

Yelina et al. show that RNA-directed DNA methylation is sufficient to locally silence Arabidopsis euchromatic crossover hot spots and is associated with increased nucleosome density and H3K9me2. This work demonstrates that DNA methylation plays a key role in establishing domains of meiotic recombination along chromosomes.
Keywords: meiosis, crossover, hot spots, epigenetics, DNA methylation, centromeres
Abstract
During meiosis, homologous chromosomes undergo crossover recombination, which is typically concentrated in narrow hot spots that are controlled by genetic and epigenetic information. Arabidopsis chromosomes are highly DNA methylated in the repetitive centromeres, which are also crossover-suppressed. Here we demonstrate that RNA-directed DNA methylation is sufficient to locally silence Arabidopsis euchromatic crossover hot spots and is associated with increased nucleosome density and H3K9me2. However, loss of CG DNA methylation maintenance in met1 triggers epigenetic crossover remodeling at the chromosome scale, with pericentromeric decreases and euchromatic increases in recombination. We used recombination mutants that alter interfering and noninterfering crossover repair pathways (fancm and zip4) to demonstrate that remodeling primarily involves redistribution of interfering crossovers. Using whole-genome bisulfite sequencing, we show that crossover remodeling is driven by loss of CG methylation within the centromeric regions. Using cytogenetics, we profiled meiotic DNA double-strand break (DSB) foci in met1 and found them unchanged relative to wild type. We propose that met1 chromosome structure is altered, causing centromere-proximal DSBs to be inhibited from maturation into interfering crossovers. These data demonstrate that DNA methylation is sufficient to silence crossover hot spots and plays a key role in establishing domains of meiotic recombination along chromosomes.
Sexually reproducing eukaryotes produce haploid gametes via the specialized meiotic cell division, where a single round of DNA replication is coupled to two rounds of chromosome segregation (Villeneuve and Hillers 2001). During meiotic prophase-I, homologous chromosomes pair and undergo programmed recombination that can result in reciprocal genetic crossover (Villeneuve and Hillers 2001; Keeney and Neale 2006). Meiotic recombination is initiated by DNA double-strand breaks (DSBs) generated by the SPO11 transesterase (Villeneuve and Hillers 2001; Keeney and Neale 2006). DSBs are then resected to yield 3′ ssDNA that is bound by the RAD51 and DMC1 recombinases, which mediate interhomolog strand invasion (Villeneuve and Hillers 2001; Keeney and Neale 2006). Strand invasion intermediates are further processed to form double Holliday junctions (dHJs) that can be resolved as crossovers or noncrossovers (Szostak et al. 1983; Schwacha and Kleckner 1995). Meiotic recombination occurs in the context of a chromatin loop–axis structure that matures into the synaptonemal complex and exerts a significant influence on crossover formation (Kleckner 2006; Storlazzi et al. 2010; Panizza et al. 2011; Ferdous et al. 2012). As a consequence of homologous recombination, chromosome segregation, and gamete fusion, meiosis strongly influences patterns of genetic diversity within sexually reproducing populations.
Eukaryotes typically initiate a larger number of meiotic DSBs than final crossover events. For example, ∼200 DSBs are estimated to form in Arabidopsis, which generate ∼10 crossovers per meiosis, with the remaining events repaired as noncrossovers or via intersister repair (Copenhaver et al. 1998; Chelysheva et al. 2010; Giraut et al. 2011; Ferdous et al. 2012; Salomé et al. 2012; Sun et al. 2012; Wijnker et al. 2013). Eighty percent to 90% of wild-type Arabidopsis crossovers are generated by an interfering pathway, which causes events to be more widely distributed than would be expected if they were independent (Copenhaver et al. 2002; Higgins et al. 2004; Mercier et al. 2005). Interfering crossovers are generated by the conserved ZMM pathway (MSH4, MSH5, MER3, HEI10, ZIP4, SHOC1, and PTD), which is thought to stabilize strand invasion intermediates and promote dHJ formation (Copenhaver et al. 2002; Higgins et al. 2004, 2008b; Chen et al. 2005; Mercier et al. 2005; Chelysheva et al. 2007, 2010, 2012; Macaisne et al. 2008). The mechanism of crossover interference is unknown but can be modeled as mechanical stress mediated via the meiotic chromosome axis (Zhang et al. 2014). The remaining minority of Arabidopsis noninterfering crossovers is dependent on MUS81 (Berchowitz et al. 2007; Higgins et al. 2008a). The FANCM helicase acts with MHF1 and MHF2 to promote formation of noncrossovers, in the absence of which most strand invasion events enter a noninterfering crossover repair pathway (Crismani et al. 2012; Knoll et al. 2012; Girard et al. 2014). FIDGITIN, RECQ4A, RECQ4B, and TOPOISOMERASE3α have been identified as additional anti-crossover factors in Arabidopsis (Girard et al. 2015; Séguéla-Arnaud et al. 2015).
Meiotic recombination is nonrandomly distributed along chromosomes and tends to occur in narrow hot spots, which are controlled by both genetic and epigenetic information (Lichten and Goldman 1995; Kauppi et al. 2004; de Massy 2013). Plant hot spots are observed at gene transcriptional start sites (TSSs) and transcriptional termination sites (TTSs) and require the histone variant H2A.Z for wild-type activity (Brown and Sundaresan 1991; Fu et al. 2001; Saintenac et al. 2011; Choi et al. 2013; Drouaud et al. 2013; Hellsten et al. 2013; Li et al. 2015). In contrast, crossovers are largely suppressed in repeat-rich heterochromatic regions in plant genomes (Copenhaver et al. 1999; Wei et al. 2009; Mayer et al. 2012; The Tomato Genome Consortium 2012; Yelina et al. 2012; Choulet et al. 2014; Rodgers-Melnick et al. 2015). Plant heterochromatin is densely epigenetically modified with DNA cytosine methylation and histone H3K9me2 methylation, which contribute to suppressed RNA polymerase II (Pol II) transcription, late DNA replication, cytological condensation, and higher-order structural organization (Fransz et al. 2002; Soppe et al. 2002; Zhang et al. 2006; Mathieu et al. 2007; Lister et al. 2008; Lee et al. 2010; Feng et al. 2014). DNA methylation occurs in CG, CHG, and CHH sequence contexts (where H = A, T, or C) in plant genomes (Law and Jacobsen 2010). CG methylation is maintained through DNA replication by METHYLTRANSFERASE1 (MET1) acting with the SWI/SNF chromatin remodeling enzyme DECREASED DNA METHYLATION1 (DDM1) (Vongs et al. 1993; Saze et al. 2003; Stroud et al. 2013). Non-CG methylation is maintained redundantly by the CHROMOMETHYLASE2 (CMT2), CHROMOMETHYLASE3 (CMT3), and DOMAINS REARRANGED METHYLASE2 (DRM2) cytosine methyltransferases (Cao et al. 2003; Stroud et al. 2013, 2014; Zemach et al. 2013; Dubin et al. 2015). DNA methylation controls meiotic recombination distributions along Arabidopsis chromosomes, as met1 and ddm1 mutants show epigenetic crossover remodeling, with increases in the gene-rich chromosome arms and decreases in the repetitive pericentromeric regions (Colomé-Tatché et al. 2012; Melamed-Bessudo and Levy 2012; Mirouze et al. 2012; Yelina et al. 2012). As total crossovers are similar between wild type and met1, this redistribution represents a homeostatic effect.
Small RNAs are able to target DNA methylation to homologous sequences via the RNA-directed DNA methylation (RdDM) pathway in plants (Wassenegger et al. 1994; Law and Jacobsen 2010). Using this mechanism, we directly DNA-methylated endogenous Arabidopsis meiotic crossover hot spots located in euchromatin and demonstrate that this is sufficient to epigenetically silence recombination. Using chromatin immunoprecipitation (ChIP), we show that RdDM at hot spots is associated with gain of H3K9me2 and increased nucleosome occupancy. To understand how loss of CG DNA methylation maintenance causes remodeling of crossovers at the chromosome scale, we combined met1 with mutations that alter interfering and noninterfering crossover repair pathways. This demonstrates that crossover remodeling is driven by loss of interfering crossovers from pericentromeric regions and gains in the euchromatic chromosome arms. We performed whole-genome bisulfite sequencing in met1/+ heterozygotes and show that crossover remodeling is driven by loss of CG context DNA methylation in the centromeric regions. Despite the extensive changes to met1 chromatin, we show that meiotic chromosomes do not form ectopic DSB foci, indicating downstream changes in crossover maturation. Together, this demonstrates that DNA methylation can potently suppress crossovers at the hot spot scale and impose regional control on recombination patterns at the chromosome scale.
Results
Silencing meiotic crossover hot spots via RdDM
Relatively low numbers of crossover events (∼10) occur per meiosis in Arabidopsis (Copenhaver et al. 1998; Giraut et al. 2011; Salomé et al. 2012), and known hot spots have genetic distances of ∼0.2–0.5 cM (Table 1; Drouaud and Mézard 2011; Yelina et al. 2012; Choi et al. 2013; Drouaud et al. 2013). Therefore, to measure >100 events at single hot spots, it is necessary to screen hundreds of thousands of meioses. To achieve this, we collected large quantities of pollen (post-meiotic male gametes) from Col/Ler F1 hybrids and extracted genomic DNA. For a given hot spot, this DNA consists of a mixture of crossover and parental molecules, distinguishable by patterns of Col/Ler sequence polymorphisms (Drouaud and Mézard 2011; Yelina et al. 2012; Choi et al. 2013; Drouaud et al. 2013). Allele-specific PCR primers that anneal to polymorphic sites can be used to amplify and titrate crossover versus parental DNA molecules (Drouaud and Mézard 2011; Yelina et al. 2012; Choi et al. 2013; Drouaud et al. 2013). Dilutions of template DNA were analyzed until approximately half of the amplifications were negative, which allowed low relative error in estimating crossover molecule numbers (Drouaud and Mézard 2011; Yelina et al. 2012; Choi et al. 2013; Drouaud et al. 2013). Single-molecule amplification products could then be genotyped via Sanger sequencing to identify internal crossover locations (Drouaud and Mézard 2011; Yelina et al. 2012; Choi et al. 2013; Drouaud et al. 2013).
Table 1.
Silencing of 3a and 3b crossover hot spots using RdDM
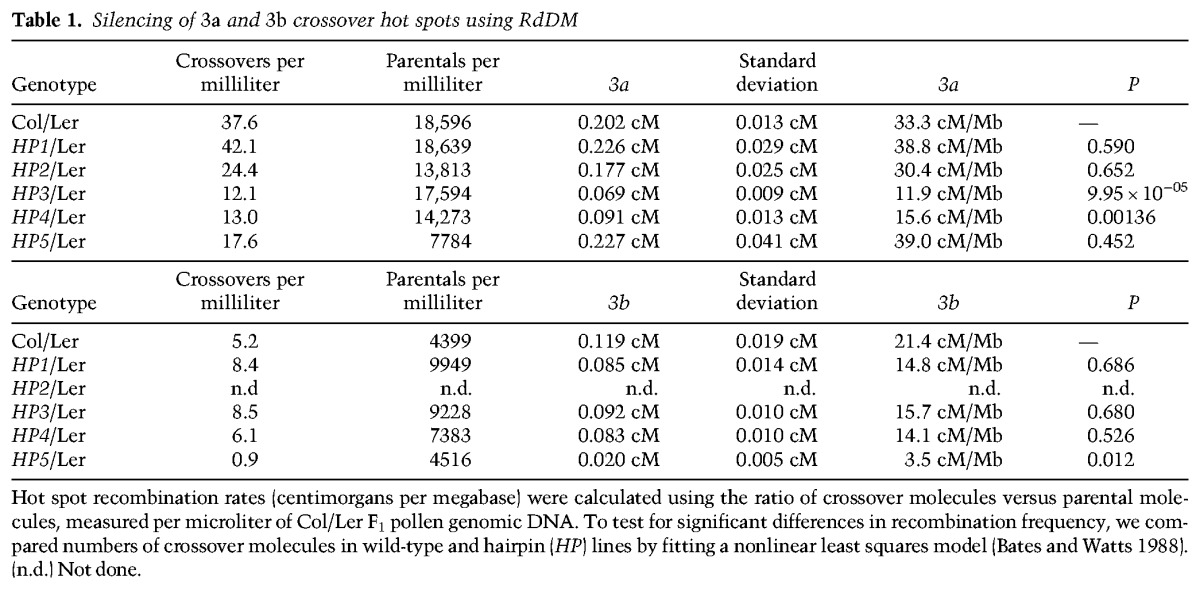
We previously mapped two hot spots, 3a and 3b, which are located subtelomerically within the first megabase of chromosome 3 (Fig. 1A; Yelina et al. 2012; Choi et al. 2013). The 3a and 3b amplicons are 5.8 and 5.7 kb, respectively, and have maximum crossover rates of 83.15 and 78.70 cM/Mb, compared with the male chromosome 3 average of 4.77 cM/Mb (Fig. 1B; Supplemental Tables 1–3; Giraut et al. 2011; Yelina et al. 2012; Choi et al. 2013). Hot spots of comparable magnitude were detected using linkage disequilibrium-based approaches that measure historical crossover activity in regions overlapping 3a and 3b (Fig. 1C; Choi et al. 2013). The 3a–3b hot spot region has low DNA methylation (CG, 4.5%; CHG, 0.3%; CHH, 0.4%) compared with the genome average (CG, 27.0%; CHG, 8.9%; CHH, 2.9%), although an intervening gene shows CG body methylation (Fig. 1A). This is consistent with genome-wide analysis of hot spots in Arabidopsis and maize, which show low levels of DNA methylation (Choi et al. 2013; Wijnker et al. 2013; Rodgers-Melnick et al. 2015).
Figure 1.

RdDM silences Arabidopsis crossover hot spots. (A, top plot) Gene (green) and transposon (blue) density (base pairs [bp] per 100 bp) along Arabidopsis chromosome 3. DNA methylation (percentage; red) is shown for wild-type Col-0. The centromere is indicated by the vertical dotted line, and the 3a–3b hot spots are indicated by the vertical black line. (Bottom plot) DNA methylation frequency (percentage) within the 3a–3b region. (Blue) CG; (green) CHG; (red) CHH. (B) 3a and 3b crossover frequency (centimorgans per megabase) measured by pollen typing. Vertical black lines indicate allele-specific primer-annealing sites. Red X-axis ticks indicate Col/Ler single-nucleotide polymorphisms (SNPs), and black arrows indicate genes. RdDM hairpins (HPs) are indicated by purple and blue bars. The horizontal dotted line indicates chromosome 3 Col/Ler male average recombination (Giraut et al. 2011). (C) Historical crossover frequency within the 3a–3b region estimated from LDhat analysis of SNPs from Eurasian (blue) and Swedish (red) Arabidopsis accessions (Cao et al. 2011; Auton and McVean 2012; Choi et al. 2013; Long et al. 2013). SNP positions are indicated by X-axis ticks. Chromosome average values are indicated by the horizontal dotted lines. (D) Inverted repeat hairpin expression via RNA Pol II from the ACTIN2 promoter produces dsRNA, which is processed by DICER-LIKE (DCL) enzymes into 21- to 24-nucleotide siRNAs, which act in trans to target DNA cytosine methylation (m) to homologous genomic target sequences. (E) Total RNA was blotted and hybridized with radioactively labelled probes complementary to hairpin (HP) sequences to detect siRNAs, and the U6 nuclear RNA was used as a loading control. (F) Total genomic DNA was untreated (−) or digested with McrBC (+) and used for PCR amplification. Methylation of HP target sequences was tested in primary T1 transformants or subsequent T2 or T3 generations. The regions analyzed were HP1a, HP2b, HP3b, HP4b, and HP5b (Supplemental Table 4). The Ta3-LTR retrotransposon and At5g13440 were used as methylated and unmethylated controls, respectively. (G) Recombination rates (centimorgans per megabase) at the 3a and 3b hot spots estimated by pollen typing in wild-type Col/Ler and HP/Ler lines. Asterisks indicate HP/Ler lines with significantly different crossover frequency compared with the Col/Ler control (Table 1).
To directly test the role of DNA methylation on hot spot activity, we used the RdDM pathway (Fig. 1D; Wassenegger et al. 1994; Law and Jacobsen 2010). Transformation of Arabidopsis with inverted repeat hairpin transgenes produces dsRNAs that are processed into 21- to 24-nucleotide (nt) siRNAs (Fig. 1D; Mette et al. 2000; Zilberman et al. 2004). These siRNAs are able to direct DNA methylation to homologous sequences in all sequence contexts (CG, CHG, and CHH) via the DRM2 de novo methyltransferase (Cao and Jacobsen 2002; Zilberman et al. 2004). To methylate the 3a and 3b hot spots, we generated hairpin constructs driven by the ACTIN2 promoter (3a: HP1–HP4; 3b: HP5) (Fig. 1B; Supplemental Table 4). HAIRPIN (HP) T1 transformants showed accumulation of 21- to 24-nt siRNAs detectable by Northern blotting and hybridization (Fig. 1E). To assess RdDM at the 3a and 3b target regions in HP transformants, we used McrBC, which digests methylated DNA, followed by PCR amplification. Using McrBC assays, we detected methylation at the boundaries of the HP targeted regions within the 3a and 3b hot spots (Fig. 1F; Supplemental Table 4). DNA methylation was detectable in HP T1 plants and increased in strength in subsequent T2 and T3 generations (Fig. 1F), consistent with promotion of RdDM during reproductive development (Teixeira et al. 2009). All McrBC assays detected DNA methylation except in the HP5a region (Fig. 1F; Supplemental Table 4). We selected HP plants with high levels of DNA methylation at 3a or 3b target regions for further analysis.
To measure 3a and 3b crossover rates after establishment of DNA methylation, we crossed HP1–HP5 Col lines to wild-type Ler plants. The resulting HP/Ler F1 plants were used for pollen collection and genomic DNA extraction alongside untransformed wild-type Col/Ler F1 controls. We used dilution of genomic DNA and amplification to measure the concentration of parental and crossover molecules at the 3a and 3b hot spots in HP lines (Fig. 1G; Table 1). For the HP1–HP4 transformants that acquired DNA methylation within 3a, we observed that only HP3 and HP4 caused significant reductions in crossover frequency (nonlinear least squares model, HP3, P = 9.95 × 10−5; HP4, P = 0.00136) (Fig. 1G; Table 1). This is consistent with the HP3 and HP4 hairpins directing DNA methylation toward 3a regions with highest recombination activity in wild type (Fig. 1B). DNA methylation within 3b directed by HP5 also caused significant suppression of recombination within this hot spot (nonlinear least squares model, P = 0.012) (Fig. 1G; Table 1). In budding yeast, hot spot activity shows compensation when adjacent hot spots are altered over distances of ∼60 kb (Robine et al. 2007). Due to the physical proximity of 3a and 3b (∼10 kb), we tested whether their activity was altered when the other hot spot was silenced. However, we did not observe significant differences in 3a when 3b was silenced or vice versa, indicating limited compensatory interactions between these loci (Fig. 1G; Table 1).
Fine-scale mapping of crossovers and chromatin at silenced hot spots
We next investigated the fine-scale relationships between DNA methylation and crossover recombination (Fig. 2). We performed sodium bisulfite sequencing using DNA from closed flower buds of HP/Ler F1 lines that showed suppressed crossover frequency (HP3, HP4, and HP5) and compared them with untransformed Col/Ler F1 plants (Supplemental Table 4). HP3, HP4, and HP5 triggered dense DNA methylation in the targeted regions, with methylation observed in all sequence contexts, consistent with the action of RdDM (Fig. 2D–F; Supplemental Tables 5–14; Cao and Jacobsen 2002; Zilberman et al. 2004; Henderson and Jacobsen 2008). Low levels of DNA methylation were observed in untransformed plants (Fig. 2D–F; Supplemental Tables 5–14). As levels of CG and CHG methylation exceeded 80% in HP/Ler F1 plants, this suggests that both Col and Ler alleles acquired DNA methylation (Fig. 2D–F; Supplemental Tables 5–14). Limited spreading (<150 base pairs [bp]) of DNA methylation was observed outside of the hairpin targeted regions (Figs. 2D–F), consistent with the known behavior of RdDM (Pélissier et al. 1999; Zilberman et al. 2004). The HP3 and HP4 hairpins directed dense DNA methylation throughout the 3a targeted regions that we tested (Fig. 2D,E). However, the HP5 hairpin caused more limited methylation, mainly within the central targeted region of 3b (Fig. 2F; Supplemental Tables 5–14).
Figure 2.

Fine-scale patterns of DNA methylation and crossover frequency at 3a and 3b (A) Recombination rates (centimorgans per megabase) within the 3a hot spot analyzed by pollen typing. Vertical black lines indicate allele-specific oligonucleotide-annealing sites. Col/Ler SNPs are shown as black X-axis ticks, and genes are indicated by black arrows. Vertical blue lines indicate the HP3 RdDM targeted region. Wild-type Col/Ler recombination rates are shown in red, and those measured in HP3/Ler plants are shown in blue. The chromosome 3 Col/Ler male average recombination rate is shown by the horizontal dotted line (Giraut et al. 2011). Red rectangles indicate regions analyzed for DNA methylation. (B) As in A but for HP4/Ler F1. (C) As in B but for HP5/Ler F1 at the 3b hot spot. (D) Sodium bisulfite sequencing data showing the percentage of DNA methylation for individual cytosines, measured in the regions corresponding to the red rectangles in wild-type Col/Ler or HP3/Ler plots. (Blue) CG; (green) CHG; (red) CHH. (E) As in D but showing bisulfite analysis of HP4/Ler F1. (F) As in D but showing bisulfite analysis of HP5/Ler F1 at the 3b hot spot.
To analyze fine-scale recombination patterns, we Sanger-sequenced ∼100–200 single-crossover-molecule PCR amplification products from HP3/Ler, HP4/Ler, and HP5/Ler F1 lines in addition to untransformed wild-type Col/Ler F1 controls (Fig. 2A–C; Supplemental Tables 1–3). This approach identifies internal crossover locations to the resolution of individual sequence polymorphisms. The HP3 and HP4 hairpins targeted DNA methylation to adjacent regions within the 3a hot spot, which correlated with shifted patterns of crossover activity (Fig. 2A,B; Supplemental Tables 1–2). The HP5 hairpin also directed localized crossover suppression within the 3b hot spot (Fig. 2C; Supplemental Table 3). This demonstrates that de novo DNA methylation causes specific and localized silencing of meiotic recombination in Arabidopsis. Interestingly, in wild type, we saw suppressed crossovers in a region of dense polymorphism 3′ to the 3b hot spot and, in HP5/Ler plants, observed increased crossovers in this region (Fig. 2C; Supplemental Table 3).
To further investigate how chromatin at the 3a and 3b hot spots changes following RdDM, we performed ChIP analysis. As DNA methylation and H3K9me2 are strongly correlated in Arabidopsis (Bernatavichute et al. 2008), we first performed ChIP for this modification within the hot spots. We assayed chromatin from inflorescences of wild-type Col, HP3 T3, HP4 T3, and HP5 T2 lines that had acquired dense DNA methylation via RdDM (Fig. 3A; Supplemental Table 4). Amplification was performed for regions overlapping DNA methylation and normalized against the Ta2 transposon (Johnson et al. 2002). We observed strong enrichment of H3K9me2 in HP3, HP4, and HP5 lines, overlapping the DNA methylated regions, but not in Col wild type (Fig. 3B). These increases were specific to the hot spot region targeted for DNA methylation (Fig. 3B). To monitor nucleosome density, we also performed ChIP using antibodies that recognize unmodified H3, analyzed the same amplicons, and normalized the values against ACTIN7 (At5g09810). We observed that the HP3, HP4, and HP5 lines showed an increase in nucleosome occupancy in the DNA/H3K9me2 methylated regions (Fig. 3C). This is consistent with methylated regions showing higher nucleosome occupancy in Arabidopsis (Chodavarapu et al. 2010). Together, these data demonstrate that establishment of DNA methylation via the RdDM pathway recruits H3K9me2 and causes an increase in nucleosome occupancy. It is likely that combination of these chromatin modifications contributes to suppression of hot spot recombination.
Figure 3.

Chromatin analysis of H3K9me2 and nucleosome density at the 3a and 3b hot spots. (A) Plots showing the HP targeted regions within the 3a (top) and 3b (bottom) hot spots, including the position of amplicons analyzed by ChIP-qPCR (red boxes) (Supplemental Table 4). Vertical black lines indicate pollen-typing allele-specific primer-binding sites, and vertical blue or purple lines indicate hairpin target regions. (B) ChIP-qPCR analysis of H3K9me2 showing normalized units of PCR enrichment in wild-type Col (light blue), HP3 T3 (orange), HP4 T3 (dark blue), and HP5 T2 (green) lines. Values were normalized against the Ta2 transposon (Johnson et al. 2002). (C) ChIP-qPCR analysis of unmodified H3 showing normalized units of enrichment in wild type, HP3 T3, HP4 T3, and HP5 T2, as in B. Values were normalized against ACTIN7 (At5g09810). Error bars represent standard deviation of biological replicates.
Epigenetic crossover remodeling along met1 chromosome arms
Using RdDM, we demonstrated that establishment of DNA methylation and H3K9me2 is sufficient to epigenetically silence crossover hot spots. However, at the chromosome scale, loss of CG DNA methylation in met1 and ddm1 mutants is associated with remodeling of crossovers, with increases in the euchromatic chromosome arms and decreases in the pericentromeres (Colomé-Tatché et al. 2012; Melamed-Bessudo and Levy 2012; Mirouze et al. 2012; Yelina et al. 2012). To further characterize epigenetic remodeling along the chromosome arms, we performed genetic mapping. Wild-type or met1-3 Col/Ler F1 was used as male parents and backcrossed onto Col/Col homozygotes. We isolated genomic DNA from 187 (wild-type) and 192 (met1-3) backcross progeny and generated sets of 96 barcoded sequencing libraries (Rowan et al. 2015). Low-depth sequencing was performed (mean 2.4× sequencing depth), and a set of 153,842 high-quality Col/Ler single-nucleotide polymorphisms (SNPs) was analyzed (Supplemental Table 15). We used the TIGER pipeline to analyze read counts supporting reference or variant positions along the chromosomes and identified 1006 and 1083 crossovers in the wild-type and met1 populations (Fig. 4A–D; Supplemental Table 16; Rowan et al. 2015). We observed that mean crossovers per individual were not significantly different between wild-type (5.38) and met1 (5.64) populations (generalized linear model [GLM] assuming a negative binomial distribution, P = 0.877) (Supplemental Fig. S1; Supplemental Table 16), which is consistent with previous genetic maps (Copenhaver et al. 1998; Giraut et al. 2011; Salomé et al. 2012; Wijnker et al. 2013; Rowan et al. 2015). However, an ∼5.15-Mb region of Col/Col homozygosity on the south arm of chromosome 5 surrounds the met1-3 allele, which was backcrossed into Ler from Col in order to generate the met1 Col/Ler F1 used for mapping (Yelina et al. 2012). As this causes artefactual crossover calls, chromosome 5 or the south arm alone was excluded from subsequent analysis.
Figure 4.

Epigenetic remodeling along chromosome arms in met1 involves interfering crossovers. (A) Normalized wild-type (Col/Ler) crossovers (black) from a male backcross population are plotted along the proportional length of chromosome arms, orientated from telomere (TEL) to centromere (CEN). Wild-type (Col) percentage of DNA methylation is plotted on the same X-axis. (Red) CG; (green) CHG; (blue) CHH. (B) As in A except that normalized crossovers are from a met1 (Col/Ler) male backcross population, and DNA methylation is from met1-3/+. (C) Normalized crossovers from wild-type (red) and met1 (blue) backcross populations plotted and overlaid as in A and B. (D) Violin plots showing the distribution of physical distances (in megabases) between double crossover (DCO) events in wild-type and met1 male backcross populations. Observed double crossovers (brown) are compared with distances for an equivalent set of randomly distributed sites (purple). (E) Gene (green) and transposon (blue) sequence density (base pairs per 100 bp) along Arabidopsis chromosome 3. Wild-type percentage of total DNA methylation (red) calculated for adjacent 10-kb windows. The centromere is indicated by the vertical dotted line and the I1b fluorescent interval is indicated by vertical black lines and colored triangles. (F) As in E but for chromosome 3 and the CEN3 interval. (G) I1b genetic distances (in centimorgans) in the indicated genotypes. Replicate measurements are indicated by black dots, and mean values are indicated by red dots. (H) As in G but for CEN3 genetic distances.
We divided and orientated chromosome arms such that each began at the telomere and ended at the centromere (Fig. 4A–C). We assigned a proportional value to each crossover along the orientated chromosome arms (excluding the south arm of chromosome 5). Crossover positional values were then binned in windows and normalized by total number of events. We also analyzed mean DNA methylation in CG, CHG, and CHH sequence contexts in 10-kb windows proportionally along the chromosome arms. In wild type, the subtelomeric (0%–0.2%) and pericentromeric (0.7%–0.9%) regions show elevated crossovers, whereas the densely methylated centromeric (>0.9%) regions are crossover-suppressed (Fig. 4A). In met1/+, we observed that CG methylation was depleted along the chromosomes and most strongly in proximity to the centromere, whereas non-CG methylation was less reduced (Fig. 4B). In met1, the pericentromeric arm (0.7%–0.9%) regions showed a significant decrease in crossovers (2 × 2 χ2 test, P = 0.01082), whereas the subtelomeric (0%–0.2%) regions significantly increased crossover frequency (2 × 2 χ2 test, P = 0.0301) (Fig. 4B,C). This genome-wide analysis confirms epigenetic remodeling of crossover frequency in met1 along the telomere–centromere axes of the chromosome arms, with the pericentromeric regions decreasing and the subtelomeric regions increasing recombination.
Crossover remodeling in met1 involves the interfering repair pathway
As total numbers of crossovers are similar between wild type and met1 but regional changes in recombination frequency are observed, we hypothesized that these phenomena are mediated via crossover interference (Yelina et al. 2012). To test this idea, we combined met1 with meiotic recombination mutants where interfering and noninterfering crossover repair is altered—specifically, the zip4 ZMM mutant, which eliminates interfering crossovers (Chelysheva et al. 2007), and fancm, where noninterfering crossovers are increased at the expense of noncrossovers (Crismani et al. 2012; Knoll et al. 2012). These mutants were combined with fluorescent FTL lines that measure crossovers within a 1.85-Mb euchromatic interstitial interval on chromosome 1 (I1b) or a 5.40-Mb centromeric/pericentromeric region of chromosome 3 (CEN3) (Fig. 4E,F; Berchowitz and Copenhaver 2008; Yelina et al. 2012). The CEN3 interval contains a central nonrecombining region (∼12,782,751–14,750,881 bp) (Copenhaver et al. 1998, 1999; Giraut et al. 2011; Salomé et al. 2012; Yelina et al. 2012) in addition to flanking 1.67- and 1.77-Mb regions that show increasing gene density and meiotic recombination (Fig. 4F; Copenhaver et al. 1998, 1999; Giraut et al. 2011; Salomé et al. 2012; Yelina et al. 2012; Choi et al. 2013). Due to the centromeric region within the CEN3 interval being crossover-suppressed, its overall recombination rate (2.11 cM/Mb) is lower than that of I1b (4.25 cM/Mb). Inbreeding met1 mutants causes progressive genome demethylation and increasingly severe developmental phenotypes, including decreased fertility (Saze et al. 2003; Mathieu et al. 2007). We also previously observed that expression of fluorescent FTL transgenes becomes stochastic in met1 homozygotes (Yelina et al. 2012). However, the effects of met1/+ heterozygosity on recombination are comparable with met1 homozygotes, consistent with demethylation of met1 gametophytes during haploid mitotic divisions (Saze et al. 2003; Yelina et al. 2012). Therefore, to avoid problems associated with analysis of met1 homozygotes, we analyzed crossovers in met1/+ heterozygotes.
Consistent with previous work, we observed that I1b showed a significant increase in crossover frequency in met1/+ (GLM, P < 2.0 × 10−16) (Fig. 4G; Supplemental Table 17; Yelina et al. 2012). In contrast, the zip4 mutant showed dramatic suppression of crossovers, consistent with the majority of wild-type recombination in this interval being ZMM-dependent (GLM, P < 2.0 × 10−16) (Fig. 4G; Supplemental Table 17; Chelysheva et al. 2007). Recombination was significantly suppressed in met1/+ zip4 double mutants compared with wild type (GLM, P < 2.0 × 10−16) (Fig. 4G; Supplemental Table 17), which demonstrates that met1/+ I1b recombination increases are dependent on interfering crossovers. In contrast, elevated I1b crossovers observed in fancm are not suppressed in fancm zip4 double mutants due to this increase being dependent on noninterfering crossovers (Fig. 4G; Supplemental Table 17; Crismani et al. 2012; Knoll et al. 2012; Yelina et al. 2013). The fancm met1/+ double mutant showed a further significant increase over fancm alone (GLM, P = 2.19 × 10−6) (Fig. 4G; Supplemental Table 17), indicating an additive effect of epigenetic remodeling by met1/+ and increased noninterfering crossovers caused by fancm. Finally, we generated fancm zip4 met1/+ mutants and observed a significant reduction in crossovers relative to fancm (GLM, P = 2.77 × 10−9) (Fig. 4G; Supplemental Table 17). We propose that, due to mutation of multiple recombination and chromatin pathways in the triple mutant, a negative interaction occurs that reduces crossovers below the fancm level.
The CEN3 interval shows significant decreases in recombination in met1/+ compared with wild type, consistent with previous observations (GLM, P < 2.0 × 10−16) (Fig. 4H; Supplemental Table 18; Yelina et al. 2012). The suppression of CEN3 crossovers observed in met1/+ was not significantly different from that observed in zip4 (GLM, P = 0.141) and remained suppressed in zip4 met1/+ double mutants (Fig. 4H). We observed a significant increase in CEN3 recombination in fancm compared with wild type (GLM, P = 3.20 × 10−7), which was significantly reduced in both fancm zip4 (GLM, P = 3.05 × 10−16) and fancm met1/+ (GLM, P < 2.0 × 10−16) (Fig. 4H). This is consistent with met1/+ causing loss of ZIP4-dependent crossovers in the CEN3 region. CEN3 crossovers were further significantly reduced in fancm zip4 met1/+ compared with fancm met1/+ (GLM, P < 2.0 × 10−16) (Fig. 4H). As for I1b, this again suggests that a negative interaction is occurring when multiple recombination and chromatin pathways are disrupted. Together, these data support that epigenetic recombination remodeling in met1 primarily reflects changes in the distribution of the interfering, ZMM-dependent crossovers.
Crossover interference is active in met1 mutants
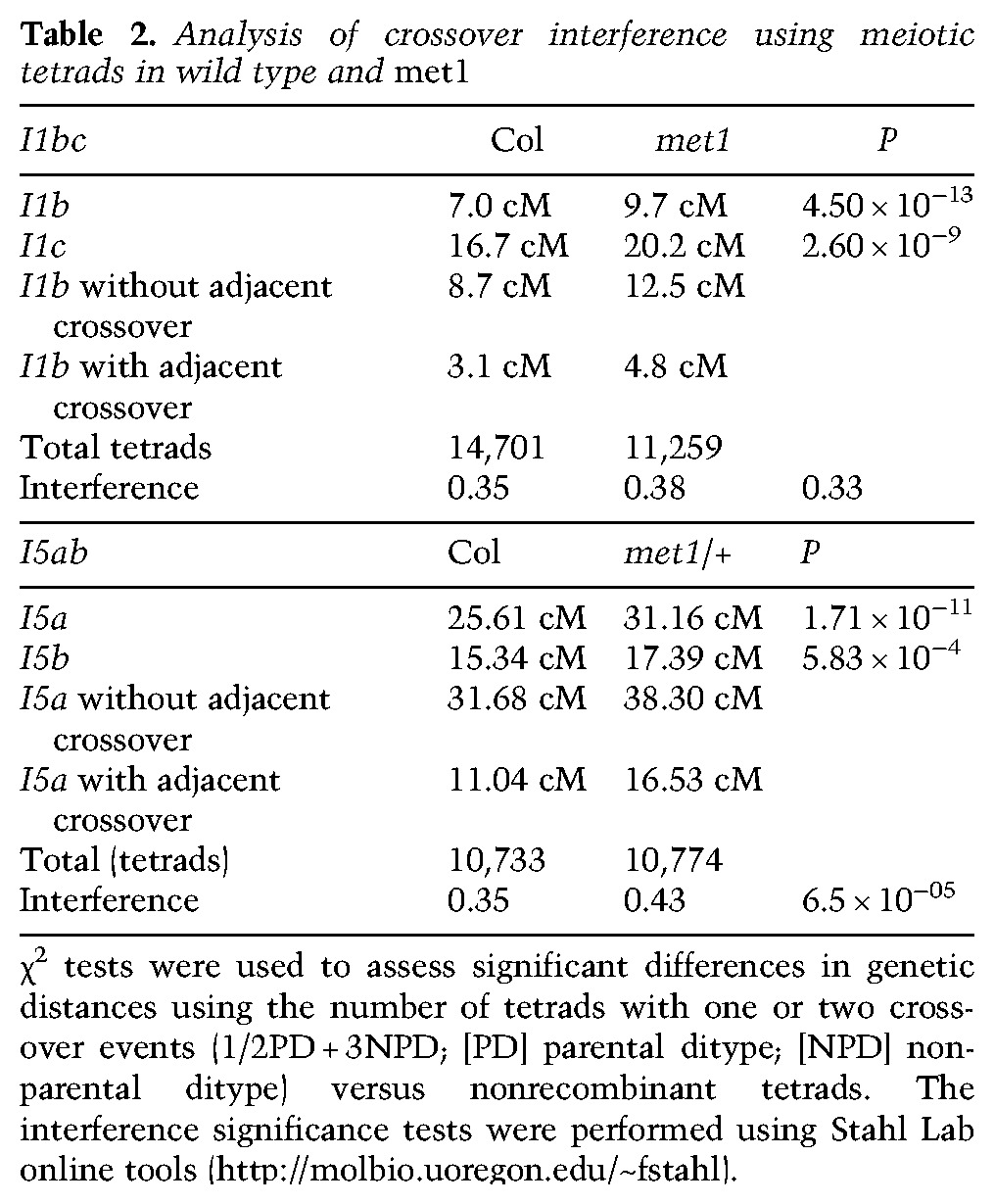
Based on our genetic analyses, we propose that the epigenetic remodeling of crossover frequency observed in met1 is driven by changes in interfering crossovers. Therefore, we next tested whether interference strength was significantly altered in met1 mutants compared with wild type. Arrangement of three linked FTL transgenes expressing different colors of fluorescent protein allows simultaneous measurement of crossovers in adjacent intervals and calculation of interference (Berchowitz and Copenhaver 2008; Yelina et al. 2013). Therefore, we crossed met1 to two independent three-color intervals on chromosomes 1 (I1bc) and 5 (I5ab), both of which are located interstitially. Due to MET1 being linked to I5ab, we performed experiments with this interval using met1/+ heterozygotes. We observed that all four single genetic intervals tested showed significant crossover frequency increases in met1, consistent with our previous observations at I1b (Table 2; Supplemental Table 19). For I1bc, we did not observe a significant difference in crossover interference in met1 relative to wild type (P = 0.33) (Table 2; Supplemental Table 19). However, for I5ab, we observed a slight but significant decrease in interference in met1/+ (P = 6.5 × 10−05) (Table 2; Supplemental Table 19).
Table 2.
Analysis of crossover interference using meiotic tetrads in wild type and met1
As an additional measure of crossover interference, we identified 239 and 183 double crossover events in the wild-type and met1 backcross low-coverage sequencing data (excluding chromosome 5) (Supplemental Table 16). Physical distances between double crossover events (mean wild type, 11.58 Mb; met1, 12.67 Mb) were greater than an equivalent set of randomly distributed events in both wild type and met1 (mean 7.41 and 6.82 Mb), as expected due to crossover interference (Fig. 4D). Observed double crossover distances were slightly but significantly greater in met1 compared with wild type (t-test, P = 0.0356) (Fig. 4D). Together, these data demonstrate comparable levels of crossover interference between wild type and met1, which is further consistent with epigenetic crossover remodeling being mediated via interference.
Loss of centromeric CG methylation drives crossover remodeling in met1
The met1 mutant shows loss of DNA methylation in centromeric repetitive sequences in addition to dispersed repeats and gene body methylation in euchromatic regions (Cokus et al. 2008; Lister et al. 2008; Stroud et al. 2013). We therefore sought to investigate which changes in DNA methylation drive epigenetic crossover remodeling in met1. To simultaneously measure euchromatic and heterochromatic recombination rates, we generated a double 420-CEN3 chromosome 3 reporter line (Melamed-Bessudo et al. 2005; Yelina et al. 2012). The seed-based 420 and pollen-based CEN3 fluorescent intervals allow euchromatic and pericentromeric regions to be measured for crossovers in the same individuals (Fig. 5; Supplemental Table 20). We crossed 420-CEN3 to naïve wild-type Col, met1/+ heterozygotes, and met1 homozygotes and measured genetic distances in the resulting F1 progeny (Fig. 5A; Supplemental Tables 20–22). For crosses to met1/+, we selected 420-CEN3 met1/+ F1 progeny for analysis. Consistent with previous observations, crossover frequency in the euchromatic 420 interval increased (GLM, met1/+, P = 2.72 × 10−4; met1, P = 9.02 × 10−3) and the pericentromeric CEN3 interval decreased (GLM, met1/+, P = 7.91 × 10−15; met1, P = 4.23 × 10−36) in met1/+ and met1 progeny compared with naïve Col (Fig. 5A; Yelina et al. 2012). Importantly, we observed a negative correlation between 420 and CEN3 crossovers when measured in the same individuals (Spearman's, r = −0.53, P = 0.0063) (Supplemental Table 20). This is consistent with compensatory recombination changes between pericentromeric and euchromatic chromosomal domains driven by interference in met1 backgrounds.
Figure 5.

Loss of centromeric CG DNA methylation drives crossover remodeling in met1. (A) 420 and CEN3 genetic distances (in centimorgans) in the indicated genotypes. Replicate measurements are indicated by black dots and mean values are indicated by red dots. (B) DNA methylation (percentage) along chromosome 3 is shown separately for CG, CHG, and CHH sequence contexts. Wild-type (red) and met1/+ (blue) controls are shown, along with 14 420-CEN3 met1/+ F1 samples that are color-coded according to the CEN3 recombination rate. (Red) Highest; (blue) lowest. The centromere is indicated by vertical dotted lines, and the 420 and CEN3 fluorescent intervals are indicated by the vertical black lines and colored triangles. (C) Average CG methylation within scaled windows across all genes and transposons and in 4-kb upstream and downstream regions. Samples are color-coded according to CEN3 centimorgans, as in B. Vertical dotted lines indicate gene TSSs and TTSs or transposon start and end coordinates. (D) Correlations between CEN3 CG methylation and CEN3 or 420 genetic distances (in centimorgans). Spearman's correlation values, r, are shown with P-values. (E) Plots show CG DNA methylation for chromosome 3 (as in B) separately for groups of 420-CEN3 F1 met1/+ individuals, according to CEN3 genetic distance (as indicated).
Greater variation in the 420 and CEN3 recombination rates was observed within the met1/+ individuals analyzed compared with Col-0 wild-type controls (Fig. 5A). This is consistent with epigenetic divergence observed during met1/+ inbreeding as a consequence of MET1 function in maintaining CG methylation during the post-meiotic gametophytic cell divisions (Saze et al. 2003). Therefore, we sought to correlate variation in methylation patterns between these met1/+ individuals, as assessed by whole-genome bisulfite sequencing and meiotic recombination rates. We generated bisulfite sequencing data for a 420-CEN3 naïve control, 14 420-CEN3 met1/+ F1 individuals, and a met1/+ heterozygote control that was the progeny of a self-fertilized met1/+ plant (referred to here as met1/+) (Supplemental Table 20). We obtained an average coverage of ∼13.7× for each cytosine, with DNA methylation false positive rates tested using the unmethylated chloroplast genome that were 0.31%, 0.34%, and 0.36% for CG, CHG, and CHH contexts, respectively (Supplemental Tables 23–24). At the chromosome scale, the 420-CEN3 met1/+ individuals showed decreased CG methylation that was intermediate between the wild-type and met1/+ controls (Fig. 5B). DNA methylation loss was less pronounced in 420-CEN3 met1/+ F1 individuals compared with the met1/+ control due to one chromosome in 420-CEN3 met1/+ F1 individuals being inherited from the wild-type parent carrying the fluorescent crossover reporters (Fig. 5B). We observed less reduction in CHG and CHH DNA methylation in 420-CEN3 met1/+ compared with CG methylation, as expected (Fig. 5B; Cokus et al. 2008; Stroud et al. 2013). A similar pattern was observed for CG methylation within transposable elements and gene bodies, where methylation was depleted in the 420-CEN3 met1/+ F1 individuals to a level intermediate between wild-type and met1/+ controls (Fig. 5C; Stroud et al. 2013). Therefore, our 420-CEN3 met1/+ individuals represent a range of methylation states intermediate between wild type and met1/+, with a predominant loss of CG methylation.
To investigate the relationship between recombination and DNA methylation, we divided the 14 420-CEN3 met1/+ F1 individuals into five groups based on CEN3 genetic distances (Fig. 5E; Supplemental Table 20). We then calculated levels of DNA methylation in CG, CHG, and CHH contexts within CEN3 and 420 and correlated these with crossover frequencies in the measured intervals (Supplemental Table 25). We observed that CEN3 recombination and CEN3 CG DNA methylation were positively correlated (Spearman's rank, r = 0.768, P = 0.0013) (Fig. 5D,E; Supplemental Table 26). Changes in recombination correlated comparably strongly with CG methylation in the core versus flanking regions within CEN3 (left flank, r = 0.729, P = 0.0029; right flank, r = 0.745, P = 0.0014; core, r = 0.786, P = 0.0008) (Supplemental Table 26). This is consistent with loss of CG methylation within the centromere and surrounding regions driving changes in interfering crossovers in met1/+. No significant correlations were detected between recombination and CG methylation outside of CEN3 or within CEN3 in non-CG sequence contexts (Supplemental Table 26). For control comparisons, we correlated CEN3 recombination with CG methylation levels in the other chromosome centromeres and with 5 Mb of randomly chosen 1-kb probes throughout the genome and observed no significant correlations (Supplemental Table 26). We observed a significant negative correlation between 420 recombination and levels of CG methylation within CEN3 (Spearman's rank, r = −0.683, P = 0.005) (Fig. 5D; Supplemental Table 26), which is again consistent with demethylation of the centromeric regions driving met1/+ crossover remodeling. We correlated 420 centimorgans with 420 DNA methylation in CG, CHG, and CHH contexts, where CG and CHH were not significant, but a positive correlation with CHG was observed (r = 0.542, P = 0.037). As a control comparison, we correlated 420 centimorgans to CG methylation levels in two comparably sized subtelomeric regions on the north and south arms of chromosome 1 and did not observe significant relationships (Supplemental Table 26). Together, this supports that epigenetic remodeling of interfering crossovers observed in met1 is driven by loss of CG context methylation within the centromeric and pericentromeric regions.
DNA DSB foci are unchanged in met1
To investigate the cause of crossover remodeling observed in met1, we considered whether the meiotic DSBs that initiate homologous recombination were altered. Meiotic DSBs can be analyzed cytologically using anther spreads and immunostaining for phosphorylated γ-H2A.X (Fig. 6A–F; Table 3; Supplemental Table 27; Sanchez-Moran et al. 2007; Ferdous et al. 2012). γ-H2A.X shows numerous foci on leptotene stage meiotic chromosomes, which are dependent on SPO11 and show numbers comparable with RAD51/DMC1 recombinase foci (Sanchez-Moran et al. 2007; Ferdous et al. 2012). At leptotene stage, meiotic chromosomes are observed to form euchromatic thin fibers connected via the meiotic axis protein ASY1, with clustered heterochromatic chromocenters containing the centromeres (Fransz et al. 2002; Ferdous et al. 2012; Yelina et al. 2012). We therefore immunostained wild-type and met1 nuclei and counted γ-H2A.X foci associated with ASY1 axial structures. In wild type, we observed a mean of 172.3 foci per nucleus with a low number of foci within the DAPI-dense chromocenters (mean = 5.8) (Fig. 6A–C; Table 3; Supplemental Fig. S2; Supplemental Table 27). We did not observe a significantly different number of foci in met1 nuclei (mean = 176.5; GLM assuming a Poisson distribution, P = 0.441) or increased numbers within the chromocenters (mean = 5.3; GLM assuming a Poisson distribution, P = 0.571) (Fig. 6A–F; Table 3; Supplemental Table 27). Therefore, epigenetic crossover remodeling in met1 is not associated with greater numbers of DSB-associated γ-H2A.X foci or changes to DSB foci in the heterochromatic regions. This indicates that changes to the meiotic recombination pathway in these regions are likely occurring downstream from DSB formation.
Figure 6.
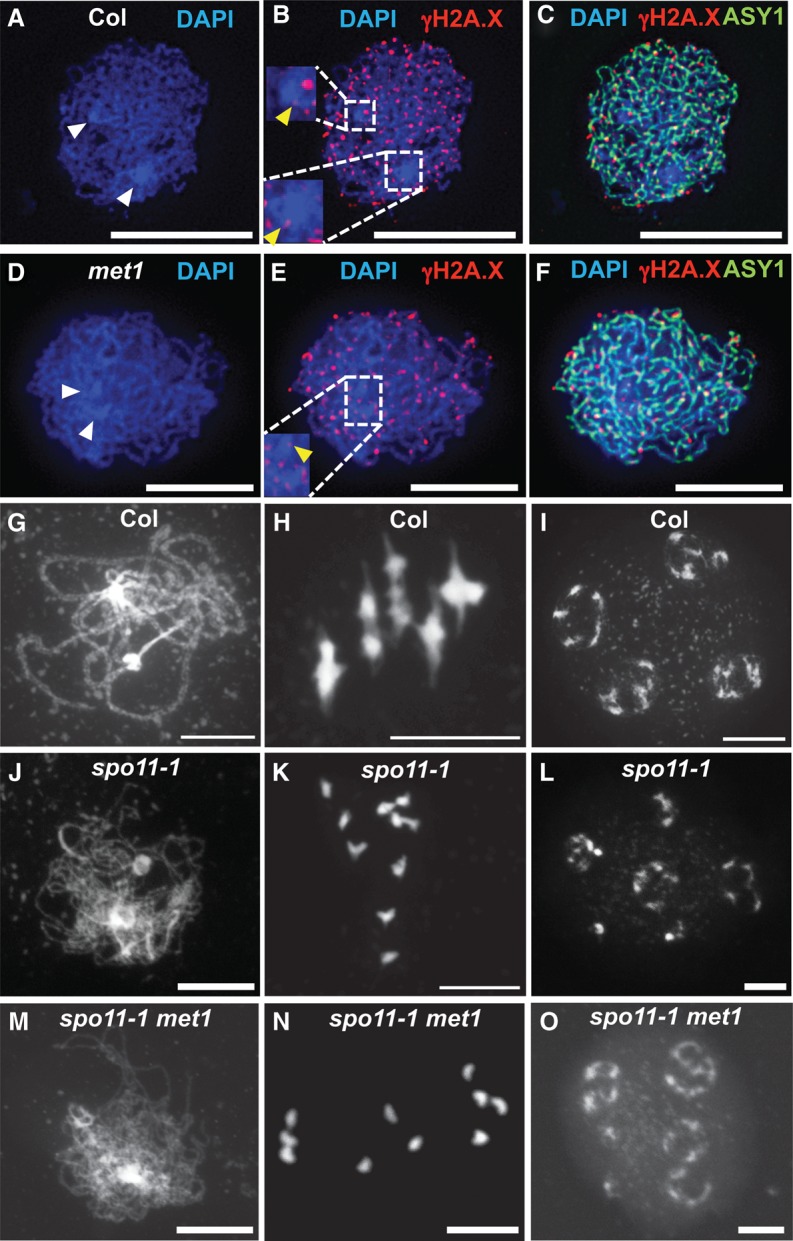
Meiotic DNA DSB foci are unchanged in met1. (A–F) Localization of ASY1 (green) and γ-H2A.X (red) in wild-type (A–C) and met1-3 (D–F) nuclei at leptotene. DNA was stained with DAPI (blue). Densely DAPI-staining chromocenters are marked with white arrowheads. The inset boxes show magnifications of the centromeric regions, with yellow arrowheads marking γ-H2A.X foci localized within the DAPI-dense regions (Supplemental Fig. 2). Bars, 10 μm. (G–O) DAPI-stained meiotic chromosome spreads of wild-type (G–I), spo11-1 (J–L), and met1 spo11-1 (M–O) nuclei at late prophase I (G,J,M), metaphase I (H,K,N), and tetrad (I,L,O) stage. Bars, 10 μm.
Table 3.
γ-H2A.X foci counts in wild-type and met1 leptotene nuclei

Loss of DNA methylation in met1 and ddm1 is known to cause transcriptional up-regulation and mobilization of transposable elements (Miura et al. 2001; Zhang et al. 2006; Lister et al. 2008). As transposition can generate DSBs, we considered whether this phenomenon could contribute to met1 meiotic phenotypes. For example, DSBs from exogenous DNA damage agents (e.g., cisplatin) or X-rays can promote crossovers in Arabidopsis and Caenorhabditis elegans, respectively (Sanchez-Moran et al. 2007; Youds et al. 2010). The spo11-1 mutant is almost completely sterile due to a failure in homologous chromosome pairing and synapsis caused by an absence of meiotic DSBs (Grelon et al. 2001). Therefore, if met1 generates ectopic meiotic DSBs that result in crossovers, then spo11-1 met1 fertility should increase. We compared seed set in spo11-1 and spo11-1 met1 double mutants and did not observe an increase in fertility (Supplemental Table 28). We also performed DAPI staining on meiotic nuclei from anther spreads (Fig. 6G–O). In wild type, paired homologous chromosomes align along the metaphase I plate as five bivalents connected via chiasmata (Fig. 6H). In spo11-1 mutants, 10 unpaired univalents are observed at the same stage (Fig. 6K; Grelon et al. 2001). The spo11-1 met1 double mutant showed patterns of univalent segregation comparable with spo11-1 single mutants (Fig. 6N). Together, this indicates that any transposition-associated DSBs occurring in met1 are unable to drive meiotic recombination and crossover formation to a significant level.
Figure 7.

Control of meiotic recombination by DNA methylation in Arabidopsis. (A) Epigenetic changes associated with RdDM at crossover hot spots. Initially, the hot spot has low DNA methylation, low H3K9me2 (orange nucleosomes), and low nucleosome occupancy and is permissive to recombination. Following establishment of RdDM, dense DNA methylation in CG (red), CHG (green), and CHH (blue) sequence contexts is acquired in addition to higher nucleosome occupancy and H3K9me2 (blue nucleosomes), together causing recombination suppression. (B) The top plots show gene density (base pairs per 100 bp, black) and percentage of DNA methylation (CG in red, CHG in green, and CHH in blue) along chromosome 3 in wild type (Col) and met1-3/+. (Bottom) Chromosome representations with euchromatin colored green and heterochromatin colored red (wild type) or purple (met1). Meiotic DSBs (stars) form in a similar pattern along the chromosome in wild type and met1. DSBs are resected to form ssDNA that invades the homologous chromosome (blue vertical lines). A subset of interhomolog invasion events become crossover-designated and impose interference on adjacent unrepaired DSBs. We propose that loss of CG methylation in the centromeric regions in met1 either (1) alters the relative timing of DSB formation between euchromatin and heterochromatin or (2) reduces the chance of interhomolog invasion events becoming crossover-designated in proximity to the centromere. Interference operates normally in met1, and, as a consequence, the euchromatic arms receive additional crossovers compared with wild type.
Discussion
We demonstrated that establishment of DNA methylation via the RdDM pathway is sufficient to potently suppress euchromatic meiotic recombination hot spots in Arabidopsis. This is consistent with low levels of DNA methylation observed at Arabidopsis and maize crossover hot spots (Choi et al. 2013; Wijnker et al. 2013; Rodgers-Melnick et al. 2015) and the widespread suppression of recombination in plant heterochromatin (Copenhaver et al. 1999; Fu et al. 2002; Liu et al. 2009; Giraut et al. 2011; Mayer et al. 2012; Salomé et al. 2012; The Tomato Genome Consortium 2012; Bauer et al. 2013; Choulet et al. 2014; Li et al. 2015; Rodgers-Melnick et al. 2015). We show that RdDM is associated with acquisition of H3K9me2 and increased nucleosome occupancy at the targeted hot spots. Therefore, the combination of these chromatin modifications is likely to suppress recombination (Fig. 7A). As hot spots associate with promoter-associated chromatin marks, including H3K4me3 and H2A.Z, RdDM may also exclude these modifications and thereby reduce crossovers (Liu et al. 2009; Choi et al. 2013; Wijnker et al. 2013). It is also important to consider that hot spots may vary in both their sensitivity to RdDM and the effect of methylation on crossovers, depending on their location in euchromatic versus pericentromeric regions.
As DNA methylation locally inhibits crossover hot spots, this leads to a prediction that loss of CG methylation in met1 centromeric/pericentromeric regions would up-regulate crossovers. However, the opposite is true, with met1 pericentromeric regions showing reduced crossovers, coupled to compensatory increases in the chromosome arms (Colomé-Tatché et al. 2012; Melamed-Bessudo and Levy 2012; Mirouze et al. 2012; Yelina et al. 2012). We explain these observations, as the hot spot–RdDM and met1 experiments are not exact reciprocals of one another in terms of epigenetic change. For example, we show that RdDM establishes CG and non-CG DNA methylation, H3K9me2, and increased nucleosome occupancy at the 3a and 3b hot spots, causing suppressed recombination (Fig. 7A). However, in met1, while CG-context DNA methylation is strongly reduced, substantial non-CG methylation and H3K9me2 remain (Fig. 7B; Cokus et al. 2008; Lister et al. 2008; Deleris et al. 2012; Stroud et al. 2013). Therefore, to understand these interactions, it will be necessary to obtain further recombination measurements in mutants with different alterations to heterochromatin.
In order to understand meiotic recombination patterns in wild type versus met1, it is also important to consider higher-order chromosome structure. For example, Arabidopsis centromeres physically interact with each other in Arabidopsis somatic nuclei, which is cytologically manifested as the DAPI-dense chromocenters from which euchromatin loops emanate (Fransz et al. 2002; Soppe et al. 2002; Probst et al. 2003; Moissiard et al. 2012; Feng et al. 2014; Grob et al. 2014; Wang et al. 2014). In met1 and ddm1, the chromocenters become cytologically decondensed and are observed to interact with euchromatin to a greater extent, as measured by Hi-C (chromosome capture followed by high-throughput sequencing) (Soppe et al. 2002; Probst et al. 2003; Mathieu et al. 2007; Moissiard et al. 2012; Feng et al. 2014). It is presently unclear how these changes to higher-order chromosome organization will interact with the chromatin loop–axis arrangement of meiotic chromosomes to influence recombination (Kleckner 2006; Storlazzi et al. 2010; Panizza et al. 2011; Ferdous et al. 2012). We propose two models to explain crossover remodeling in met1 (Fig. 7B). First, although the spatial distribution of DSB foci is unchanged in met1, the relative timing of DSB formation between euchromatin and heterochromatin may be altered. For example, it is known that replication occurs later in Arabidopsis heterochromatin (Lee et al. 2010) and that replication is tightly coupled to progression of meiotic recombination in budding yeast (Borde et al. 2000; Murakami and Keeney 2014). Therefore, it is possible that met1 could alter the relative timing of replication between heterochromatin and euchromatin and thereby influence patterns of crossover designation. Second, we propose that loss of CG methylation alters met1 chromosome structure, including its interaction with the meiotic axis, such that interhomolog invasion sites closer to the centromere are inhibited from crossover designation. As total crossovers are similar in number in met1 and as interference is active, crossovers are favored in the gene-rich arms (Fig. 7B).
Together, our results highlight the importance of epigenetic information for determining distributions of meiotic recombination along eukaryotic chromosomes. We demonstrate that analyzing the roles of chromatin marks at both the whole chromosome and hot spot scales is important. Although DNA methylation is widely conserved, its distribution and function between eukaryotes vary, and it has been lost from several model species (Feng et al. 2010; Zemach et al. 2010). For example, within fungi, DNA methylation strongly suppresses crossovers in Ascobolus immersus (Maloisel and Rossignol 1998), whereas this modification is absent in Schizosaccharomyces pombe and Saccharomyces cerevisiae. However, in fission yeast, other heterochromatic marks, including H3K9me2, mediate a repressive effect on meiotic crossovers (Ellermeier et al. 2010). Therefore, it will be important to study the interactions of DNA methylation and additional markers of heterochromatin in multiple species to more completely understand control of meiotic recombination by epigenetic information.
Materials and methods
Plant material
Arabidopsis lines used in this study were Col-0, Ler-0, met1-3 (Saze et al. 2003), fancm-1 zip4-2 (Crismani et al. 2012), and spo11-1-3 (SALK_146172) (Stacey et al. 2006). Plants were grown at 20°C and 60% humidity with a long day photoperiod (16 h light) and light intensity of 150 μmol. Genotyping of met1-3 was performed by PCR amplification using met1-3-F and met1-3-R oligonucleotides for wild type and met1-3-F and met1-3-T for met1-3 (oligonucleotide sequences are provided in Supplemental Table 29). Genotyping of fancm-1 was carried out by PCR amplification using fancm-F and fancm-R oligonucleotides followed by MboII restriction endonuclease digestion. zip4-2 genotyping was carried out by PCR amplification using oligonucleotides zip4-F and zip4-R for the wild type and zip4-R and SALK-LB for the T-DNA alleles. Genotyping for spo11-1-3 was performed as described (Stacey et al. 2006).
RdDM
PCR fragments corresponding to HP1–HP5 were amplified using overlap PCR from Col-0 genomic DNA such that a unique restriction site was introduced into the hairpin (Supplemental Tables 4, 29). The resulting amplification products were cloned into pENTR/D-TOPO (Life Technologies) and then into pJawohl-Act2 using Gateway LR Clonase II (Life Technologies) to produce pJawohl-Act2-HP plasmids. Arabidopsis plants were transformed using floral dipping with Agrobacterium GV3101 carrying pMP90 helper plasmids.
Northern blot and hybridization analysis of small RNAs
Five to seven inflorescences were collected per genotype, and total RNA was extracted using TRI reagent (Sigma). Ten micrograms of total RNA was separated on 15% polyacrylamide/8 M urea gels, blotted on Zeta probe membrane (Bio-Rad), and hybridized with 32P-labeled probes synthesized using Rediprime II kits (Amersham Biosciences). Hybridization was carried out overnight at 39°C in PerfectHyb Plus hybridization buffer (Sigma). Templates for 32P-labeled probes were obtained by PCR amplification of Arabidopsis genomic DNA.
DNA methylation analysis
DNA isolated from Arabidopsis closed flower buds was used for McrBC analysis. One-hundred nanograms of DNA was digested with 10 U of McrBC (New England Biolabs) for 6 h at 37°C. The enzyme was heat-inactivated for 20 min at 65°C. Two nanograms of McrBC-digested DNA was used as a template for PCR amplification. For Sanger sequencing analysis, DNA sodium bisulfite conversion was carried out using an EZ DNA methylation gold kit (Zymo Research). Prior to conversion, DNA was digested at the restriction sites introduced into the hairpin arms in order to enrich for endogenous target sequences. Following conversion, DNA was PCR-amplified using oligonucleotides designed as described (Henderson et al. 2010). PCR products were cloned into pGem-T Easy (Promega), and 15–26 individual clones were Sanger-sequenced to determine the percentage of DNA methylation for cytosines in the amplified regions (Gruntman et al. 2008). For genome-wide bisulfite sequencing, DNA was isolated from leaves of 3- to 5-wk-old Arabidopsis plants. One microgram of genomic DNA was sheared to yield fragments with an average size of 340 bp using a Covaris E220 instrument and purified using Agencourt AMPure XP beads (ratio 1.8×; Beckman Coulter). DNA was end-repaired and A-tailed using T4 DNA polymerase and Klenow fragment (New England Biolabs) and purified again using XP beads (ratio 1.8×). Finally, double-stranded paired-end adapters were ligated using quick-stick ligase (Bioline), and DNA was purified using XP beads (ratio 1.8×). Four-hundred-fifty nanograms of DNA was used for sodium bisulfite conversion using an EZ DNA methylation gold kit. DNA was quantified with a Bioanalyzer 2100 using the high-sensitivity DNA kit (Agilent Technologies) and size-selected to yield fragments with the average size of 400 bp using XP beads (ratio 0.8×). DNA was barcoded using PCR amplification with KAPA HiFi HotStart uracil+ ready mix (Kapabiosystems) with PE1.0 and one of the iPCRtag 1–17 oligonucleotides and then purified using XP beads (ratio 1×), and libraries were quantified on a Bioanalyzer 2100. Final libraries were mixed in equal molar ratios to a final concentration of 10 nM and used for 150-bp paired-end sequencing on a HiSeq 2000 instrument. FastQ files were analyzed and DNA methylation was identified using Bismark (Krueger and Andrews 2011) and SeqMonk (http://www.bioinformatics.bbsrc.ac.uk/projects/seqmonk).
Pollen typing
Pollen typing was performed as previously described (Drouaud and Mézard 2011; Yelina et al. 2012; Choi et al. 2013; Drouaud et al. 2013). Genomic DNA was isolated from Col/Ler F1 or HP/Ler F1 pollen and used for nested PCR amplifications using parental or crossover configurations of allele-specific oligonucleotide primers (Yelina et al. 2012; Choi et al. 2013). To test for significant differences in recombination frequency, we compared numbers of crossover molecules in wild-type and hairpin lines by fitting a nonlinear least squares model (Bates and Watts 1988). The model fit is given by Xijk ∼ Sk + P × Iparental(j) + P[1 − Iparental(j)] [Z − z × Imutant(k)], where Xijk is the log of the number of estimated molecules for the ith observation, with the index j identifying whether the estimation is for the parental or crossover molecules, and the index k indicating whether the estimation is for wild-type or HP strains. The model thus describes for each observation a strain effect (Sk). If the observation is for the parental molecules, a parental effect (P) is added; if the observation is for crossover molecules, the parental effect is modified by a crossover factor [Z − z Imutant(k)], where z represents the change in the crossover factor attributed to the HP line. The key result in the model fit for evaluating whether the number of crossovers is influenced by the hairpin is thus to evaluate whether z significantly deviates from 0.
H3 and H3K9me2 ChIP
ChIP was performed as described (Johnson et al. 2002). Five micrograms of antibodies ab1220 (Abcam) and ab1791 (Abcam) was used to ChIP H3K9me2 and unmodified H3, respectively. Primer sequences used for ChiP-qPCR are provided in Supplemental Table 29. Two biological replicates were performed for each experiment, and each was analyzed using two technical replicates.
Mapping crossovers via low-coverage sequencing
We applied a method to identify crossovers from low-coverage (∼0.5×–2×) sequencing, using 96 barcoded genomic DNA libraries generated from recombinant individuals (Rowan et al. 2015) with the following modifications. DNA was digested with 0.3 U of dsDNA Shearase (Zymo Research) in a final volume of 15 µL. The resulting DNA fragments were end-repaired with 3 U of T4 DNA polymerase (New England Biolabs), 10 U of T4 polynucleotide kinase (Thermo Fisher Scientific), and 1.25 U of Klenow fragment (New England Biolabs) in the presence of 0.4 mM dNTPs in a reaction volume of 30 µL for 30 min at 20°C. DNA fragments were cleaned as described (Rowan et al. 2015), and the protocol was followed until the DNA fragment size selection step. To size-select, following barcoded Illumina adaptor ligation, 30 µL of a mixture of eight concentrated DNA libraries was combined in a tube containing 48 µL of a 1:1 mix of AMPure XP magnetic SPRI beads (Beckman-Coulter; referred to as SPRI beads) in water. After 5 min of incubation at room temperature, the samples were placed on a magnetic rack and allowed to clear before supernatant was transferred to a fresh tube and mixed with 0.12 vol of undiluted SPRI beads. After 5 min of incubation at room temperature, the tubes were placed on a magnetic rack and allowed to clear. The supernatants were discarded, and the beads were washed twice with 80% ethanol. DNA was eluted in 20 µL of 10 mM Tris (pH 8.0). Twelve microliters of the eluate was used for PCR amplification in a reaction volume of 50 µL using KAPA HiFi HotStart ReadyMix PCR kit (Kapabiosystems) and the reported DNA oligonucleotides (Rowan et al. 2015). Twelve cycles of amplification were performed, and PCR products were then purified on SPRI beads (1× ratio) and quantified with a Bioanalyzer. The resulting libraries were subjected to paired-end 150-bp sequencing on an Illumina NextSeq instrument.
We pooled data from 94 wild-type libraries and aligned reads to the TAIR10 Col genome assembly using Bowtie 2 (Langmead and Salzberg 2012). Variant sites were called using SAMtools and BCFtools (Li et al. 2009). Sites were filtered to remove those with qualities <100 and those with >2.5× mean coverage and repeat-masked as described previously (Choi et al. 2013). Sites were also intersected with an independent set of high-quality Ler SNP calls provided by K. Schneeberger, yielding a final set of 153,842 SNPs. For each barcoded library, the number of reads supporting reference versus variant bases at these sites was obtained and used as input for the TIGER pipeline, and the crossovers identified were used for subsequent analysis (Rowan et al. 2015). To test for significant differences in crossovers between wild type and met1, we used a GLM assuming a negative binomial distribution. To analyze chromosomal distributions, chromosome arms were split and orientated such that they began at the telomere and ended at the centromere. Each crossover was assigned a proportional distance value along the chromosome arms. Events were binned into 100 windows, counted, and normalized by total crossovers. A cubic smoothing spline was fitted to these data using the R function smooth.spline and used for plotting. To test for significant differences between regions of the chromosome arms, the counts inside and outside the region being considered in wild type and met1 were used to construct 2 × 2 contingency tables, and χ2 tests were performed. To analyze double crossovers, we identified chromosomes with more than one crossover and measured physical distances between the crossovers. For each double crossover chromosome, we generated the same number of randomly distributed sites and used the distances between these positions for random comparisons.
Seed and pollen fluorescence measurement of crossovers
Fluorescent tetrad scoring and crossover interference calculations were carried out as described (Berchowitz and Copenhaver 2008). Flow cytometry on fluorescent pollen was performed as described (Yelina et al. 2012, 2013). Fluorescent seed scoring was carried out as described (Melamed-Bessudo et al. 2005; Yelina et al. 2012). To test whether recombinant and nonrecombinant counts were significantly different between replicate groups, we used a GLM. We assumed the count data are binomially distributed—Yi ∼ B(ni,pi), where Yi represents the recombinant counts, and ni is the total counts—and modeled the proportions Yi/ni. Then, E(Yi/ni) = pi and
Thus, our variance function is V(µi) = µi(1 − µi), and our link function must map from (0,1) → (−∞, ∞). We used a logistic link function, which is
where εi ∼ N(0,σ2). Both replicates and genotypes were treated as independent variables (x) in our model. We considered P-values <0.05 as significant.
Cytological analysis
Chromosome spreads of Arabidopsis pollen mother cells (PMCs) were immunostained as described (Ferdous et al. 2012). The following antibodies were used for immunostaining: α-ASY1 (rat, 1/500 dilution) (Armstrong et al. 2002) and α-γH2A.X (Ser 139, rabbit, 1/100 dilution; Upstate Biotechnology, catalog no. 07-164). Microscopy was conducted using a DeltaVision Personal DV microscope (Applied Precision/GE Healthcare) equipped with a CCD Coolsnap HQ2 camera (Photometrics). Image capture, image analysis, and processing were performed using SoftWoRx software version 5.5 (Applied Precision/GE Healthcare). The images were deconvolved using the function “Mexican hat” of SoftWoRx software version 5.5. Deconvolution improved the contrast and resolution of images, allowing better visualization of the signal.
Accession numbers
Accession numbers for the next-generation sequencing data in this study have been uploaded to EMBL European Bioinformatics Institute Annotare (https://www.ebi.ac.uk/fg/annotare) and are as follows: for whole-genome bisulphite sequencing of met1-3/+, ArrayExpress accession E-MTAB-3927; for mapping meiotic crossovers in Arabidopsis using low-coverage whole-genome sequencing, ArrayExpress accession E-MTAB-3926.
Supplementary Material
Acknowledgments
We thank Korbinian Schneeberger and Beth Rowan for advice implementing TIGER and Ler polymorphism data, Donna Bond for pJawohl-Act2, Quentin Gouil for the bisulfite sequencing protocol, Simon Andrews and Felix Krueger for advice using SeqMonk, Gregory Copenhaver and Avi Levy for fluorescent lines, Raphael Mercier for zip4-2 fancm-1, Chris Franklin for the ASY1 antibody, and the Gurdon Institute Imaging Facility for access to microscopes. Research was supported by a Broodbank Fellowship (to N.E.Y.), a Royal Society University Research Fellowship (to I.R.H.), grant GAT2962 from the Gatsby Charitable Foundation (to I.R.H.), and Biotechnology and Biological Sciences Research Council grant BB/L006847/1 (to I.R.H.).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.270876.115.
References
- Armstrong SJ, Caryl AP, Jones GH, Franklin FCH. 2002. Asy1, a protein required for meiotic chromosome synapsis, localizes to axis-associated chromatin in Arabidopsis and Brassica. J Cell Sci 115: 3645–3655. [DOI] [PubMed] [Google Scholar]
- Auton A, McVean G. 2012. Estimating recombination rates from genetic variation in humans. Methods Mol Biol 856: 217–237. [DOI] [PubMed] [Google Scholar]
- Bates DM, Watts DG. 1988. Nonlinear regression analysis and its applications. John Wiley & Sons, New York. [Google Scholar]
- Bauer E, Falque M, Walter H, Bauland C, Camisan C, Campo L, Meyer N, Ranc N, Rincent R, Schipprack W, et al. 2013. Intraspecific variation of recombination rate in maize. Genome Biol 14: R103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchowitz LE, Copenhaver GP. 2008. Fluorescent Arabidopsis tetrads: a visual assay for quickly developing large crossover and crossover interference data sets. Nat Protoc 3: 41–50. [DOI] [PubMed] [Google Scholar]
- Berchowitz LE, Francis KE, Bey AL, Copenhaver GP. 2007. The role of AtMUS81 in interference-insensitive crossovers in A. thaliana. PLoS Genet 3: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernatavichute YV, Zhang X, Cokus S, Pellegrini M, Jacobsen SE. 2008. Genome-wide association of histone H3 lysine nine methylation with CHG DNA methylation in Arabidopsis thaliana. PLoS One 3: e3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borde V, Goldman AS, Lichten M. 2000. Direct coupling between meiotic DNA replication and recombination initiation. Science 290: 806–809. [DOI] [PubMed] [Google Scholar]
- Brown J, Sundaresan V. 1991. A recombination hotspot in the maize A1 intragenic region. Theor Appl Genet 81: 185–188. [DOI] [PubMed] [Google Scholar]
- Cao X, Jacobsen SE. 2002. Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr Biol 12: 1138–1144. [DOI] [PubMed] [Google Scholar]
- Cao X, Aufsatz W, Zilberman D, Mette MF, Huang MS, Matzke M, Jacobsen SE. 2003. Role of the DRM and CMT3 methyltransferases in RNA-directed DNA methylation. Curr Biol 13: 2212–2217. [DOI] [PubMed] [Google Scholar]
- Cao J, Schneeberger K, Ossowski S, Günther T, Bender S, Fitz J, Koenig D, Lanz C, Stegle O, Lippert C, et al. 2011. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet 43: 956–963. [DOI] [PubMed] [Google Scholar]
- Chelysheva L, Gendrot G, Vezon D, Doutriaux M-P, Mercier R, Grelon M. 2007. Zip4/Spo22 is required for class I CO formation but not for synapsis completion in Arabidopsis thaliana. PLoS Genet 3: e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelysheva L, Grandont L, Vrielynck N, le Guin S, Mercier R, Grelon M. 2010. An easy protocol for studying chromatin and recombination protein dynamics during Arabidopsis thaliana meiosis: immunodetection of cohesins, histones and MLH1. Cytogenet Genome Res 129: 143–153. [DOI] [PubMed] [Google Scholar]
- Chelysheva L, Vezon D, Chambon A, Gendrot G, Pereira L, Lemhemdi A, Vrielynck N, Le Guin S, Novatchkova M, Grelon M. 2012. The Arabidopsis HEI10 is a new ZMM protein related to Zip3. PLoS Genet 8: e1002799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Zhang W, Timofejeva L, Gerardin Y, Ma H. 2005. The Arabidopsis ROCK-N-ROLLERS gene encodes a homolog of the yeast ATP-dependent DNA helicase MER3 and is required for normal meiotic crossover formation. Plant J 43: 321–334. [DOI] [PubMed] [Google Scholar]
- Chodavarapu RK, Feng S, Bernatavichute YV, Chen P-Y, Stroud H, Yu Y, Hetzel JA, Kuo F, Kim J, Cokus SJ, et al. 2010. Relationship between nucleosome positioning and DNA methylation. Nature 466: 388–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K, Zhao X, Kelly KA, Venn O, Higgins JD, Yelina NE, Hardcastle TJ, Ziolkowski PA, Copenhaver GP, Franklin FCH, et al. 2013. Arabidopsis meiotic crossover hot spots overlap with H2A.Z nucleosomes at gene promoters. Nat Genet 45: 1327–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choulet F, Alberti A, Theil S, Glover N, Barbe V, Daron J, Pingault L, Sourdille P, Couloux A, Paux E, et al. 2014. Structural and functional partitioning of bread wheat chromosome 3B. Science 345: 1249721. [DOI] [PubMed] [Google Scholar]
- Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE. 2008. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452: 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colomé-Tatché M, Cortijo S, Wardenaar R, Morgado L, Lahouze B, Sarazin A, Etcheverry M, Martin A, Feng S, Duvernois-Berthet E, et al. 2012. Features of the Arabidopsis recombination landscape resulting from the combined loss of sequence variation and DNA methylation. Proc Natl Acad Sci 109: 16240–16245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copenhaver GP, Browne WE, Preuss D. 1998. Assaying genome-wide recombination and centromere functions with Arabidopsis tetrads. Proc Natl Acad Sci 95: 247–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copenhaver GP, Nickel K, Kuromori T, Benito MI, Kaul S, Lin X, Bevan M, Murphy G, Harris B, Parnell LD, et al. 1999. Genetic definition and sequence analysis of Arabidopsis centromeres. Science 286: 2468–2474. [DOI] [PubMed] [Google Scholar]
- Copenhaver GP, Housworth EA, Stahl FW. 2002. Crossover interference in Arabidopsis. Genetics 160: 1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crismani W, Girard C, Froger N, Pradillo M, Santos JL, Chelysheva L, Copenhaver GP, Horlow C, Mercier R. 2012. FANCM limits meiotic crossovers. Science 336: 1588–1590. [DOI] [PubMed] [Google Scholar]
- Deleris A, Stroud H, Bernatavichute Y, Johnson E, Klein G, Schubert D, Jacobsen SE. 2012. Loss of the DNA methyltransferase MET1 Induces H3K9 hypermethylation at PcG target genes and redistribution of H3K27 trimethylation to transposons in Arabidopsis thaliana. PLoS Genet 8: e1003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Massy B. 2013. Initiation of meiotic recombination: how and where? Conservation and specificities among eukaryotes. Annu Rev Genet 47: 563–599. [DOI] [PubMed] [Google Scholar]
- Drouaud J, Mézard C. 2011. Characterization of meiotic crossovers in pollen from Arabidopsis thaliana. Methods Mol Biol 745: 223–249. [DOI] [PubMed] [Google Scholar]
- Drouaud J, Khademian H, Giraut L, Zanni V, Bellalou S, Henderson IR, Falque M, Mézard C. 2013. Contrasted patterns of crossover and non-crossover at Arabidopsis thaliana meiotic recombination hotspots. PLoS Genet 9: e1003922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin MJ, Zhang P, Meng D, Remigereau M-S, Osborne EJ, Paolo Casale F, Drewe P, Kahles A, Jean G, Vilhjálmsson B, et al. 2015. DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. Elife 4: e05255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellermeier C, Higuchi EC, Phadnis N, Holm L, Geelhood JL, Thon G, Smith GR. 2010. RNAi and heterochromatin repress centromeric meiotic recombination. Proc Natl Acad Sci 107: 8701–8705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Cokus SJ, Zhang X, Chen P-Y, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME, et al. 2010. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci 107: 8689–8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Cokus SJ, Schubert V, Zhai J, Pellegrini M, Jacobsen SE. 2014. Genome-wide Hi-C analyses in wild-type and mutants reveal high-resolution chromatin interactions in Arabidopsis. Mol Cell 55: 694–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdous M, Higgins JD, Osman K, Lambing C, Roitinger E, Mechtler K, Armstrong SJ, Perry R, Pradillo M, Cuñado N, et al. 2012. Inter-homolog crossing-over and synapsis in Arabidopsis meiosis are dependent on the chromosome axis protein AtASY3. PLoS Genet 8: e1002507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransz P, De Jong JH, Lysak M, Castiglione MR, Schubert I. 2002. Interphase chromosomes in Arabidopsis are organized as well defined chromocenters from which euchromatin loops emanate. Proc Natl Acad Sci 99: 14584–14589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Park W, Yan X, Zheng Z, Shen B, Dooner HK. 2001. The highly recombinogenic bz locus lies in an unusually gene-rich region of the maize genome. Proc Natl Acad Sci 98: 8903–8908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Zheng Z, Dooner HK. 2002. Recombination rates between adjacent genic and retrotransposon regions in maize vary by two orders of magnitude. Proc Natl Acad Sci 99: 1082–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard C, Crismani W, Froger N, Mazel J, Lemhemdi A, Horlow C, Mercier R. 2014. FANCM-associated proteins MHF1 and MHF2, but not the other Fanconi anemia factors, limit meiotic crossovers. Nucleic Acids Res 42: 9087–9095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard C, Chelysheva L, Choinard S, Froger N, Macaisne N, Lehmemdi A, Mazel J, Crismani W, Mercier R. 2015. AAA–ATPase FIDGETIN-LIKE 1 and helicase FANCM antagonize meiotic crossovers by distinct mechanisms. PLoS Genet 11: e1005369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraut L, Falque M, Drouaud J, Pereira L, Martin OC, Mézard C. 2011. Genome-wide crossover distribution in Arabidopsis thaliana meiosis reveals sex-specific patterns along chromosomes. PLoS Genet 7: e1002354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grelon M, Vezon D, Gendrot G, Pelletier G. 2001. AtSPO11-1 is necessary for efficient meiotic recombination in plants. EMBO J 20: 589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grob S, Schmid MW, Grossniklaus U. 2014. Hi-C analysis in Arabidopsis identifies the KNOT, a structure with similarities to the flamenco locus of Drosophila. Mol Cell 55: 678–693. [DOI] [PubMed] [Google Scholar]
- Gruntman E, Qi Y, Slotkin RK, Roeder T, Martienssen RA, Sachidanandam R. 2008. Kismeth: analyzer of plant methylation states through bisulfite sequencing. BMC Bioinformatics 9: 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellsten U, Wright KM, Jenkins J, Shu S, Yuan Y, Wessler SR, Schmutz J, Willis JH, Rokhsar DS. 2013. Fine-scale variation in meiotic recombination in Mimulus inferred from population shotgun sequencing. Proc Natl Acad Sci 110: 19478–19482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson IR, Jacobsen SE. 2008. Tandem repeats upstream of the Arabidopsis endogene SDC recruit non-CG DNA methylation and initiate siRNA spreading. Genes Dev 22: 1597–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson IR, Chan SR, Cao X, Johnson L, Jacobsen SE. 2010. Accurate sodium bisulfite sequencing in plants. Epigenetics 5: 47–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JD, Armstrong SJ, Franklin FCH, Jones GH. 2004. The Arabidopsis MutS homolog AtMSH4 functions at an early step in recombination: evidence for two classes of recombination in Arabidopsis. Genes Dev 18: 2557–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JD, Buckling EF, Franklin FCH, Jones GH. 2008a. Expression and functional analysis of AtMUS81 in Arabidopsis meiosis reveals a role in the second pathway of crossing-over. Plant J 54: 152–162. [DOI] [PubMed] [Google Scholar]
- Higgins JD, Vignard J, Mercier R, Pugh AG, Franklin FCH, Jones GH. 2008b. AtMSH5 partners AtMSH4 in the class I meiotic crossover pathway in Arabidopsis thaliana, but is not required for synapsis. Plant J 55: 28–39. [DOI] [PubMed] [Google Scholar]
- Johnson L, Cao X, Jacobsen S. 2002. Interplay between two epigenetic marks. DNA methylation and histone H3 lysine 9 methylation. Curr Biol 12: 1360–1367. [DOI] [PubMed] [Google Scholar]
- Kauppi L, Jeffreys AJ, Keeney S. 2004. Where the crossovers are: recombination distributions in mammals. Nat Rev Genet 5: 413–424. [DOI] [PubMed] [Google Scholar]
- Keeney S, Neale MJ. 2006. Initiation of meiotic recombination by formation of DNA double-strand breaks: mechanism and regulation. Biochem Soc Trans 34: 523–525. [DOI] [PubMed] [Google Scholar]
- Kleckner N. 2006. Chiasma formation: chromatin/axis interplay and the role(s) of the synaptonemal complex. Chromosoma 115: 175–194. [DOI] [PubMed] [Google Scholar]
- Knoll A, Higgins JD, Seeliger K, Reha SJ, Dangel NJ, Bauknecht M, Schröpfer S, Franklin FCH, Puchta H. 2012. The Fanconi anemia ortholog FANCM ensures ordered homologous recombination in both somatic and meiotic cells in Arabidopsis. Plant Cell 24: 1448–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR. 2011. Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics 27: 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law JA, Jacobsen SE. 2010. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11: 204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T-J, Pascuzzi PE, Settlage SB, Shultz RW, Tanurdzic M, Rabinowicz PD, Menges M, Zheng P, Main D, Murray JAH, et al. 2010. Arabidopsis thaliana chromosome 4 replicates in two phases that correlate with chromatin state. PLoS Genet 6: e1000982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Li L, Yan J. 2015. Dissecting meiotic recombination based on tetrad analysis by single-microspore sequencing in maize. Nat Commun 6: 6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichten M, Goldman AS. 1995. Meiotic recombination hotspots. Annu Rev Genet 29: 423–444. [DOI] [PubMed] [Google Scholar]
- Lister R, O'Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. 2008. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133: 523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Yeh C-T, Ji T, Ying K, Wu H, Tang HM, Fu Y, Nettleton D, Schnable PS. 2009. Mu transposon insertion sites and meiotic recombination events co-localize with epigenetic marks for open chromatin across the maize genome. PLoS Genet 5: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Q, Rabanal FA, Meng D, Huber CD, Farlow A, Platzer A, Zhang Q, Vilhjálmsson BJ, Korte A, Nizhynska V, et al. 2013. Massive genomic variation and strong selection in Arabidopsis thaliana lines from Sweden. Nat Genet 45: 884–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaisne N, Novatchkova M, Peirera L, Vezon D, Jolivet S, Froger N, Chelysheva L, Grelon M, Mercier R. 2008. SHOC1, an XPF endonuclease-related protein, is essential for the formation of class I meiotic crossovers. Curr Biol 18: 1432–1437. [DOI] [PubMed] [Google Scholar]
- Maloisel L, Rossignol JL. 1998. Suppression of crossing-over by DNA methylation in Ascobolus. Genes Dev 12: 1381–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu O, Reinders J, Caikovski M, Smathajitt C, Paszkowski J. 2007. Transgenerational stability of the Arabidopsis epigenome is coordinated by CG methylation. Cell 130: 851–862. [DOI] [PubMed] [Google Scholar]
- Mayer KFX, Waugh R, Brown JWS, Schulman A, Langridge P, Platzer M, Fincher GB, Muehlbauer GJ, Sato K, Close TJ, et al. 2012. A physical, genetic and functional sequence assembly of the barley genome. Nature 491: 711–716. [DOI] [PubMed] [Google Scholar]
- Melamed-Bessudo C, Levy AA. 2012. Deficiency in DNA methylation increases meiotic crossover rates in euchromatic but not in heterochromatic regions in Arabidopsis. Proc Natl Acad Sci 109: E981–E988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed-Bessudo C, Yehuda E, Stuitje AR, Levy AA. 2005. A new seed-based assay for meiotic recombination in Arabidopsis thaliana. Plant J 43: 458–466. [DOI] [PubMed] [Google Scholar]
- Mercier R, Jolivet S, Vezon D, Huppe E, Chelysheva L, Giovanni M, Nogué F, Doutriaux M-P, Horlow C, Grelon M, et al. 2005. Two meiotic crossover classes cohabit in Arabidopsis: one is dependent on MER3,whereas the other one is not. Curr Biol 15: 692–701. [DOI] [PubMed] [Google Scholar]
- Mette MF, Aufsatz W, van der Winden J, Matzke MA, Matzke AJ. 2000. Transcriptional silencing and promoter methylation triggered by double-stranded RNA. EMBO J 19: 5194–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirouze M, Lieberman-Lazarovich M, Aversano R, Bucher E, Nicolet J, Reinders J, Paszkowski J. 2012. Loss of DNA methylation affects the recombination landscape in Arabidopsis. Proc Natl Acad Sci 109: 5880–5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura A, Yonebayashi S, Watanabe K, Toyama T, Shimada H, Kakutani T. 2001. Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature 411: 212–214. [DOI] [PubMed] [Google Scholar]
- Moissiard G, Cokus SJ, Cary J, Feng S, Billi AC, Stroud H, Husmann D, Zhan Y, Lajoie BR, McCord RP, et al. 2012. MORC family ATPases required for heterochromatin condensation and gene silencing. Science 336: 1448–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami H, Keeney S. 2014. Temporospatial coordination of meiotic DNA replication and recombination via DDK recruitment to replisomes. Cell 158: 861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panizza S, Mendoza MA, Berlinger M, Huang L, Nicolas A, Shirahige K, Klein F. 2011. Spo11-accessory proteins link double-strand break sites to the chromosome axis in early meiotic recombination. Cell 146: 372–383. [DOI] [PubMed] [Google Scholar]
- Pélissier T, Thalmeir S, Kempe D, Sänger HL, Wassenegger M. 1999. Heavy de novo methylation at symmetrical and non-symmetrical sites is a hallmark of RNA-directed DNA methylation. Nucleic Acids Res 27: 1625–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst AV, Fransz PF, Paszkowski J, Mittelsten Scheid O. 2003. Two means of transcriptional reactivation within heterochromatin. Plant J 33: 743–749. [DOI] [PubMed] [Google Scholar]
- Robine N, Uematsu N, Amiot F, Gidrol X, Barillot E, Nicolas A, Borde V. 2007. Genome-wide redistribution of meiotic double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol 27: 1868–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers-Melnick E, Bradbury PJ, Elshire RJ, Glaubitz JC, Acharya CB, Mitchell SE, Li C, Li Y, Buckler ES. 2015. Recombination in diverse maize is stable, predictable, and associated with genetic load. Proc Natl Acad Sci 112: 3823–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan BA, Patel V, Weigel D, Schneeberger K. 2015. Rapid and inexpensive whole-genome genotyping-by-sequencing for crossover localization and fine-scale genetic mapping. G3 (Bethesda) 5: 385–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saintenac C, Faure S, Remay A, Choulet F, Ravel C, Paux E, Balfourier F, Feuillet C, Sourdille P. 2011. Variation in crossover rates across a 3-Mb contig of bread wheat (Triticum aestivum) reveals the presence of a meiotic recombination hotspot. Chromosoma 120: 185–198. [DOI] [PubMed] [Google Scholar]
- Salomé PA, Bomblies K, Fitz J, Laitinen RAE, Warthmann N, Yant L, Weigel D. 2012. The recombination landscape in Arabidopsis thaliana F2 populations. Heredity (Edinb) 108: 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Moran E, Santos J-L, Jones GH, Franklin FCH. 2007. ASY1 mediates AtDMC1-dependent interhomolog recombination during meiosis in Arabidopsis. Genes Dev 21: 2220–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saze H, Mittelsten Scheid O, Paszkowski J. 2003. Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat Genet 34: 65–69. [DOI] [PubMed] [Google Scholar]
- Schwacha A, Kleckner N. 1995. Identification of double Holliday junctions as intermediates in meiotic recombination. Cell 83: 783–791. [DOI] [PubMed] [Google Scholar]
- Séguéla-Arnaud M, Crismani W, Larchevêque C, Mazel J, Froger N, Choinard S, Lemhemdi A, Macaisne N, Van Leene J, Gevaert K, et al. 2015. Multiple mechanisms limit meiotic crossovers: TOP3α and two BLM homologs antagonize crossovers in parallel to FANCM. Proc Natl Acad Sci 112: 4713–4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soppe WJJ, Jasencakova Z, Houben A, Kakutani T, Meister A, Huang MS, Jacobsen SE, Schubert I, Fransz PF. 2002. DNA methylation controls histone H3 lysine 9 methylation and heterochromatin assembly in Arabidopsis. EMBO J 21: 6549–6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey NJ, Kuromori T, Azumi Y, Roberts G, Breuer C, Wada T, Maxwell A, Roberts K, Sugimoto-Shirasu K. 2006. Arabidopsis SPO11-2 functions with SPO11-1 in meiotic recombination. Plant J 48: 206–216. [DOI] [PubMed] [Google Scholar]
- Storlazzi A, Gargano S, Ruprich-Robert G, Falque M, David M, Kleckner N, Zickler D. 2010. Recombination proteins mediate meiotic spatial chromosome organization and pairing. Cell 141: 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud H, Greenberg MVC, Feng S, Bernatavichute YV, Jacobsen SE. 2013. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152: 352–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud H, Do T, Du J, Zhong X, Feng S, Johnson L, Patel DJ, Jacobsen SE. 2014. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat Struct Mol Biol 21: 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Ambrose JH, Haughey BS, Webster TD, Pierrie SN, Muñoz DF, Wellman EC, Cherian S, Lewis SM, Berchowitz LE, et al. 2012. Deep genome-wide measurement of meiotic gene conversion using tetrad analysis in Arabidopsis thaliana. PLoS Genet 8: e1002968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW. 1983. The double-strand-break repair model for recombination. Cell 33: 25–35. [DOI] [PubMed] [Google Scholar]
- Teixeira FK, Heredia F, Sarazin A, Roudier F, Boccara M, Ciaudo C, Cruaud C, Poulain J, Berdasco M, Fraga MF, et al. 2009. A role for RNAi in the selective correction of DNA methylation defects. Science 323: 1600–1604. [DOI] [PubMed] [Google Scholar]
- The Tomato Genome Consortium. 2012. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485: 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve AM, Hillers KJ. 2001. Whence meiosis? Cell 106: 647–650. [DOI] [PubMed] [Google Scholar]
- Vongs A, Kakutani T, Martienssen RA, Richards EJ. 1993. Arabidopsis thaliana DNA methylation mutants. Science 260: 1926–1928. [DOI] [PubMed] [Google Scholar]
- Wang C, Liu C, Roqueiro D, Grimm D, Schwab R, Becker C, Lanz C, Weigel D. 2014. Genome-wide analysis of local chromatin packing in Arabidopsis thaliana. Genome Res 25: 246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassenegger M, Heimes S, Riedel L, Sänger HL. 1994. RNA-directed de novo methylation of genomic sequences in plants. Cell 76: 567–576. [DOI] [PubMed] [Google Scholar]
- Wei F, Zhang J, Zhou S, He R, Schaeffer M, Collura K, Kudrna D, Faga BP, Wissotski M, Golser W, et al. 2009. The Physical and genetic framework of the maize B73 genome. PLoS Genet 5: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijnker E, Velikkakam James G, Ding J, Becker F, Klasen JR, Rawat V, Rowan BA, de Jong DF, de Snoo CB, Zapata L, et al. 2013. The genomic landscape of meiotic crossovers and gene conversions in Arabidopsis thaliana. Elife 2: e01426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelina NE, Choi K, Chelysheva L, Macaulay M, de Snoo B, Wijnker E, Miller N, Drouaud J, Grelon M, Copenhaver GP, et al. 2012. Epigenetic remodeling of meiotic crossover frequency in Arabidopsis thaliana DNA methyltransferase mutants. PLoS Genet 8: e1002844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelina NE, Ziolkowski PA, Miller N, Zhao X, Kelly KA, Muñoz DF, Mann DJ, Copenhaver GP, Henderson IR. 2013. High-throughput analysis of meiotic crossover frequency and interference via flow cytometry of fluorescent pollen in Arabidopsis thaliana. Nat Protoc 8: 2119–2134. [DOI] [PubMed] [Google Scholar]
- Youds JL, Mets DG, McIlwraith MJ, Martin JS, Ward JD, ONeil NJ, Rose AM, West SC, Meyer BJ, Boulton SJ. 2010. RTEL-1 enforces meiotic crossover interference and homeostasis. Science 327: 1254–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A, McDaniel IE, Silva P, Zilberman D. 2010. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328: 916–919. [DOI] [PubMed] [Google Scholar]
- Zemach A, Kim MY, Hsieh P-H, Coleman-Derr D, Eshed-Williams L, Thao K, Harmer SL, Zilberman D. 2013. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 153: 193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SW-L, Chen H, Henderson IR, Shinn P, Pellegrini M, Jacobsen SE, et al. 2006. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 126: 1189–1201. [DOI] [PubMed] [Google Scholar]
- Zhang L, Liang Z, Hutchinson J, Kleckner N. 2014. Crossover patterning by the beam-film model: analysis and implications. PLoS Genet 10: e1004042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman D, Cao X, Johansen LK, Xie Z, Carrington JC, Jacobsen SE. 2004. Role of Arabidopsis ARGONAUTE4 in RNA-directed DNA methylation triggered by inverted repeats. Curr Biol 14: 1214–1220. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.