llambi et al. 2011; Czabotar et al. 2013; Brouwer et al. 2014), indicating that this property is not unique to BH3 domains of BH3-only proteins.
An earlier model suggested that BH3-only proteins predominantly neutralize anti-apoptotic family members such as BCL2, MCL1, and BCLXL, which otherwise bind preactivated BAX and BAK to prevent MOMP (Willis et al. 2007). Formulated to account for BAX and BAK activation in the face of difficulty in detecting complexes between BH3-only proteins and BAX or BAK in intact cells (Wei et al. 2000; Willis et al. 2007), this model fell out of favor as interactions between BH3-only family members and BAX or BAK were demonstrated through biochemical (Gavathiotis et al. 2008; Kim et al. 2009; Dai et al. 2011) and genetic (Merino et al. 2009) approaches. Nonetheless, earlier studies clearly indicated that anti-apoptotic BCL2 family members neutralize overexpressed BAX (Oltvai et al. 1993) and BAK (Chittenden et al. 1995) in addition to BH3-only family members (Willis et al. 2007; Llambi et al. 2011).
Despite the growing consensus that BH3-only family members can bind BAX and BAK to directly activate them, the possibility that BAX or BAK is preactivated within cells, as postulated by the indirect activation model, has not been systematically examined. Here we show that purified recombinant BAKΔTM (BAK lacking its transmembrane domain to facilitate purification) can spontaneously oligomerize and permeabilize membranes under cell-free conditions. We also demonstrate that endogenous BAK is constitutively activated in many lymphohematopoietic cell lines, show that the extent of constitutive BAK activation correlates with BAK expression levels, and assess the impact of this constitutive BAK activation on sensitivity to therapeutic agents.
Results
Requirement for BAK activation prior to BCLXL and MCL1 binding
To provide a background for examining BAK binding and neutralization in intact cells, interactions between BAK and anti-apoptotic BCL2 family members were studied under cell-free conditions. Consistent with earlier results (Willis et al. 2005), surface plasmon resonance (SPR) indicated that MCL1 bound to immobilized BAKΔTM protein or BAK BH3 peptide with similar, low nanomolar affinities (Fig. 1A–D). In the reciprocal experiment, recombinant BAKΔTM protein bound 100-fold less tightly than BAK BH3 peptide to immobilized MCL1 (Fig. 1B–D) or BCLXL (Fig. 1E; Supplemental Fig. S1A).
Figure 1.

MCL1 and BCLXL preferentially bind activated BAK. (A) SPR of 300 nM MCL1ΔTM binding to immobilized BAKΔTM or BAK BH3 peptide. (B) SPR (relative units) of different concentrations of BAK BH3 peptide binding to immobilized MCL1ΔTM. (C) Direct comparison of 320 nM BAKΔTM and 320 nM BAK BH3 peptide binding to immobilized MCL1ΔTM. (D,E) Based on the SPR assays, the affinities of MCL1ΔTM (D) and BCLXLΔTM (E) for BAKΔTM and BAK BH3 peptide were calculated. Error bars indicate ±sd of three independent experiments using different chips and different protein batches. (F) SPR of MCL1ΔTM binding to 500 nM BAKΔTM, 500 nM BIM BH3, or 500 nM BAKΔTM + 500 nM BIM BH3. The double arrow indicates increased resonance due to BAK activation. The box at the top of F indicates peptides used in this figure.
To confirm that tight binding of BAK to anti-apoptotic BCL2 family members requires BAK activation, BAKΔTM protein was bound to immobilized MCL1 in the absence or presence of a low concentration of BIM BH3 domain peptide, which activates BAK (Dai et al. 2011). More BAKΔTM bound tightly to MCL1 in the presence of the BIM BH3 domain than in its absence (Fig. 1F, arrow), suggesting that low BAK binding reflects lack of activation. In further experiments, BAK binding to anti-apoptotic BCL2 family members was also examined in the absence and presence of 1% Triton X-100, which artificially activates BAX and BAK (Hsu and Youle 1997; Llambi et al. 2011), or 1% CHAPS. Triton X-100 enhanced binding of BAKΔTM to immobilized MCL1 (Supplemental Fig. S1B), binding of BAKΔN20ΔTM to purified MCL1 and BCLXL in solution (Supplemental Fig. S1C, red box), and recovery of full-length endogenous BAK with anti-apoptotic proteins in lysates from nonapoptotic cells (Supplemental Fig. S1D, red box). In contrast, 1% CHAPS did not detectably increase binding of BAK species to anti-apoptotic BCL2 family members in these assays (Supplemental Fig. S1B–D), consistent with previous reports that CHAPS fails to activate BAX in cell lysates (Hsu and Youle 1997).
Constitutive BAK oligomerization varies among cell lines
In subsequent studies, oligomerization of endogenous BAK was analyzed by size exclusion chromatography of CHAPS lysates from 23 human leukemia and lymphoma cell lines. Purified recombinant BAKΔTM, which migrates as a monomer, served as a control (Fig. 2A). In several lines, including ML-1, Jeko, U937, WSU, and Daudi, endogenous BAK was predominantly monomeric (Fig. 2B, orange box; Supplemental Fig. S2A,B). In contrast, BAK was more extensively oligomerized in other lines (Fig. 2B, green box; Supplemental Fig. S2C,D). Similar results were obtained using lysates prepared in 1% digitonin (Supplemental Fig. S3), another detergent used when spurious BAK activation is to be avoided (Ma et al. 2013). No oligomerization of BAX was detectable in the same chromatography fractions (Fig. 2B, right panels). Collectively, these observations suggested that BAK, but not BAX, might be constitutively activated in some cell lines but not others.
Figure 2.
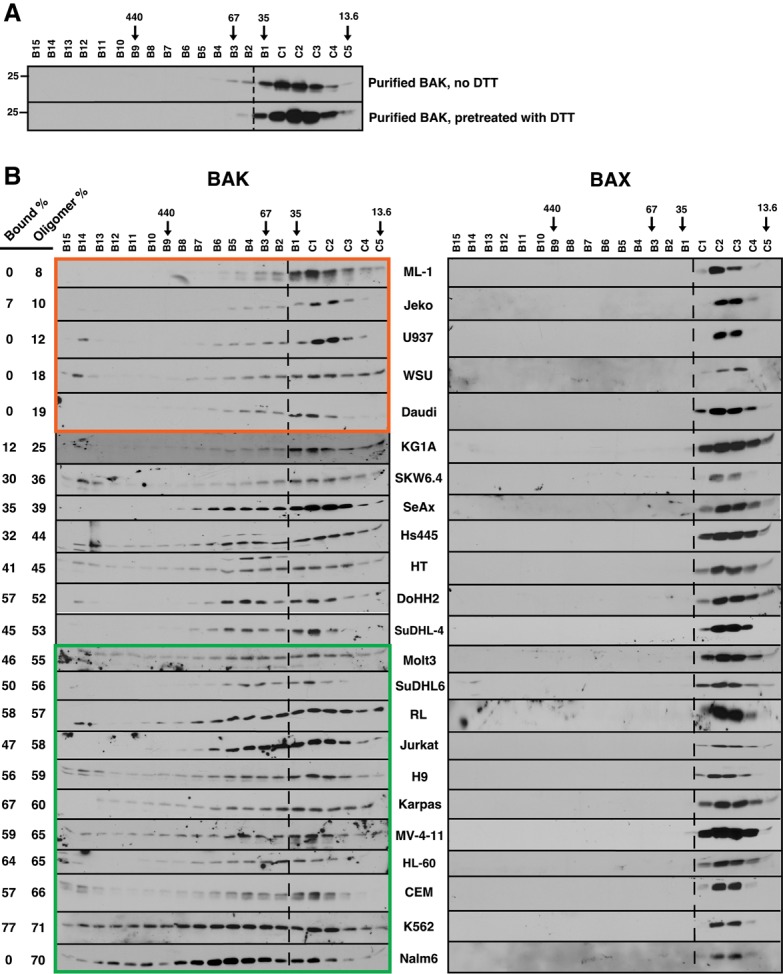
BAK, but not BAX, is constitutively oligomerized in malignant lymphohematopoietic cell lines. (A) Purified BAKΔTM (20 µM) without or with 5 mM DTT was incubated in isotonic buffer containing 1% CHAPS for 1 h and subjected to size exclusion chromatography. (B) Lysates prepared from the indicated log-phase cells in 1% CHAPS were subjected to size exclusion chromatography and blotted with antibodies to BAK (left) or BAX (right). Numbers at the left indicate the sum of the total percentage of BAK that is detected as bound to BCLXL + MCL1 + BCL2 in pull-down assays (“Bound %”) (Fig. 3; Supplemental Fig S5), calculated as described (Dai et al. 2009), and on FPLC assays (running to the left of the dashed line) (“Oligomer %”), calculated as indicated in Supplemental Figure S2. Arrows indicate size markers in kilodaltons. Orange and green boxes indicate cell lines with low (<20%) and high (>55%) constitutive BAK oligomerization, respectively.
Consistent with this possibility, we observed that BAK in cell lines with high levels of oligomerization reacted with an antibody recognizing an epitope that is rendered accessible during BAK activation (Supplemental Fig. S4A), that MCL1 and BCLXL were present with (Supplemental Fig. S4B,C) and bound to (Supplemental Fig. S4A) BAK migrating with greater than monomer molecular weight on gel filtration, and that addition of antibody to MCL1 (Supplemental Fig. S4D) or BCLXL (Supplemental Fig. S4E,F) shifted the BAK to even high molecular weight fractions. To further examine BAK binding to anti-apoptotic BCL2 family members, endogenous BCLXL, MCL1, and BCL2 were immunoprecipitated from all of the lines. Blotting for BAK revealed four patterns. First, in some lines (e.g., ML-1 and WSU) BAK was not detectably bound to any of the three anti-apoptotic proteins (Fig. 3A; Supplemental Fig. S5A), consistent with the paucity of BAK oligomers in these cells (Fig. 2B). Second, in several lines (e.g., K562) BAK was predominantly bound to BCLXL rather than MCL1 (Fig. 3B; Supplemental Fig. S5B). Third, in SUDHL-6 cells, BAK was bound to MCL1 in preference to BCLXL and BCL2 (Fig. 3C). Finally, BAK was bound to multiple anti-apoptotic proteins, including BCLXL and MCL1, in several lines (Fig. 3D; Supplemental Fig. S5C). In addition, BAK was extensively bound to BCL2 in RL cells (Fig. 3E), which contain a mutant BCL2 protein with enhanced affinity for proapoptotic BCL2 family members (Smith et al. 2011), and less extensively but detectably bound to BCL2 in lines such as DoHH2, Jeko, and HL-60 (Supplemental Fig. S5), which contain high levels of BCL2 protein (see below). Collectively, up to 77% of BAK was recovered in the BCLXL, MCL1, and BCL2 immunoprecipitates, in good agreement with the FPLC assay for oligomerization (Fig. 3F). Reciprocal immunoprecipitations likewise indicated that BAK is bound to BCLXL, MCL1, and (in the case of RL cells) BCL2 (Supplemental Fig. S6A,B). Although BAK is constitutively bound to VDAC2 in some cell lines (Cheng et al. 2003), we did not detect VDAC2 in BAK immunoprecipitates in the cells used in this study.
Figure 3.

BAK is constitutively bound to various anti-apoptotic BCL2 family members. (A–E,G) CHAPS lysates of WSU (A), K562 (B), Su-DHL-6 (C), Jurkat (D), RL (E), and Nalm6 (G) cells were immunoprecipitated (IP) with antibodies to BCLXL, MCL1, or BCL2, and compared with serial dilutions of the input. Pull-downs with normal rabbit IgG served as a control. (F) The relative amount of BAK bound to BCL2 + BCLXL + MCL1 (% Bound) compared with the relative amount of oligomerized BAK (% Oligomerized), calculated as indicated in Supplemental Figure S2. Cell lines used here and in Figure 5 and Supplemental Figure S8 are ML-1 (1), Jeko (2), U937 (3), WSU (4), Daudi (5), KG1A (6), SKW6.4 (7), SeAx (8), Hs445 (9), HT (10), DoHH2 (11), SuDHL-4 (12), Molt3 (13), SuDHL-6 (14), RL (15), Jurkat (16), H9 (17), Karpas422 (18), MV-4-11 (19), HL-60 (20), CEM (21), Nalm6 (22), and K562 (23). As indicated in the text, Nalm6 was excluded from subsequent calculations.
Curiously, even though the acute lymphocyte leukemia cell line Nalm6 contained large amounts of BAK oligomers, BAK was not detectable in BCLXL, MCL1, or BCL2 immunoprecipitates (Fig. 3G). Likewise, BAK immunoprecipitates from Nalm6 cells lacked detectable MCL1, BCLXL, BCL2A1, BCLW, BCL2, BCLB, or VDAC2 (Supplemental Fig. S6A), the known BAK binding partners. Because this cell line might contain an additional, currently unknown BAK binding partner, we excluded this line from further analysis.
Sensitivity to navitoclax or A1210477 reflects constitutively oligomerized BAK
Further experiments examined the impact of constitutive BAK oligomerization on drug sensitivity. Each lymphohematopoietic line was assayed for sensitivity to three BH3 mimetics (Fig. 4A,B; Supplemental Fig. S7A): navitoclax, which neutralizes BCL2, BCLXL, and BCLW (Tse et al. 2008); venetoclax (ABT-199), which selectively neutralizes BCL2 (Souers et al. 2013); and A1210477, which preferentially neutralizes MCL1 (Leverson et al. 2015). As indicated in Figure 4, A and C, cells in which BAK was predominantly bound to BCLXL (blue symbols) or a combination of BCLXL and other anti-apoptotic family members (green symbols) were sensitive to navitoclax, with nine of 16 cell lines exhibiting IC50 values <500 nM. In contrast, lines in which BAK was not bound to BCLXL exhibited lower sensitivity, with a median IC50 of 3500 nM (Fig. 4A,C, red and yellow symbols). Similarly, lines in which BAK was extensively bound to MCL1 alone (Fig. 4B,D, yellow symbols) or in combination with other anti-apoptotic family members (Fig. 4B,D, green symbols) were sensitive to A1210477 (median IC50 4.4 µM) (Fig. 4B,D), whereas lines in which BAK was not bound to MCL1 (Fig. 4B,D, blue and red symbols) were less sensitive (median IC50 11 µM). Cells with constitutive binding of BAK to MCL1 were also more sensitive to pharmacological down-regulation of MCL1 with sorafenib (Rahmani et al. 2005; Yu et al. 2005) than cells with BAK constitutively bound to BCLXL or not bound at all (Supplemental Fig. S7C,D). Finally, cell lines with BAK constitutively bound to BCL2 were also more sensitive to venetoclax (Supplemental Fig. S7A,B), although the results were of borderline statistical significance. Collectively, these results suggest that cells with constitutive BAK•BCLXL interactions are sensitive to navitoclax, whereas cells with constitutive BAK•MCL1 interactions are sensitive to MCL1 inhibition or down-regulation, providing functional validation of the constitutive binding detected in Figure 3 and Supplemental Figure S5.
Figure 4.

Sensitivities of cell lines to navitoclax and A1210477 correlate with BAK binding status. (A,B) After the cell lines were treated with navitoclax (A) or A1210477 (B), the percentages of Annexin V-positive cells were assessed by flow cytometry. Graphs show the average of three independent experiments. Symbols and line colors represent different BAK binding statuses. (Red) No binding; (blue) BAK bound to BCLXL only; (yellow) BAK bound to MCL1 only; (purple) unknown binding partner; (green) BAK bound to multiple partners. (C,D) Concentrations of navitoclax (C) or A1210477 (D) required to induce apoptosis in 50% of cells (IC50) grouped according to BAK binding status as determined in Figure 3 and Supplemental Figure S5. Symbols for each cell line correspond to the key between A and B. Heavy horizontal lines represent the mean, and error bars indicate the SD of the indicated groups of lines.
Role of BAK expression level in constitutive BAK activation
Previous studies suggested that BH3 peptide-induced permeabilization of isolated mitochondria (Letai et al. 2002) reflects displacement of preactivated BIM from anti-apoptotic BCL2 family members, causing BIM to bind and activate BAK or BAX (Certo et al. 2006). To assess a possible relationship between BIM or other BH3-only proteins and constitutive BAK oligomerization, we examined BCL2 family protein levels (Fig. 5A). There was no correlation between the extent of BAK oligomerization (Fig. 2B) and levels of BIM, BID, NOXA, PUMA, BAX, BCL2, BCLXL, or MCL1 (Fig. 5B,C; Supplemental Fig. S8). Moreover, knockdown of BIM or BIM + PUMA had no effect on the amount of BAK constitutively bound to BCLXL or MCL1 (Fig. 6A,B; Supplemental Fig. S9), reinforcing the concept that constitutive BAK activation does not depend on BIM or PUMA.
Figure 5.

Constitutive BAK oligomerization correlates with relative BAK content. (A) Whole-cell lysates, along with different concentrations of Molt3, were blotted with the indicated antibodies. Orange and green letters indicate the cell lines in the orange and green boxes, respectively, in Figure 2. (B–D) The percentage of BAK that is constitutively oligomerized (% BAK oligomer), calculated as shown in Supplemental Figure S2, versus the amount of BIMEL (B), BID (C), or BAK (D) in each line relative to an equal amount of Molt3 whole-cell lysate, determined as described in the Materials and Methods.
Figure 6.

Constitutive BAK oligomerization is inhibited by BAK knockdown. (A) Jurkat sublines stably expressing BCLXL or MCL1 tagged at the N terminus with S peptide (S-BCLXL or S-MCL1). (B) Forty-eight hours after cells in A were transfected with the indicated siRNAs, proteins were pulled down on S peptide-agarose and, along with aliquots of the lysates (“input”), subjected to immunoblotting. (C) After the indicated concentrations of BAKΔTM without or with BH3 peptides were incubated with liposomes for 30 min at 37°C, BAK oligomers were cross-linked with bismaleimidohexane and detected by immunoblotting. Note the increasing dimer and trimer with increasing BAK concentrations. (D) Release of encapsulated FITC-dextran from liposomes during a 1-h incubation as illustrated in Supplemental Figure S11A. (E) Forty-eight hours after Jurkat cells were transfected with BAK shRNA, CHAPS lysates were fractioned by FPLC and blotted with BAK antibody. (Inset) Western blot of whole-cell lysates. (F) Forty-eight hours after the cells shown in A were transfected with BAK siRNA or nontargeting control, the percentages of BAK bound to S-BCLXL and S-MCL1 were calculated from data shown in Supplemental Figure S12, B and C. (G) Twenty-four hours after Jurkat cells were transfected with the indicated siRNAs, cells were treated with navitoclax for another 24 h and stained with Annexin V. Error bars indicate ±sd of three independent assays (D,G) or a range of two independent assays (F). (*) Nonspecific band in A and B.
In contrast, cells with higher total BAK expression had, on average, a higher degree of BAK oligomerization (Fig. 5D). This correlation was observed when BAK expression was normalized to either β-Actin (Fig. 5D) or the mitochondrial chaperone HSP60 (Supplemental Fig. S8G).
Because these differences in cellular BAK expression seemed to parallel differences in mitochondrial BAK content (Supplemental Fig. S10), we examined the possibility that BAK could be activated by increased BAK expression independently of BH3-only proteins. When purified BAKΔTM was increased twofold to fourfold over concentrations previously used in cell-free assays (Dai et al. 2011), BAKΔTM spontaneously oligomerized (Fig. 6C) and permeabilized vesicles composed of MOM lipids (Fig. 6D; Supplemental Fig. S11A) in the absence of BH3 peptides or BH3-only proteins. Likewise, purified BAKΔTM permeabilized mitochondria from Bax−/− Bak−/− double-knockout mouse embryonic fibroblasts (Supplemental Fig. S11B). These observations suggested that elevated BAK expression by itself might contribute to constitutive BAK activation.
To further explore this possibility, cell lysates from Jurkat cells with or without BAK knockdown (Fig. 6E, inset; Supplemental Fig. S12A, inset) were subjected to FPLC. BAK knockdown by shRNA (Fig. 6E) or siRNA (Supplemental Fig. S12A) diminished the proportion of total BAK found in oligomers. Likewise, BAK knockdown markedly diminished the percentage of total BAK constitutively bound to BCLXL and MCL1 (Fig. 6F; Supplemental Fig. S12B,C, lane 2 vs. 9). Consistent with these results, BAK knockdown markedly diminished sensitivity to BH3 mimetics, whereas BIM siRNA or BIM + PUMA siRNA had a minimal effect (Fig. 6G; Supplemental Fig. S12D). Similarly, BAK knockdown diminished the percentage of total BAK found in oligomers in CEM cells, whereas BIM and PUMA knockdown did not (Supplemental Fig. S12E).
Modulating BAK activation for therapeutic advantage
As indicated in Figure 4, cells that lack constitutively active BAK are relatively resistant to navitoclax. In an attempt to reverse this resistance, we examined whether sublethal BAK activation might sensitize cells. Treatment of U937 and Daudi with the MEK inhibitor CI-1040 (PD184352) not only up-regulated BIM (Fig. 7A, lanes 7,8; Supplemental Fig. S13A) but also increased the amount of BAK bound to BCLXL (Fig. 7A, lane 2 vs. 5; Supplemental Fig. S13A) and navitoclax sensitivity (Fig. 7B; Supplemental Fig. S13B). Similarly, treatment of ML-1 cells with TRAIL enhanced BAK oligomerization (Fig. 7C) and navitoclax sensitivity (Fig. 7D). Both the increased BAK oligomerization and navitoclax sensitization induced by TRAIL were inhibited by BID siRNA (Supplemental Fig. S13C–E), implicating BID in these processes. Importantly, sensitization was observed at CI-1040 and TRAIL concentrations that by themselves induced little killing of U937, Daudi, and ML-1 cells but nonetheless markedly increased navitoclax-induced apoptosis (Fig. 7B,D; Supplemental Fig. S13B). Moreover, these effects required expression of BAK and/or BAX. The same treatment of WSU cells, which express much less BAX and BAK (Fig. 5A), failed to enhance navitoclax-induced apoptosis (Fig. 7E).
Figure 7.

Drug-induced BAK oligomerization sensitizes to navitoclax. (A) After U937 cells were treated with 10 µM CI-1040 for 24 h, BCLXL and MCL1 were immunoprecipitated and compared with 1/10 of input. Normal rabbit IgG antibody served as an immunoprecipitation control. (B,D,E) After U937 (B), ML-1 (D), or WSU (E) cells were treated with the indicated drugs for 24 h, apoptosis was detected by Annexin V staining. (C) Twenty-four hours after ML-1 cells were treated with 12.5 ng/mL TRAIL, CHAPS lysates were subjected to size exclusion FPLC and blotted for BAK. (F) Clinical acute myelogenous leukemia (AML) samples harvested at initial diagnosis together with different concentrations of Molm16 cell lines as control were blotted with the indicated antibodies. (+) Attainment of complete remission (CR); (−) no CR; (NE) not evaluable due to death before remission status determined. (G) Relative BAK expression, calculated as BAKx ÷ RAFx, where BAKx and RAFx are the signals for BAK and the housekeeping protein cRAF in 5 × 105 leukemia cells from patient X relative to the serial dilution of Molm16 cells (see F), compared in two groups according to patient outcome. (H) Sequential AML samples harvested at diagnosis and relapse were blotted with the indicated antibodies. (I) Model of concentration-dependent BAK activation described in the Discussion. Equilibrium dissociation constants (KDs) for various complexes were derived from SPR experiments shown in Figure 1 and Supplemental Figure S15 or as previously published (Dai et al. 2009).
Relationship between BAK expression and response in acute myelogenous leukemia (AML)
To begin to assess the relationship between BAK levels and response in a clinical setting, we examined BCL2 family member expression in marrow aspirates (>80% blasts) from 27 newly diagnosed AML patients, 16 of whom subsequently achieved complete remission (CR), and 11 of whom did not. As indicated in Figure 7F, BCL2 family member expression varied among the specimens. Consistent with previous results (Kaufmann et al. 1998; Pan et al. 2014), baseline levels of BCL2, BCLXL, MCL1, PUMA, BID, BIM, and BAX failed to correlate with therapeutic response (Fig. 7F; Supplemental Fig. S14). In contrast, BAK levels, on average, were twofold higher in samples from patients who achieved CR compared with those who did not (Fig. 7G). Further immunoblotting in serial leukemia samples harvested during the course of disease also demonstrated that levels of BAK, but not BAX, were decreased at the time of leukemic relapse compared with diagnosis in three of 10 pairs (Fig. 7H).
Discussion
Although recent studies provide substantial insight into the molecular alterations that occur during BAK activation (Dewson et al. 2008, 2009; Moldoveanu et al. 2013; Brouwer et al. 2014), factors that control BAK activation in intact cells remain incompletely understood. In the present study, we examined the oligomerization state of BAX and BAK in lymphohematopoietic cells, determined the binding partners of BAK in cell lines where it is oligomerized, and tested the hypothesis that basal expression of BH3-only proteins versus other factors governs constitutive BAK activation. This analysis demonstrated that BAX is monomeric in all cell lines examined, that BAK shows differences in oligomerization among various lines, that endogenous BAK expression levels play a major role in constitutive BAK activation, and that the binding partners of constitutively activated BAK determine sensitivity to BH3 mimetics.
Previous studies have demonstrated that a fraction of BAK in various cell lines is constitutively bound to MCL1, BCLXL, and BCL2 (Willis et al. 2005; Meng et al. 2007; Dai et al. 2009; Llambi et al. 2011). It has been unclear whether this reflects partial activation of BAK in these cell lines as opposed to the intrinsic affinity of unactivated BAK for the anti-apoptotic proteins. In the present study, SPR and GST pull-down assays both demonstrate that recombinant human BAK in its native state has low affinity for BCLXL, MCL1, or BCL2 (Fig. 1C–E; Supplemental Fig. S1C), likely because its BH3 domain is inaccessible (Dewson et al. 2008). Instead, BAK is tightly bound to these proteins only after immobilization for SPR (Fig. 1A,D,E), activation by BIM BH3 peptide (Fig. 1F), or exposure to Triton X-100 (Supplemental Fig. S1B–D), which is known to activate BAX and BAK (Hsu and Youle 1997; Willis et al. 2007; Llambi et al. 2011). These observations suggest that BAK requires activation in order to bind anti-apoptotic BCL2 family proteins.
In further studies, whole-cell lysates prepared from lymphohematopoietic cell lines using the nonactivating detergent CHAPS (Supplemental Fig. S1C) or digitonin exhibited varying degrees of BAK oligomerization (Fig. 2B; Supplemental Figs. S2, S3). Immunoprecipitation (Fig. 3A–E) indicated that the oligomers reflected a variety of binding patterns, including predominant BAK binding to MCL1 (SUDHL-6 cells), BCLXL (K562 cells), multiple anti-apoptotic BCL2 family members (e.g., Jurkat cells), or mutant BCL2 (RL cells). Importantly, cells with constitutive BAK•BCLXL complexes were more sensitive to navitoclax (Fig. 4A,C), and cells with constitutive BAK•MCL1 complexes were more sensitive to A1210477 (Fig. 4B,D), providing functional validation of the constitutive binding interactions detected by immunoprecipitation.
The direct activation model suggests that oligomerization of BAX and BAK results from interaction with activator BH3-only proteins. Because BID is not constitutively active (cleaved) in these cells (Meng et al. 2007, 2010), our studies examined the potential role of BIM and PUMA. If these activator proteins were responsible for constitutive BAK activation, one would expect (1) that BIM and PUMA knockdown would diminish constitutive BAK activation and (2) that BAX, which is preferentially activated by these BH3-only proteins (Edwards et al. 2013; Sarosiek et al. 2013), would also be activated. Contrary to these predictions, down-regulating BIM and PUMA had little effect on basal BAK oligomerization (Fig. 6B; Supplemental Fig. S12E), and there was no evidence of constitutive BAX activation (Fig. 2B). Instead, a number of observations pointed to endogenous BAK expression as a driver of constitutive BAK activation. In particular, studies under cell-free conditions demonstrated that BAKΔTM is able to self-oligomerize, permeabilize membranes, and cause cytochrome c release from isolated mitochondria without exogenous BH3-only proteins (Fig. 6C,D; Supplemental Fig. S11), consistent with earlier experiments showing that BAK transfection by itself is sufficient to kill a wide variety of cells (e.g., Chittenden et al. 1995; Farrow et al. 1995; Dai et al. 2013). Conversely, BAK down-regulation diminishes not only the total amount of BAK within cells but also the fraction of that total found in oligomers (Fig. 6E,F; Supplemental Fig. S12). These observations suggest that concentration-dependent BAK autoactivation contributes to formation of the BAK-containing oligomers described here.
Our previous studies (Meng et al. 2007) indicate that constitutive BAK•BCLXL interactions involve the BH3-binding groove of BCLXL. On the other hand, biochemical and crystallographic studies suggest that BAK forms a symmetric homodimer, with the BH3 domain of one BAK monomer occupying the BH3-binding groove of the other, en route to higher-order oligomers (Dewson et al. 2008; Brouwer et al. 2014). In the homodimeric configuration, the BAK BH3 domain would be inaccessible (Dewson et al. 2008), precluding binding of BAK to anti-apoptotic BCL2 family members (Sattler et al. 1997). Accordingly, some other configuration of BAK must be constitutively bound to BCLXL and MCL1. SPR (Supplemental Fig. S15) indicates that the affinity of BAKΔTM for immobilized BAKΔTM is similar to that of BAKΔTM for BCLXL. Although we cannot rule out the possibility that immobilization loosens the binding of Bak in dimers, our affinity measurements raise the possibility that BAK dimerization might be somewhat reversible in the absence of BH3-only proteins, leading to sequestration of BAK monomers by anti-apoptotic BCL2 family members (Fig. 7I).
A growing body of literature suggests that sensitivity to BH3 mimetics and possibly other agents can be assessed using an assay termed mitochondrial profiling (Certo et al. 2006; Deng et al. 2007; Ni Chonghaile et al. 2011; Vo et al. 2012; Pan et al. 2014; Montero et al. 2015). This assay, which involves assessment of mitochondrial health after treatment with purified BH3 peptides, indicates that mitochondria from some cells or tumors appear “primed” for MOMP. Although this primed state has been attributed to constitutive binding of BH3-ony proteins such as BIM to anti-apoptotic BCL2 family members (Deng et al. 2007) or BAX activation (Schellenberg et al. 2013), the present results suggest that high levels of endogenous BAK result in a state that is primed for apoptosis, with high sensitivity to one set of stimuli (e.g., navitoclax or Bad-like molecules) if BAK is constitutively bound to BCLXL and high sensitivity to another set (e.g., A1210477 or Noxa-like molecules) if BAK is constitutively bound to MCL1. These observations potentially shed new light on the interpretation of BH3-profiling assays.
On the other hand, our results do not exclude the possibility that displacement of direct activators from anti-apoptotic BCL2 family members also contributes to BH3 mimetic-induced death. Cells with low endogenous BAK expression and oligomerization can clearly be killed by navitoclax and A1210477, although higher concentrations are required (Fig. 4A,B).
Finally, the present results indicate that endogenous BAK levels vary widely among cases of newly diagnosed AML (Fig. 7F) and correlate with response to induction chemotherapy (Fig. 7G). If high BAK levels sensitize cells to chemotherapy, then one would predict that BAK might sometimes be down-regulated in chemoresistant cells that repopulate the bone marrow at relapse. Consistent with this possibility, we observed BAK down-regulation at relapse (Fig. 7H) that was mutually exclusive with the MCL1 up-regulation previously reported (Kaufmann et al. 1998). These observations identify endogenous BAK levels as a potentially important determinant of AML chemosensitivity that requires further study.
In short, the demonstration that BAK can oligomerize in the absence of active BH3-only proteins provides additional insight into one of the roles of anti-apoptotic BCL2 family members, identifies a previously unappreciated source of apoptotic drive when cells are treated with BH3 mimetics, and suggests a new factor that affects AML sensitivity to chemotherapy.
Materials and methods
Materials
Reagents were obtained as follows: glutathione (GSH)-agarose from Thermo Scientific, Ni2+-NTA-agarose from Novagen, navitoclax and venetoclax from ChemieTek, A1210477 from Active Biochem, digitonin from Sigma-Aldrich, and obatoclax from Selleck Chemicals. BAK BH3 and BIM BH3 peptides were produced by solid-phase synthesis in the Mayo Clinic Proteomics Core. All other reagents were obtained as described (Dai et al. 2011, 2014).
Antibodies were from the following suppliers: murine monoclonal anti-cytochrome c from BD Biosciences; murine monoclonal anti-BCL2 from Dako; goat anti-β-Actin and rabbit anti-PUMA antibodies from Santa Cruz Biotechnology; rabbit anti-BAK, mouse anti-BAK Ab-1, and rabbit anti-VDAC1 from Millipore; mouse anti-BCLB and rabbit monoclonal anti-BCL2A1 from Abcam; and rabbit antibodies to BAX, BCLXL, BIM, MCL1, BCLW, VDAC2, HSP60, GAPDH, and GFP from Cell Signaling Technology. Anti-S peptide antibody was raised as described (Hackbarth et al. 2004). Rat monoclonal anti-BID antibody was a kind gift from David Huang (Walter and Eliza Hall Institute, Melbourne, Australia).
Protein expression and purification
cDNAs encoding BCL2ΔTM, BCLXLΔTM, or MCL1ΔTM were cloned in-frame with GST in pGEX-4T-1 as described (Smith et al. 2011). Plasmids encoding BAKΔTM (residues 1–186; GenBank accession no. BC004431) in pET29b(+) and pGEX-4T-1 (Moldoveanu et al. 2006) were subjected to site-directed mutagenesis to yield BAK 21–186 (BAKΔN20ΔTM). All alterations were verified by automated sequencing.
Escherichia coli BL21 harboring the plasmids were grown to optical density 0.8, incubated in 1 mM isopropyl 1-β-d-thiogalactopyranoside for 24 h at 16°C, washed, and sonicated on ice in calcium- and magnesium-free Dulbecco's phosphate-buffered saline (PBS) containing 1 mM PMSF (GST-tagged proteins) or TS buffer (150 mM NaCl containing 10 mM Tris-HCl at pH 7.4, 1 mM PMSF, His6-tagged proteins). All further steps were performed at 4°C. His6-tagged proteins were applied to Ni2+- NTA-agarose and washed with aliquots of TS buffer containing 0 and 40 mM imidazole before elution in TS buffer containing 200 mM imidazole. GST-tagged proteins were incubated with GSH-agarose for 4 h at 4°C and then washed with PBS and eluted with PBS containing 20 mM GSH.
Affinity measurements by SPR
Proteins for SPR were further purified by FPLC on a Superdex S200 size exclusion column, concentrated using a 10-kDa cutoff (Centricon, Millipore), dialyzed against Biacore running buffer (10 mM HEPES at pH 7.4, 150 mM NaCl, 0.05 mM EDTA, 0.005% [w/v] Polysorbate 20), and stored for <48 h at 4°C before use.
Binding assays were performed at 25°C on a Biacore 3000 or T200 biosensor (Biacore). His6-tagged BAKΔTM or BAK BH3 peptide was immobilized on a CM5 sensor chip. After washing with Biacore running buffer, GST-MCL1ΔTM, GST-BCLXLΔTM, or GST was injected at 30 μL/min for 1 min. Bound polypeptide was allowed to dissociate in Biacore running buffer at 30 μL/min for 10 min. Residual bound proteins were desorbed with 2 M MgCl2. Binding kinetics were derived from sensorgrams using Biacore BIA evaluation software.
Alternatively, MCL1ΔTM or BCLXLΔTM (cleaved by thrombin from GST-MCL1ΔTM or GST-BCLXLΔTM) was immobilized on a CM5 chip. His6-tagged BAKΔTM or BAK BH3 peptide was injected at 30 μL/min for 1 min, and dissociation was allowed for 10 min.
Cell culture and drug sensitivity
Leukemia and lymphoma cell lines were obtained from the sources indicated in Supplemental Table S1. All cell lines were maintained at densities below 106 cells per milliliter in RPMI 1640 containing 100 U/mL penicillin G, 100 µg/mL streptomycin, 2 mM glutamine, and 15% FBS (SeAx, H9, and Hs445) or 10% FBS (all other lines). Jurkat sublines stably expressing BCL2, BCLXL, or MCL1 with an N-terminal S peptide tag were generated by electroporation followed by selection in 800 µg/mL geneticin, cloning by limiting dilution, and analysis by immunoblotting (Meng et al. 2007).
Log-phase cells were treated for 24 h (navitoclax or venetoclax) or 48 h (A1210477 or obatoclax), washed twice with PBS, and stained with APC-coupled Annexin V. After 20,000 events were collected on a BD Biosciences FACSCanto II flow cytometer, Annexin V-positive cells were quantitated using BD CellQuest software.
Analytical gel filtration
Aliquots containing 5 × 107 log-phase cells were lysed in CHAPS lysis buffer (1% [w/v] CHAPS, 20 mM HEPES at pH 7.4, 150 mM NaCl, 1% [v/v] glycerol, 1 mM PMSF, 10 μg/mL leupeptin, 10 μg/mL pepstatin, 100 mM NaF, 10 mM sodium pyrophosphate, 1 mM sodium vanadate, 20 nM microcystin) for 30 min at 4°C. Samples (200 µL; 10 mg of protein per milliliter) were injected onto a Superdex S200 size exclusion column, and 500-µL fractions were collected for immunoblotting. In some experiments, digitonin lysis buffer (1% [w/v] digitonin, 20 mM Bis-Tris at pH 7.4, 50 mM NaCl, 10 mM EDTA, 10% [v/v] glycerol with the same protease and phosphatase inhibitors as CHAPS lysis buffer) was substituted. Molecular markers (Sigma-Aldrich) were dissolved in the same buffer and chromatographed on the same column.
Immunoblotting
Whole-cell lysates (Kaufmann et al. 1997), pull-downs, or column fractions were subjected to SDS-PAGE and immunoblotting with enhanced chemiluminscent detection (Kaufmann 2001). Where indicated, films were scanned on a Hewlett-Packard Scanjet 4C and quantified using ImageJ software (http://rsb.info.nih.gov). Protein levels were compared with serial dilutions of Molt3 or Molm16 lysate as indicated in the figures and then normalized based on the levels of housekeeping protein. Thus, in Figure 5A, relative BAK levels were calculated according to the formula BAKx/Actinx, where BAKx and Actinx are the BAK and β-actin levels in cell line X as determined by comparison with a standard curve constructed using values from the serial dilution of Molt3 cells (Fig. 5A). An identical approach was applied to BAX and BH3-only proteins.
S protein pull-down assay
Log-phase Jurkat cells growing in antibiotic-free medium were transiently transfected with plasmids encoding S peptide-tagged BCL2, BCLXL, and MCL1 (Meng et al. 2007; Smith et al. 2011) using a BTX 830 square wave electroporator delivering a 10-msec pulse at 240 mV. After a 24-h incubation, cells were lysed in CHAPS lysis buffer for 30 min at 4°C and sedimented at 14,000g for 15 min. S protein-agarose beads were added to the supernatants and incubated overnight at 4°C. Following four washes with isotonic wash buffer containing 1% CHAPS, bound proteins were solubilized in SDS sample buffer.
Immunoprecipitation
All steps were at 4°C. Log-phase cells were lysed in CHAPS lysis buffer for 30 min. After removal of insoluble material at 14,000g for 15 min, lysates were precleared by incubation for 1 h with protein G-agarose beads. Precleared lysates (600 µg of protein) were incubated overnight with antibodies to BCL2, MCL1, or BCLXL that were precoupled to protein G-agarose using dimethyl pimelimidate (Cliby et al. 2002). Following four washes with isotonic wash buffer containing 1% CHAPS, bound polypeptides were solubilized at 65°C in SDS sample buffer for immunoblotting. The amounts of BCL2, BCLXL, and MCL1 bound to BAK were compared with a serial dilution of whole-cell lysates and calculated as described previously (Dai et al. 2009).
siRNA transfection
Log-phase cells growing in antibiotic-free medium were transiently transfected with siRNAs targeting BIM, PUMA, BID, or BAK (Supplemental Table S2) at 1 µM by electroporation at 280 V for 10 msec in a BTX830 square wave electroporator. Studies using FAM-labeled nontargeting siRNA demonstrated a transfection efficiency of >95%.
Statistical analysis
The relationships between constitutively oligomerized BAK and levels of BCL2 family proteins or percent BAK bound were analyzed using Spearman's rank correlation. Differences between groups were analyzed using Student's t-tests with n − 2 degrees of freedom. The correlations between clinical response and BCL2 family protein in AML were analyzed by ANOVA.
Supplementary Material
Acknowledgments
We thank past and present members of the Kaufmann laboratory for stimulating discussions, Philip Burke and Ken Hall for assistance in AML sample acquisition, Nicole Vincelette for advice regarding electroporation of AML cell lines, David Huang for anti-Bid antibody, and Deb Strauss for editorial assistance. This study was supported in part by R01 CA166741.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.267997.115.
References
- Antonsson B, Montessuit S, Sanchez B, Martinou JC. 2001. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J Biol Chem 276: 11615–11623. [DOI] [PubMed] [Google Scholar]
- Brouwer JM, Westphal D, Dewson G, Robin AY, Uren RT, Bartolo R, Thompson GV, Colman PM, Kluck RM, Czabotar PE. 2014. Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers. Mol Cell 55: 938–946. [DOI] [PubMed] [Google Scholar]
- Certo M, Moore Vdel G, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A. 2006. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9: 351–365. [DOI] [PubMed] [Google Scholar]
- Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. 2003. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 301: 513–517. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Green DR. 2008. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol 18: 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittenden T, Harrington EA, O'Connor R, Flemington C, Lutz RJ, Evan GI, Guild BC. 1995. Induction of apoptosis by the Bcl-2 homologue Bak. Nature 374: 733–736. [DOI] [PubMed] [Google Scholar]
- Cliby WA, Lewis KA, Lilly KK, Kaufmann SH. 2002. S phase and G2 arrests induced by topoisomerase I poisons are dependent on ATR kinase function. J Biol Chem 277: 1599–1606. [DOI] [PubMed] [Google Scholar]
- Cragg MS, Jansen ES, Cook M, Harris C, Strasser A, Scott CL. 2008. Treatment of B-RAF mutant human tumor cells with a MEK inhibitor requires Bim and is enhanced by a BH3 mimetic. J Clin Invest 118: 3582–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, et al. 2013. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 152: 519–531. [DOI] [PubMed] [Google Scholar]
- Czabotar PE, Lessene G, Strasser A, Adams JM. 2014. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15: 49–63. [DOI] [PubMed] [Google Scholar]
- Dai H, Meng XW, Lee S-H, Schneider PA, Kaufmann SH. 2009. Context-dependent Bcl-2/Bak interactions regulate lymphoid cell apoptosis. J Biol Chem 284: 18311–18322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H, Smith A, Meng XW, Schneider PA, Pang Y-P, Kaufmann SH. 2011. Transient binding of an activator BH3 domain to the Bak BH3-binding groove initiates Bak oligomerization. J Cell Biol 194: 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H, Ding H, Meng XW, Lee S-H, Kaufmann SH. 2013. Contribution of Bcl-2 phosphorylation to Bak binding and drug resistance. Cancer Res 73: 6998–7008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H, Pang Y-P, Kaufmann SH. 2014. Evaluation of the BH3-only protein Puma as a direct Bak activator. J Biol Chem 289: 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. 2007. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 12: 171–185. [DOI] [PubMed] [Google Scholar]
- Dewson G, Kratina T, Sim HW, Puthalakath H, Adams JM, Colman PM, Kluck RM. 2008. To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3: groove interactions. Mol Cell 30: 369–380. [DOI] [PubMed] [Google Scholar]
- Dewson G, Kratina T, Czabotar P, Day CL, Adams JM, Kluck RM. 2009. Bak activation for apoptosis involves oligomerization of dimers via their α6 helices. Mol Cell 36: 696–703. [DOI] [PubMed] [Google Scholar]
- Du H, Wolf J, Schafer B, Moldoveanu T, Chipuk JE, Kuwana T. 2011. BH3 domains other than Bim and Bid can directly activate Bax/Bak. J Biol Chem 286: 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AL, Gavathiotis E, LaBelle JL, Braun CR, Opoku-Nsiah KA, Bird GH, Walensky LD. 2013. Multimodal interaction with BCL-2 family proteins underlies the proapoptotic activity of PUMA BH3. Chem Biol 20: 888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrow SN, White JH, Martinou I, Raven T, Pun KT, Grinham CJ, Martinou JC, Brown R. 1995. Cloning of a bcl-2 homologue by interaction with adenovirus E1B 19K. Nature 374: 731–733. [DOI] [PubMed] [Google Scholar]
- Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu HC, Kim H, Cheng EH, Tjandra N, et al. 2008. BAX activation is initiated at a novel interaction site. Nature 455: 1076–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M, Wahner Hendrickson A, Seongseouk Y, Han JJ, Schneider PA, Stenson M, Wellik L, Shing JC, Peterson KL, Flatten KS, et al. 2012. Dual TORC1/TORC2 inhibition diminishes Akt activation and induces Puma-dependent apoptosis in lymphoid malignancies. Blood 119: 476–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackbarth JS, Lee S-H, Meng XW, Vroman BT, Kaufmann SH, Karnitz LM. 2004. S-peptide epitope tagging for protein purification, expression monitoring and localization in mammalian cells. BioTechniques 37: 835–839. [PubMed] [Google Scholar]
- Hsu YT, Youle RJ. 1997. Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem 272: 13829–13834. [DOI] [PubMed] [Google Scholar]
- Jiang X, Wang X. 2004. Cytochrome C-mediated apoptosis. Annu Rev Biochem 73: 87–106. [DOI] [PubMed] [Google Scholar]
- Johnstone RW, Ruefli AA, Lowe SW. 2002. Apoptosis. A link between cancer genetics and chemotherapy. Cell 108: 153–164. [DOI] [PubMed] [Google Scholar]
- Kaufmann SH. 2001. Reutilization of immunoblots after chemiluminescent detection. Anal Biochem 296: 283–286. [DOI] [PubMed] [Google Scholar]
- Kaufmann SH, Earnshaw WC. 2000. Induction of apoptosis by cancer chemotherapy. Exp Cell Res 256: 42–49. [DOI] [PubMed] [Google Scholar]
- Kaufmann SH, Svingen PA, Gore SD, Armstrong DK, Cheng Y-C, Rowinsky EK. 1997. Altered formation of topotecan-stabilized topoisomerase I-DNA adducts in human leukemia cells. Blood 89: 2098–2104. [PubMed] [Google Scholar]
- Kaufmann SH, Karp JE, Svingen PA, Krajewski S, Burke PJ, Gore SD. 1998. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood 91: 991–1000. [PubMed] [Google Scholar]
- Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. 2009. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell 36: 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda J, Puthalakath H, Cragg MS, Kelly PN, Bouillet P, Huang DCS, Kimura S, Ottmann OG, Druke BJ, Villunger A, et al. 2006. Bim and Bad mediate imatinib-induced killing of Bcr/Abl leukemic cells and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad 103: 14907–14912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. 2002. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111: 331–342. [DOI] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. 2002. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2: 183–192. [DOI] [PubMed] [Google Scholar]
- Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, Nimmer P, Jin S, Smith M, Xiao Y, et al. 2015. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis 6: e1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL, Dillon CP, Green DR. 2011. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell 44: 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, Andrews DW. 2008. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135: 1074–1084. [DOI] [PubMed] [Google Scholar]
- Ma S, Hockings C, Anwari K, Kratina T, Fennell S, Lazarou M, Ryan MT, Kluck RM, Dewson G. 2013. Assembly of the Bak apoptotic pore: a critical role for the Bak protein α6 helix in the multimerization of homodimers during apoptosis. J Biol Chem 288: 26027–26038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinou JC, Youle RJ. 2011. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell 21: 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng XW, Lee SH, Dai H, Loegering D, Yu C, Flatten K, Schneider P, Dai NT, Kumar SK, Smith BD, et al. 2007. Mcl-1 as a buffer for proapoptotic Bcl-2 family members during TRAIL-induced apoptosis: a mechanistic basis for sorafenib (Bay 43-9006)-induced TRAIL sensitization. J Biol Chem 282: 29831–29846. [DOI] [PubMed] [Google Scholar]
- Meng XW, Heldebrant MP, Flatten KS, Loegering DA, Dai H, Schneider PA, Akazawa Y, Trushin S, Smith BD, Karp JE, et al. 2010. Protein kinase Cβ in modulates ligand-induced cell surface death receptor accumulation: a mechanistic basis for enzastaurin-death ligand synergy. J Biol Chem 285: 888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino D, Giam M, Hughes PD, Siggs OM, Heger K, O'Reilly LA, Adams JM, Strasser A, Lee EF, Fairlie WD, et al. 2009. The role of BH3-only protein Bim extends beyond inhibiting Bcl-2-like prosurvival proteins. J Cell Biol 186: 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldoveanu T, Liu Q, Tocilj A, Watson M, Shore G, Gehring K. 2006. The X-ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol Cell 24: 677–688. [DOI] [PubMed] [Google Scholar]
- Moldoveanu T, Grace CR, Llambi F, Nourse A, Fitzgerald P, Gehring K, Kriwacki RW, Green DR. 2013. BID-induced structural changes in BAK promote apoptosis. Nat Struct Mol Biol 20: 589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero J, Sarosiek KA, DeAngelo JD, Maertens O, Ryan J, Ercan D, Piao H, Horowitz NS, Berkowitz RS, Matulonis U, et al. 2015. Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell 160: 977–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore Vdel G, Deng J, Anderson KC, Richardson P, Tai YT, et al. 2011. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 334: 1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh KJ, Singh P, Lee K, Foss K, Lee S, Park M, Aluvila S, Kim RS, Symersky J, Walters DE. 2010. Conformational changes in BAK, a pore-forming proapoptotic Bcl-2 family member, upon membrane insertion and direct evidence for the existence of BH3-BH3 contact interface in BAK homo-oligomers. J Biol Chem 285: 28924–28937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. 1993. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74: 609–619. [DOI] [PubMed] [Google Scholar]
- Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, Cortes J, DeAngelo DJ, Debose L, Mu H, et al. 2014. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov 4: 362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmani M, Davis EM, Bauer C, Dent P, Grant S. 2005. Apoptosis induced by the kinase inhibitor Bay 43-9006 in human leukemia cells involves downregulation of MCL-1 through inhibition of translation. J Biol Chem 280: 35217–35227. [DOI] [PubMed] [Google Scholar]
- Sarosiek KA, Chi X, Bachman JA, Sims JJ, Montero J, Patel L, Flanagan A, Andrews DW, Sorger P, Letai A. 2013. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol Cell 51: 751–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, et al. 1997. Structure of Bcl-xL–Bak peptide complex: recognition between regulators of apoptosis. Science 275: 983–986. [DOI] [PubMed] [Google Scholar]
- Schellenberg B, Wang P, Keeble JA, Rodriguez-Enriquez R, Walker S, Owens TW, Foster F, Tanianis-Hughes J, Brennan K, Streuli CH, et al. 2013. Bax exists in a dynamic equilibrium between the cytosol and mitochondria to control apoptotic priming. Mol Cell 49: 959–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AJ, Dai H, Correia C, Lee S-H, Takahashi R, Kaufmann SH. 2011. Noxa/Bcl-2 interactions contribute to bortezomib resistance in human lymphoid cells. J Biol Chem 286: 17682–17692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, et al. 2013. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 19: 202–208. [DOI] [PubMed] [Google Scholar]
- Strasser A, Cory S, Adams JM. 2011. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J 30: 3667–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan T-T, Degenhardt K, Nelson DA, Beaudoin B, Nieves-Neira W, Bouillet P, Villunger A, Adams JM, White E. 2005. Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell 7: 227–238. [DOI] [PubMed] [Google Scholar]
- Taylor RC, Cullen SP, Martin SJ. 2008. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol 9: 231–241. [DOI] [PubMed] [Google Scholar]
- Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et al. 2008. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 68: 3421–3428. [DOI] [PubMed] [Google Scholar]
- van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen LB, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, et al. 2006. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 10: 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A. 2003. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 302: 1036–1038. [DOI] [PubMed] [Google Scholar]
- Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, Deangelo DJ, Frattini MG, Letai A. 2012. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 151: 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL, Korsmeyer SJ. 2006. A stapled BID BH3 helix directly binds and activates BAX. Mol Cell 24: 199–210. [DOI] [PubMed] [Google Scholar]
- Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB, Korsmeyer SJ. 2000. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome C. Genes Dev 14: 2060–2071. [PMC free article] [PubMed] [Google Scholar]
- Westphal D, Dewson G, Menard M, Frederick P, Iyer S, Bartolo R, Gibson L, Czabotar PE, Smith BJ, Adams JM, et al. 2014. Apoptotic pore formation is associated with in-plane insertion of Bak or Bax central helices into the mitochondrial outer membrane. Proc Natl Acad Sci 111: E4076–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC. 2005. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev 19: 1294–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, et al. 2007. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 315: 856–859. [DOI] [PubMed] [Google Scholar]
- Yu C, Bruzek LM, Meng XW, Gores GJ, Carter CA, Kaufmann SH, Adjei AA. 2005. The role of Mcl-1 down-regulation in the pro-apoptotic activity of the Raf kinase inhibitor BAY 43-9006. Oncogene 24: 6861–6869. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.