

In this study, Bankaitis et al. investigate how niche environments are organized within the pancreas and identify and functionally characterize an epithelial endocrine progenitor niche that guides the production of the endocrine lineage during the secondary transition wave of pancreatic epithelial differentiation. These findings provide novel insight into how differentiation is coordinated with the growth and morphogenesis of the developing pancreatic epithelium.
Keywords: plexus, organogenesis, niche, endocrine, progenitor
Abstract
In the mammalian pancreas, endocrine cells undergo lineage allocation upon emergence from a bipotent duct/endocrine progenitor pool, which resides in the “trunk epithelium.” Major questions remain regarding how niche environments are organized within this epithelium to coordinate endocrine differentiation with programs of epithelial growth, maturation, and morphogenesis. We used EdU pulse-chase and tissue-reconstruction approaches to analyze how endocrine progenitors and their differentiating progeny are assembled within the trunk as it undergoes remodeling from an irregular plexus of tubules to form the eventual mature, branched ductal arbor. The bulk of endocrine progenitors is maintained in an epithelial “plexus state,” which is a transient intermediate during epithelial maturation within which endocrine cell differentiation is continually robust and surprisingly long-lived. Within the plexus, local feedback effects derived from the differentiating and delaminating endocrine cells nonautonomously regulate the flux of endocrine cell birth as well as proliferative growth of the bipotent cell population using Notch-dependent and Notch-independent influences, respectively. These feedback effects in turn maintain the plexus state to ensure prolonged allocation of endocrine cells late into gestation. These findings begin to define a niche-like environment guiding the genesis of the endocrine pancreas and advance current models for how differentiation is coordinated with the growth and morphogenesis of the developing pancreatic epithelium.
While we know much about the transcriptional and signaling cues that balance cell differentiation against progenitor maintenance during organogenesis, little is known about how developmental programs are coordinated to guide formation of complex organs. A strategy involved in many if not all developing organs is to localize and maintain progenitors within “niche” signaling environments in which there is coordination of cell communication and feedback signaling mechanisms that regulate progression through various developmental processes (Fuchs et al. 2004; Li and Xie 2005). The present study is concerned with the spatiotemporal and functional characterization of an epithelial niche that directs feedback control of progenitor maintenance, differentiation, and morphogenesis during the secondary transition wave of pancreatic endocrine cell differentiation.
The pancreas develops from part of the foregut endoderm around embryonic day 8.5 (E8.5)–E9.0. During the primary transition (E9.0–E12.5), a pancreas-specified and relatively homogeneous multipotent progenitor cell (MPC) population evaginates from the naïve endodermal tube and proliferates to form dorsal and ventral buds (Pan and Wright 2011). Around E11.0, scattered groups of cells undergo apicobasal polarization to initiate formation of microlumens, which expand and coalesce to build a web-like network (or plexus) of epithelial tubes (Hick et al. 2009; Kesavan et al. 2009; Villasenor et al. 2010). Within the forming epithelium (approximately E11–E13.5), progenitors become segregated into “tip” and “trunk” epithelial domains (Zhou et al. 2007), the latter containing a pool of Sox9+ bipotent endocrine/duct progenitors (Solar et al. 2009; Kopinke et al. 2011; Kopp et al. 2011). Tip and trunk populations undergo a wave of differentiation during the secondary transition (E13.5–E17.5). The trunk progenitor pool derives endocrine progeny that delaminate and cluster into the nascent islets of Langerhans. Cells that remain behind ultimately form the mature ductal system, with each ductal tube terminally capped by digestive enzyme-secreting acinar cells. How the growth, maintenance, and differentiation behaviors of endocrine progenitors, particularly those that give rise to insulin-secreting β cells, are patterned and controlled within the dynamic trunk environment remains under intense study.
Mounting evidence indicates that Notch-dependent feedback determines the balance between endocrine differentiation and progenitor maintenance (Serup 2012). While perturbation studies specifically within the trunk are still needed, it is evident that down-regulation of Notch activity in pancreatic progenitors will permit endocrine commitment. Constitutive activation of Notch in Pdx1-expressing epithelial cells blocks differentiation in favor of an undifferentiated progenitor state (Murtaugh et al. 2003), whereas Notch inactivation results in “default” acquisition of an endocrine phenotype (Apelqvist et al. 1999). Genetic inactivation of the transcription factor and endocrine lineage determinant Neurogenin3 (Ngn3) (Gradwohl et al. 2000; Schwitzgebel et al. 2000; Gu et al. 2002; Smith et al. 2004) results in reduced Notch activity and derepression of Ngn3 promoter activity throughout the trunk, suggesting that inhibitory Notch signals are derived from the differentiating endocrine cells (Lammert et al. 2000; Magenheim et al. 2011a; Shih et al. 2012; Qu et al. 2013). Finally, studies in zebrafish suggest that stepwise decreases in Notch activity regulate states of mitotic quiescence, progenitor replication, and endocrine differentiation, respectively (Ninov et al. 2012). Substantial gaps remain regarding whether, during diversification of duct and endocrine lineages, Notch activity operates broadly throughout the trunk or is deployed within a discrete endocrine progenitor niche.
The existence of an endocrine progenitor niche is supported by studies showing that the endocrine differentiation program can directly affect and is reciprocally affected by alterations to the morphological state of the pancreatic epithelium. Blocking endocrine differentiation by genetically ablating Ngn3 or accelerating endocrine differentiation via inhibiting Notch results in altered epithelial branching patterns (Magenheim et al. 2011a). Conversely, perturbation of the RhoGTPase Cdc42 showed that misregulated apicobasal polarization results in incorrect allocation of progenitors to a proacinar “tip” over “trunk” state and an inability to generate normal endocrine cell numbers (Kesavan et al. 2009). These defects were attributed to a non-cell-autonomous function for Cdc42 in positioning initially multipotent progenitors within environments conducive for endocrine differentiation. Finally, tissue-specific inactivation of transcription factors expressed in the trunk—such as HNF6 (Pierreux et al. 2006), Prox1 (Wang et al. 2005; Westmoreland et al. 2012), or HNF1b (De Vas et al. 2015)—results in epithelial malformations and concurrent deficits in endocrine differentiation. While these outcomes are interpreted largely on the basis of upstream transcriptional effects on Ngn3, they are consistent with the idea that misregulated epithelial morphology precludes efficient engagement of the endocrine differentiation program. We previously proposed that the interdependency between progenitor maintenance, cell differentiation, and trunk morphogenesis reflects a programmed niche framework that coordinates the birth of endocrine cells during organogenesis (Rieck et al. 2012).
The pancreas poses challenges in identifying niche environments. It was recently determined that pancreatic epithelial morphogenesis—unlike the branching morphogenesis observed, for example, in the lungs and salivary glands (Metzger et al. 2008; Hick et al. 2009)—proceeds through asynchronous branching and remodeling of an irregular, web-like plexus intermediate before yielding a typical arborized ductal system (Villasenor et al. 2010). As such, the process of trunk morphogenesis remains poorly understood. There also remains little understanding of how endocrine lineage flux is controlled spatiotemporally within the trunk. Here, we define three-dimensional (3D) morphological patterns that comprise trunk epithelial morphogenesis during the secondary transition and employ EdU pulse-chase analyses and endocrine progenitor mapping to pinpoint locations where endocrine birth is enriched. We found that endocrine progenitors are maintained in an epithelial “plexus state,” from which the bulk of the endocrine lineage is derived. Experimental perturbation of endocrine differentiation and Notch pathway activity uncovered how feedback interactions that are organized locally within the plexus state coordinate endocrine progenitor maintenance and differentiation and epithelial morphogenesis to ensure prolonged endocrine cell production. Our results assign niche-like properties to the plexus state and provide a foundation for understanding how genetic regulation of progenitor behavior interfaces with niche development and maintenance during pancreas development.
Results
Ngn3+ populations show nonrandom localization patterns within the trunk
To address whether nascent endocrine cells arise preferentially in different locations within the secondary transition trunk, we evaluated the localization of Ngn3+ cells relative to the Muc1+ epithelial lumen. Because the bulk of the endocrine mass at birth lies relatively interiorly (data not shown), we compared core and peripheral regions to assess whether the later endocrine mass distribution reflects an earlier bias in the location of newly born endocrine cells. As previously described, Ngn3+ cells at E15.5 were broadly distributed in a salt and pepper pattern across the Muc1+ lumen in both the core and periphery and were absent in Muc1+amylase+ proacinar tips (Fig. 1A,C; Zhou et al. 2007). At E17.5, significant portions of epithelium showed a reduction or lack of Ngn3+ cells (Fig. 1B,D). Diminished Ngn3+ cell numbers were evident in the core, especially where lumen diameters were large, and in the periphery where lumens joined with amylase+ tips. However, there remained frequent instances along the lumen where the density of Ngn3+ cells was comparable with the E15.5 stage. These data outline a nonrandom localization pattern of endocrine commitment from the trunk and suggest that endocrine cell birth might be coordinated with specific epithelial morphogenetic states.
Figure 1.
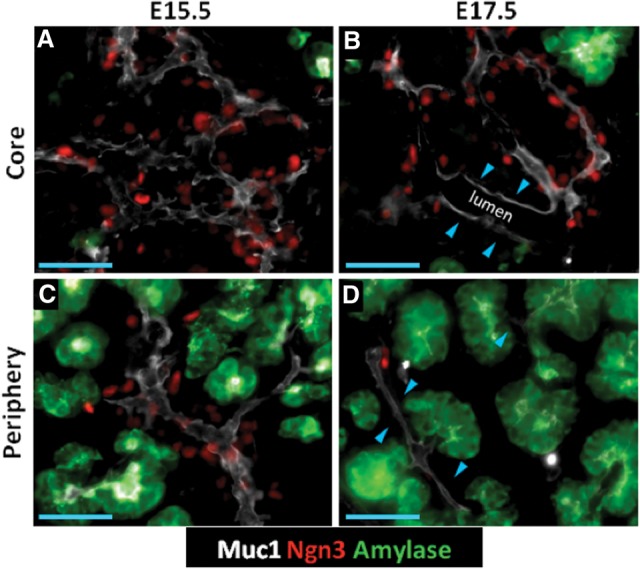
Ngn3+ populations show nonrandom localization patterns within the trunk. (A–D) Epifluorescence images of core and peripheral regions in 10-µm cryosections showing Muc1, Ngn3, and amylase. (A,C) Ngn3+ cells distributed over the Muc1+ lumen surface. (B,D) Regions of lumen where Ngn3+ cells are reduced in number. (Blue arrowheads in B) Enlarged lumen in the core; (blue arrowheads in D) lumen interconnecting amylase+ tips. Bars, 50 μm.
Plexus expansion and plexus-to-duct transformation in the organ core
As a first step toward analyzing relationships between endocrine differentiation and epithelial morphogenesis, we used a whole-mount and thick section-based approach to categorize epithelial states (derived via lumen morphology) existing as the trunk remodels from the initial plexus to the arborized ductal system (Fig. 2A). At E14.5–E15.5, consistent with a previous report (Villasenor et al. 2010), the majority of the core was composed of a complex, web-like plexus (Fig. 2B). Within the plexus, intersecting segments of epithelium varied in length and diameter (Fig. 2H). In traced representations, cell fields bounded by the Muc1+ lumen (represented as “holes” in the plexus web) varied in size and shape but showed a uniform heterogeneity across the core (Fig. 2K). There was no evidence of an extensive arborized ductal structure at these stages. We found little evidence of multibranched epithelia interconnecting proacinar tips except at the outer edges of the plexus. Most proacinar tips (demarcated by a bulb-like morphology and intense Muc1 positivity) appeared as small, individual tips interspersed throughout the plexus. We conclude that the principal epithelial state in the core from E14–15.5 is plexus.
Figure 2.

Epithelial morphogenesis comprises plexus remodeling in the core and epithelial branching in the periphery. (A) Composite 10× images of a 45-µm-thick cryosectioned dorsal pancreas at E16.75. Green and red dashed lines bound the core and periphery, respectively. (B–D) Muc1-labeled 35-µm-thick cryosections showing typical progression through plexus expansion and duct transformation in the core. (E–G) Epithelial branching in the periphery. (White dashed line) Outer organ boundary. (H–M) Confocal z-stacks show features of plexus (H,K), duct (I,L), and ductal branch (DB) (J,M) states. (I,J) Core (green dashed line) and periphery (red dashed line) are shown. Small, unclefted DBs in the core (blue arrowheads), clefted DBs in the periphery (yellow arrowheads in H), lobes of DBs (blue dashed line), Muc1+ lumen of proacinar tips (white arrowheads in J), and connecting lumen (red arrowheads in J) are shown. (K–M) Line traces of lumen from H–J. Individual segments of plexus (dotted light blue lines) and core duct states (red dashed line) are shown. Note the increase in lumen diameter and the continuous directionality across a representative 40× field of the duct state in L relative to the plexus in K. (Green arrowheads) Unlabeled cell fields (epithelial and parenchymal) bounded by Muc1 signal. (N,O) Traced representations of the processes of plexus-to-duct transformation in the core and epithelial branching in the periphery. Bars: B,E,H-J, 20 μm; C,F, 50 μm; A,D,G, 100 μm.
At E16.5, there was an expansion of the plexus state concomitant with the increase in organ size. This was accompanied by a transformation of the plexus into ductal states representing the beginnings of an arborized ductal system. Ductal states in the core were discernible by their increased lumen diameter and extended, continuous directional orientation compared with the web-like plexus (Fig. 2C,D). This “plexus-to-duct transformation” occurred asynchronously across the organ and was evident first with the appearance of the central duct (E16.5). Cells in the core ductal state showed measurably reduced Sox9 immunoreactivity and a flattening of cells and their nuclei (Supplemental Fig. 1A–F), consistent with an acquisition of a ductal phenotype (Grapin-Botton 2005). At E17–E18.5, connecting interlobular ducts were observed to form, oriented toward the periphery (Supplemental Fig. 1 G–I). This process remained incomplete even late into the secondary transition, and abundant plexus remained in scattered locations at E18.5 (Supplemental Fig. 2). These analyses show that trunk morphogenesis in the core is dominated by an initial phase of growth and expansion in a plexus state followed by plexus-to-duct transformation to generate the core of the ductal arbor (Fig. 2N).
Epithelial branch remodeling in the organ periphery
In contrast to the organ core, a pervasive epithelial branching process was evident in the periphery (Fig. 2A,E–G). From E14.5 to E18.5, proacinar tips exhibited a variety of clefted morphologies and appeared to grow and multiply progressively to generate lobes of interconnected ductal branches (DBs) linking multiple tips (Fig. 2J,M). Intervening DB segments multiplied and lengthened over time, at late stages forming CK19+DBA+ intralobular ducts and distal CK19+DBA− intercalating ducts (Supplemental Fig. 1J–L; Reichert and Rustgi 2011). DBs were most advanced in peripheral regions devoid of plexus but were less so in regions within or approaching the plexus. Taken together, we detect two disparate patterns of trunk morphogenesis during the secondary transition. There is a long-lived plexus expansion followed by plexus-to-duct transformation in the core, in contrast to pervasive epithelial branching in the periphery (Fig. 2N,O). These observations on the spatiotemporal and morphological features of plexus, duct, and branched (DB) epithelial states provide a basis to map correlations between endocrine lineage flux and states of epithelial morphology.
In vivo endocrine progenitor dynamics and establishment of the Ngn3:Sox9 ratio
In order to identify locations within the trunk that are enriched for endocrine progenitor activity, we devised a method, based on the ratio of [differentiating Ngn3+ cells]:[Sox9+ epithelial cells], to quantify the relative local flux from the Sox9+ trunk toward the Ngn3+ endocrine lineage. Sox9 expression is lost upon up-regulation of Ngn3 (Seymour et al. 2008; Kopp et al. 2011). The transient up-regulation of Ngn3 in turn indicates commitment to a post-mitotic endocrine precursor state (Supplemental Fig. 3A; Desgraz and Herrera 2009; Miyatsuka et al. 2011). Therefore, the Ngn3:Sox9 observed at a given time is largely determined by the temporal relationship between (1) the average cell cycle period of the Sox9+ population (epithelial growth/maintenance) and (2) the average duration of the Ngn3+ period of endocrine commitment (differentiation) (Supplemental Fig.3B,C). Provided the time frames for these parameters are comparable, the Ngn3:Sox9 ratio reflects the magnitude of endocrine differentiation (endocrine flux) from the trunk. To validate the Ngn3:Sox9 ratio, we adapted EdU pulse-chase methods (Salic and Mitchison 2008) to measure and relate progenitor replication and endocrine differentiation parameters in vivo (Supplemental Fig. 3D).
Dividing Sox9+ populations progress continuously through an estimated 12-h cell cycle period
When Sox9+ cells enter mitosis, we invariably observed subcellular segregation of Sox9 immunoreactivity away from condensed DAPI+ DNA. These Sox9+ mitotic figures (MFs) are EdU+ in cells that have recently undergone S phase (Fig. 3A–C). To quantify the average cell cycle period, we timed the appearance and disappearance of pulse-administered EdU within the DAPI+ DNA of Sox9+ progenitors as they passed though rounds of mitosis (Fig. 3G; Fox et al. 2011). Because EdU bioavailability after single-pulse administration is short (distribution half-life 1.4 min ± 1.7 min; elimination half-life 24 min ± 2.9 min at 100 mg/kg) (Cheraghali et al. 1994) and because the MF state represents a short, transient event during the cell cycle, this method estimates cell cycle periods at a population level.
Figure 3.
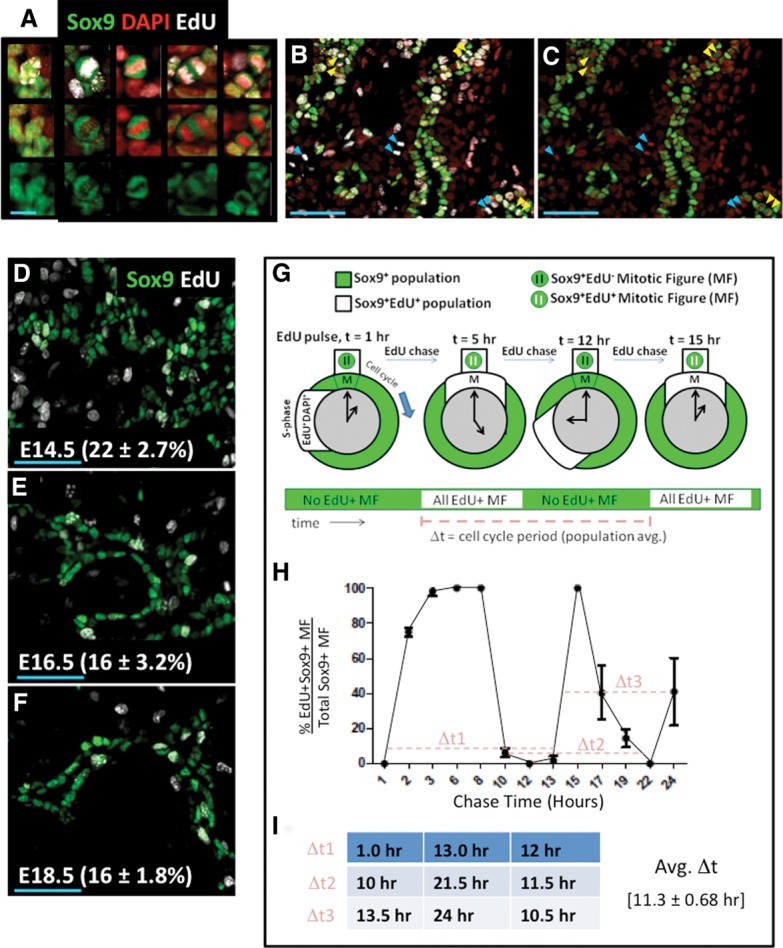
EdU pulse-chase analysis measured average cell cycle period in Sox9+ cells. (A) Individual Sox9+EdU+ cells captured before and during mitosis. (Left to right) Sox9+ cells not in mitosis and MFs in prophase, metaphase, anaphase, and cytokinesis. Bar, 10 μm. (B,C) MFs observed in a representative 20× field at E14.5, 3 h after EdU injection. (Yellow arrowheads) Sox9+EdU+ MFs; (cyan arrowheads) Sox9–EdU+ MFs; (doublet arrowheads) MFs after DNA segregation into daughters. Bars, 50 μm. (D–F) One-hour S-phase indices were similar from E14.5–E18.5. Bar, 50 μm. (G) Diagram of EdU pulse chase in replicating Sox9+ populations. Sox9+ cells undergoing S phase were labeled by EdU. EdU+Sox9+ populations (white) undergo and complete mitosis (t = 5 h) before undergoing a subsequent round of mitosis (t = 12–15 h). Rounds of mitosis can be tracked by monitoring waves of EdU positivity in the Sox9+ MF states (green and white bar over time). Time differences (“Δt”s) are determined from intervals between waves of EdU+ MFs. (H) Twenty-four-hour EdU pulse-chase analysis of cell cycle period in Sox9+ populations, with “Δt”s represented by a dashed light-red line. (I) Calculation of average Δt from pulse-chase measurements in H. Error bars are SEM.
In the Sox9+ epithelium, S-phase indices determined after 1 h of EdU pulse are similar at E14.5, E16.5, and E18.5 (22% ± 2.7%, 16% ± 3.2%, and 16% ± 1.8%, respectively) (Fig. 3D–F). We chose stage E14.5 for a 24-h EdU pulse-chase analysis of the Sox9+ cell cycle period. All Sox9+ MFs were EdU− 1 h after EdU pulse (Fig. 3H), indicating that cells incorporating EdU at S phase had not yet entered M phase. At 2 h, the percentage of Sox9+EdU+ MFs remained unchanged, as did the S-phase index in the Sox9+ population, consistent with a ≤1-h bioavailability of EdU under these conditions. Between 2–3 h after EdU pulse, essentially all of the detected Sox9+ MFs became EdU+ (75.3% ± 2.7% at 2 h; 98.3% ± 2.5% at 3 h), consistent with an asynchronously cycling Sox9+ population incorporating EdU at different periods along the duration of S phase and then proceeding over time into mitosis. EdU saturation in the Sox9+ MFs was maintained for ∼5 h before decreasing essentially to 0% between the 8- to 13-h period (6.3% ± 2.5% at 8 h; 0% ± 0% at 12 h; 1.7% ± 2.6% at 13 h), consistent with the EdU+Sox9+ population completing M phase of the cell cycle. Subsequent time points (13–15 h) showed rapid and saturating re-entry of EdU+ DNA in Sox9+ MFs. After this population passed through mitosis (indicated by a second reduction in EdU positivity in Sox9+ MFs), a third M phase was detected at 24 h. At late time points, EdU saturation of the Sox9+ MF population was shorter in duration, reflecting an expected DNA replication-dependent dilution of EdU signal. These results demonstrate that EdU pulse chase reproducibly detects a labeled portion of the Sox9+ population moving through three consecutive and largely coherent cell cycle periods with average of ∼11.3-h ± 0.68-h time intervals.
Endocrine-committing populations take an average 12 h to move through the Ngn3+ state
To determine the average duration of the Ngn3+ period of endocrine commitment, we used EdU pulse chase to monitor EdU+ DNA from initial incorporation in the replicating Ngn3–Sox9+ progenitors through marker-defined, chronological stages of endocrine commitment (Fig. 4A–A′′; Gouzhi et al. 2011). These include the initial trunk-resident Sox9+Ngn3+ state, the post-delamination Sox9− Ngn3HI state, and the late Sox9−Ngn3LO state (Seymour et al. 2008; Kopp et al. 2011; Shih et al. 2012). The baseline EdU+ index in all Ngn3+ states at 1 h of EdU pulse was ∼1%, consistent with their largely post-mitotic status. At 2 h of EdU pulse, the percentage of Sox9+Ngn3+EdU+ cells remained unchanged (Fig. 4F), indicating that Sox9+Ngn3– cells incorporating EdU at S phase had not yet progressed into the initial Sox9+Ngn3+ state. At 3 h, we observed an accumulation of EdU+ DNA in the Sox9+Ngn3+ population. At 6 h, the EdU+ index in Sox9+Ngn3+ cells reached 22% ± 2.7%, as expected, because this quantity reflects the baseline EdU+ index in Ngn3−Sox9+ progenitors from which Ngn3+ cells arise. Similar accumulation of EdU in the Sox9−Ngn3HI state reached 22% at 10 h, followed by the late Sox9−Ngn3LO state at 15 h (Fig. 4B,C). Finally, at 17 h, EdU+ DNA was detected in Pdx1HINgn3− cells, which represent a state (likely β cell, the principle cell type produced at this stage) arising after down-regulation of Ngn3 (Fig. 4D,E; Supplemental Fig. 4A–F; Gradwohl et al. 2000; Wang et al. 2010). By measuring the time difference (Δt) between the curves demarcating the early and late Ngn3+ state (between Sox9+Ngn3+ and Sox9−Ngn3LO) bounded within the range defining the minimum and maximum baselines for EdU incorporation in the Ngn3−Sox9+ and Sox9+Ngn3LO cell states (Sox9+Ngn3+ ≤ y ≥ Ngn3–Sox9+) (Fig. 4F, gray shaded area), these data indicate an average duration of ∼12.3 h ± 1.03 h (Fig. 4G) for the Ngn3+ period of endocrine commitment.
Figure 4.

EdU pulse chase measures duration of the Ngn3+ period of endocrine commitment. (A–A′′) Pulsed EdU was incorporated in Sox9+ cells at S phase and chased (EdU+ DNA in white) through marker-defined stages of Ngn3 positivity. (B,C) EdU captured in cells of various Ngn3+ states. (Yellow arrowheads) Early Sox9+Ngn3+ cells; (red arrowheads) Sox9–Ngn3HI cells; (white arrowheads) late Sox9−Ngn3LO cells. (D,E) EdU in Ngn3−Pdx1HI cells (blue arrowheads). (F) Time course for EdU pulse-chase analysis of the Ngn3+ period. The graph indicates the percentage of EdU+ cells in Sox9+Ngn3+ (yellow line), Sox9−Ngn3HI (red line), or Sox9−Ngn3LO (orange line) states at the time points indicated. The blue dashed line shows a 1-h EdU incorporation baseline for Sox9+Ngn3− cells (22% ± 2.7%). (Black dashed line) One-hour EdU incorporation baseline for Sox9+Ngn3+ cells (8.5% ± 2.8%). Blue shading with arrows indicates the period when EdU+ DNA becomes increasingly observed in Ngn3−Pdx1HI populations (indicating the end of the Ngn3+ period). The gray shaded area indicates the domain and range of data points used to estimate the Ngn3+ period. (G) Calculation of Δt (brown dashed lines in F) from the lines demarcating Sox9+Ngn3+ and Sox9−Ngn3LO cell states. Error bars indicate standard deviations. N = 3 pancreata. (H–H′′) Derivation of endocrine yield as a quantitative measure of the relative magnitude of endocrine differentiation from the trunk domain.
These analyses indicate that the Sox9+ cell cycle period and the duration of the Ngn3+ period of endocrine commitment are similar (11.3 h ± 0.68 h and 12.3 h ± 1.03 h, respectively). Therefore, the Ngn3:Sox9 ratio estimates quantitatively the relative magnitude of endocrine differentiation occurring from the trunk at a given sampling time. For simplicity, we converted Ngn3:Sox9 here to the fractional term “endocrine yield” (given as total Ngn3+ cells/total Sox9+ cells) (Fig. 4H–H′′).
Endocrine yield is robust throughout the secondary transition
Previous measurements from whole pancreas mRNA or from limited quantifications of Ngn3+ cell numbers suggest that endocrine birth peaks around E14.5–E15.5 and reduces thereafter (Gradwohl et al. 2000; Schwitzgebel et al. 2000; Villasenor et al. 2008). However, these measurements do not take into account the direct relationship between cells undergoing endocrine lineage commitment (Ngn3+) versus maintenance and growth as Sox9+ progenitors. To address this, we quantified endocrine yield in sections representing ∼33% of the dorsal pancreas from the E14.5 to E18.5 stages as a bulk measure of endocrine differentiation dynamics. We detected essentially a constant endocrine yield throughout the E14.5–E17.5 stages; this trend reversed—relatively sharply—only at E18.5 (Fig. 5A). At all time points, we detected no significant change in the relative number of early (Ngn3+Sox9+) versus late (Ngn3+Sox9−) endocrine-committing cell states, indicating that new endocrine cells are being generated at all stages (Supplemental Fig. 5). These results show that Sox9+ trunk populations continuously allocate a large fraction of their progeny toward the endocrine lineage over essentially the entire mid to late gestational period (Supplemental Fig. 6A–L).
Figure 5.

Endocrine differentiation is enriched in the plexus state. (A) Bulk endocrine yields determined from analysis of 10-µm sections. Thirty percent of the dorsal pancreas was scored. Error bars are SEM. (*) P = 0.1161; (**) P = 0.0249, Student's t-test. (B) Comparison of endocrine yield in the plexus, duct, and DB states. Each point represents endocrine yield summed from individual 35- to 50-µm-thick 40× z-stacks. E14.5–E18.5 plexus versus DB, P < 0.0001; E17.5 and E18.5 plexus versus duct, P < 0.0001; E16.5 plexus versus duct, P = 0.0019. Error bars are SEM; Student's t-test. Bulk endocrine yield as determined in A (black dashed line). (C–J) Representative images of Ngn3+ cells in relation to the plexus (blue dashed box), duct (red dashed box), and DB (green dashed box) states. Bars, 20 μm.
Endocrine differentiation is enriched in the plexus
To ascertain whether Ngn3+ populations have specific localization patterns within the remodeling epithelium, we compared endocrine yield between the plexus, duct, and DB states from E14.5 to E18.5. Epithelial cells in the plexus, duct, and DB states showed similar Sox9 immunoreactivity (Supplemental Fig. 7A–F). Low Sox9 signal was observed in proacinar tip cells, which were excluded from our analysis. From E14.5 to E18.5, endocrine yield was highest in the plexus and significantly lower or absent in ducts and DBs (Fig. 5B–J). Endocrine yields measured selectively from samples of the plexus consistently exceeded those measured from bulk sectional analyses, indicating that endocrine yield is enriched in the plexus. At E18.5, there were scattered yet extensive remainders of the plexus, which were invariably associated with large numbers of Ngn3+ cells, indicating that a high level of endocrine differentiation is consistently maintained in the plexus, independent of stage (Supplemental Fig. 8A–D). Sharp boundaries demarcating high and low endocrine yields were commonly observed at the interface between the plexus and duct or DB states, respectively, suggesting a rapid cessation of endocrine cell birth concomitant with loss of plexus morphology. These data put forth the view that endocrine differentiation is robust and enriched within the plexus during the entire secondary transition and is rapidly down-regulated upon plexus-to-duct transformation in the core or engagement of epithelial branching in the periphery.
Notch-responsive endocrine progenitors are enriched in the plexus state
Consistent with a proposed model for lateral Notch pathway activation by Ngn3-producing cells (Metzger et al. 2012; Shih et al. 2012; Qu et al. 2013), genetic ablation of Ngn3, as reported previously (Magenheim et al. 2011a), caused loss of Hes1 (a Notch transcriptional effector) signal in Sox9+ epithelial cells, suggesting that Notch pathway activity is Ngn3-dependent and is engaged locally in the plexus state (Supplemental Fig. 9A–D). Consistent with this notion, immunodetection of Hes1 showed relatively pervasive expression in the plexus compared with regions of fully resolved core ducts and peripheral DBs, suggesting that Notch-responding cells are enriched within the plexus (Fig. 6A,B). Because Notch–Hes1 signaling blocks endocrine differentiation, we hypothesized that Sox9+ populations in the plexus would show an increased endocrine differentiation response, compared with duct and DB states, under conditions where Notch is inhibited. To test this, we compared endocrine yield in E17.5 embryos from pregnant dams injected each day for 2 d with the γ-secretase inhibitor DBZ, which blocks Notch signaling (Milano et al. 2004; van Es et al. 2005). Similar to a previous report showing dose-dependent reductions in Hes1 and Sox9 expression in Notch-inhibited pancreatic explants (Shih et al. 2012), increasing DBZ doses elicited reproducible, progressive reductions in pancreatic Hes1 and Sox9 expression, as measured by quantitative RT–PCR (qRT–PCR). Hes1 decreased at a lower DBZ dose (0.15 ± 0.06 and 0.09 ± 0.07 at 12 and 20 µmol/kg DBZ relative to control, respectively) as compared with Sox9 (0.88 ± 0.18 and 0.43 ± 0.18 at 12 and 20 µmol/kg DBZ relative to control) (Fig. 6H). We chose the 12 µmol/kg DBZ condition for analysis of endocrine yield due to the minimal effect registered on Sox9. Endocrine yield from the plexus was robustly increased in response to DBZ (Fig. 6C–F) and consistently approached a near threefold increase relative to the vehicle-treated condition. Assuming the absence of a proliferation effect on Sox9+ cells, this increase indicates that approximately half of the plexus population (endocrine yield = 1 indicates 1:1 Ngn3:Sox9) defaults to the endocrine lineage under these conditions (Fig. 6G). In contrast, Sox9+ cells in the duct and DB states showed marginal and variable responses to DBZ, indicating that Sox9+ cells in these states are largely refractory to endocrine differentiation. These expression data and inhibition studies suggest that the plexus state is enriched for a Notch-responsive Sox9+ progenitor pool that is poised to adopt the endocrine lineage under normal conditions.
Figure 6.

Notch-responsive progenitors are enriched in the plexus state. (A) Detection of Hes1 relative to the Muc1+ duct state (yellow arrowheads), plexus state (blue arrowheads), and DBs (green arrowheads). Bar, 50 μm. (B) Mean nuclear intensity of Hes1 signal in individual Sox9+ cells. N = 102 plexus; N = 91 DBs, ([*] P < 0.0001); N = 52 duct (P < 0.0001) from three separate z-stack volumes. Error bars are SEM; Student's t-test. (C–F) Representative images of Muc1 and Ngn3 in the plexus and duct states under vehicle or DBZ treatment. Blue arrowheads delineate the plexus in C and D, yellow arrowheads indicate the surface of ductal states in E and F, and dashed magenta lines delineate locations where the Ngn3-rich plexus intersects with the duct state. Bar, 20 μm. (G) Endocrine yield in the plexus, duct, and DB states under vehicle or DBZ treatment. Each point represents endocrine yields summed from individual 40× z-stack volumes (∼35–50 µm thick). N = 11 plexus; N = 12 duct; N = 15 DB in vehicle control. N = 16 plexus; N = 11 duct; N = 15 DB in the DBZ-treated group. n = 3 for both conditions. Error bars are SEM. (**) P < 0.0001; (***) P = 0.0149; (****) P = 0.0013, Student's t-test. (H) Normalized gene expression for Sox9 and Hes1 under control, Ngn3-deficient, and DBZ-treated conditions. Error bars are SEM. n ≥ 3 for each condition. (*) P = 0.0373. (I,J) One-hour EdU indices in Sox9+ populations under control or DBZ treatment. Error bars are SEM. n = 3.
Sox9+ cell replication is uncoupled from Ngn3-dependent Notch inhibition of endocrine differentiation
Notch signaling has been linked to the regulation of mitogenic cell cycle progression (Kageyama et al. 2008; Pierfelice et al. 2011; Ninov et al. 2012). We analyzed whether the Notch function in maintaining the undifferentiated progenitor state is coupled with the maintenance of proliferative growth in Sox9+ cells. We evaluated EdU uptake in embryonic pancreata taken from DBZ-treated dams. For both the 12 and 20 µmol/kg doses, we observed no change in the proportion of Sox9+ cells replicating DNA after 2 d of DBZ administration (Fig. 6I,J). Because these DBZ doses reduced Notch activity by an amount similar to or beyond that caused by Ngn3 deficiency (Fig. 6H), we conclude that bulk epithelial proliferation behaviors are not significantly affected by reduced Notch activity under these conditions.
Ngn3 deficiency causes reduced Sox9+ progenitor replication and precocious remodeling of the plexus
To evaluate cell-nonautonomous functions for Ngn3 in regulating the growth of the epithelial progenitor pool, we compared cell replication behaviors in Ngn3EGFP/+ and Ngn3EGFP/EGFP pancreata. Analysis of S-phase indices indicated a 50% decrease in EdU labeling of Sox9+ progenitors when Ngn3 is lost. This decrease was evident at E13.5, when the vast majority of the trunk is in the plexus state, and also at E16.5, when plexus-to-duct transformation begins in the organ core (Fig. 7A–C). This reduction in EdU labeling was not a general feature of all pancreatic epithelial cells. S-phase indices in proacinar cells were unchanged, and because they represent the major cell type during mid to late gestation, no gross organ hypoplasia was evident at any stage (Supplemental Fig 10A–C; data not shown). These results demonstrate a previously unreported function for Ngn3 in supporting robust replication in the Sox9+ trunk that is, based on the DBZ treatment results above, largely independent of Notch pathway activity.
Figure 7.
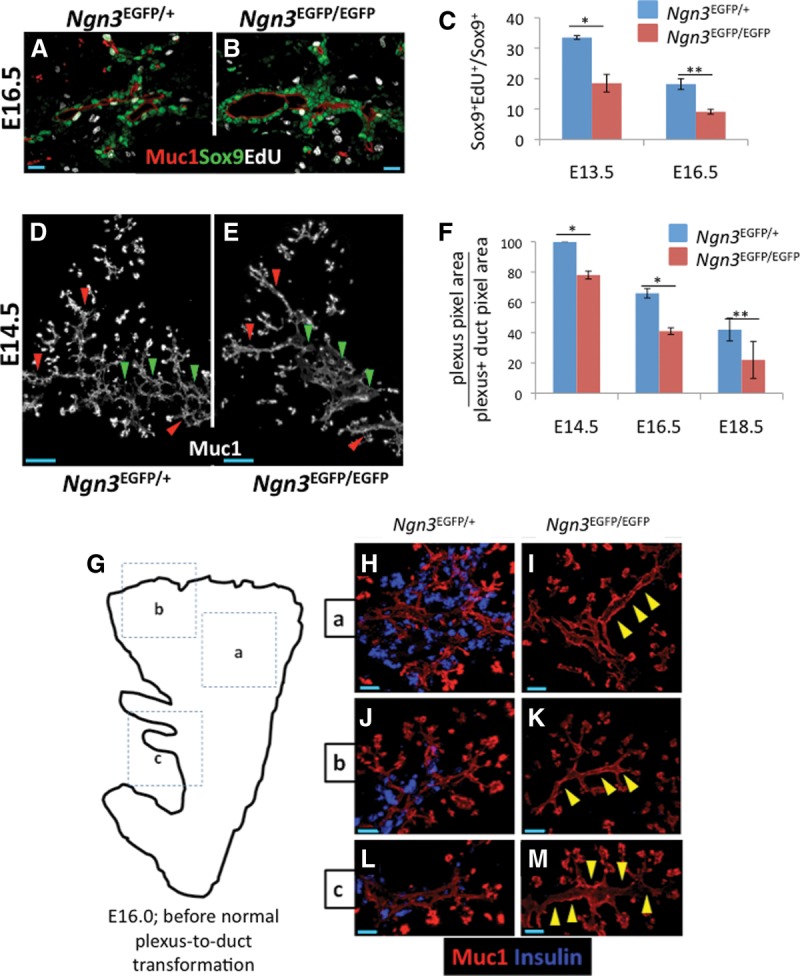
Ngn3 deficiency causes reduced cell replication and precocious loss of the plexus state. (A,B) S-phase indices in Sox9+ cells from Ngn3EGFP/+ and Ngn3EGFP/EGFP samples. (C) Quantification of A and B (E16.5) plus measurements at E13.5. Error bars are SEM. n = 2 E13.5 ([*] P = 0.0277); n = 3 E16.5 ([**] P = 0.0048) with 30% of dorsal pancreas scored, Student's t-test. (D,E) Dorsal pancreas from E14.5 Ngn3EGFP/+ and Ngn3EGFP/EGFP samples at E14.5. (Red arrowheads) Regions where the plexus is lost in Ngn3EGFP/EGFP animals compared with control; (green arrowheads) core locations where the plexus becomes dysmorphic in Ngn3EGFP/EGFP animals compared with control. (F) Quantification of the plexus-to-duct transformation in core Ngn3EGFP/+ and Ngn3EGFP/EGFP pancreata. Error bars are SEM. n = 3 all stages. (*) P < 0.01; (**) P = 0.119. The entire dorsal pancreas core was analyzed in serial thick sections (40 µm). (G) Analysis of precocious loss of the plexus at E16.0. (G) Regions a, b, and c in the cartoon correspond to the images in H–M. (H–M) Muc1 and insulin immune detection in similar locations in Ngn3EGFP/+ and Ngn3EGFP/EGFP littermates. Bars: A,B, 20 μm; D,E, 50 μm; H–M, 40 μm.
Given the reduction in progenitor replication observed under Ngn3 deficiency, we assessed potential defects in the maintenance of the plexus by comparing the time course for Ngn3-deficient plexus remodeling into the duct and DB states. Analyses at E14.5 confirmed previously reported increases in lumen diameters and moderate decreases in peripheral branching (Fig. 7D,E; Magenheim et al. 2011a). Surprisingly, we also found frequent DB-like structures emerging in peripheral locations normally comprised of the plexus at this stage, indicating a precocious remodeling of plexus morphology into nonplexus states under Ngn3-deficient conditions. At E16.0, a time point just before the plexus normally undergoes transformation into core duct and peripheral DB states, we observed multiple regions where plexus morphology was diminished or completely lost relative to control (Fig. 7G–M). Consistent with this, the relative area of the plexus compared with duct-like states was significantly decreased at E14.5, E16.5, and E18.5 in Ngn3-null tissue (Fig. 7F). These results indicate that Ngn3 functions to not only determine a normal plexus morphology but also maintain and propagate the plexus by limiting its remodeling into nonplexus states.
Late stage autonomous epithelial remodeling in the Ngn3-deficient plexus
While a limited amount of precocious remodeling and alteration of the Ngn3-null plexus morphology was observed at midgestation, there was no grossly aberrant phenotype at late gestation, suggesting that the mechanisms guiding epithelial remodeling could eventually override the earlier defect. Consistent with this, Muc1 and insulin (to label the endocrine cell mass derived from the epithelium) detection in equivalent organ regions from E14.5 to E17.5 showed a progressive normalization of the Ngn3-null epithelium (Supplemental Fig. 11A–F). Specifically, the expansive enlargements of Ngn3EGFP/EGFP plexus lumens seen at E14.5 underwent remodeling toward a morphology largely indistinguishable from control by E17.5. Furthermore, the previously reported moderate reduction in peripheral branching in Ngn3-nulls (Magenheim et al. 2011a) was also corrected (Supplemental Fig. 11M,N). The plexus morphology at E12.5 was similar between Ngn3EGFP/+ and Ngn3EGFP/EGFP (data not shown), indicating that the perturbed morphology of the Ngn3-deficient plexus manifests most severely during the peak of endocrine (β-cell) birth (Johansson et al. 2007). These analyses show that a late stage process of autonomous epithelial remodeling occurs in the absence of Ngn3-dependent differentiation, delamination, and Notch-regulated processes.
Discussion
Understanding how niche environments direct progenitor activities is fundamental to uncovering the basic principles underlying complex organ formation and function and predisposition to disease. In studying endocrine progenitor activity during the secondary transition of pancreatic organogenesis, we discovered that endocrine progenitors are largely constrained within an epithelial plexus state. Although the plexus is transient, it is maintained through all stages of the secondary transition and is as productive at generating endocrine cells during late gestation as it is at earlier stages. Genetic and pharmacological interference tests indicate that endocrine progenitor expansion, differentiation, and morphogenesis processes are coordinated by local feedback effects within the plexus. These effects derive largely from differentiating Ngn3+ cells but can be uncoupled on the basis of Ngn3-dependent differentiation/delamination functions supporting progenitor replication and Notch-dependent functions in maintaining an undifferentiated progenitor state. Together, these feedback interactions serve to propagate the plexus and the large numbers of endocrine progenitor cells maintained therein late into gestation. Ultimately, as the plexus remodels into the duct and DB states, endocrine differentiation and progenitor maintenance behaviors are reduced and then lost. We propose that the epithelial plexus functions as a niche environment where endocrine progenitors are assembled and maintained and where local differentiation-dependent feedback interactions are deployed to direct the genesis of the endocrine pancreas (Supplemental Fig. 12).
Distinct processes of plexus-to-duct remodeling and epithelial branching in the trunk
Our characterization of pancreatic epithelial remodeling indicates distinct modes of morphogenesis in largely separate zones of the organ. In the core, plexus expansion and growth from E12.5 to E15.5, accompanied by plexus-to-duct transformation from E16.5 to E18.5, represent the principal mode by which ductal tissues analogous to the trunk and main limbs of a tree are generated. In the periphery, however, an “epithelial branching” mode remodels the epithelium, apparently by a clefting and outgrowth process, to eventually generate tightly packed peripheral lobules of interconnected DB structures (Fig. 2). This dual-mode program, especially the major contribution from plexus expansion and resolution, diverges greatly from the classical and more stereotypically defined branching morphogenesis involved in the growth of other endodermal organs (Hogan 1999; Metzger et al. 2008; Hick et al. 2009).
Because the plexus remodeling process occurs asynchronously across the organ, it will be important to determine what influences (cellular or otherwise) cause some plexus regions to undertake expansion while remaining unresolved (and in a high endocrine yield state), while others begin transformation toward the mature duct-like states. We did not find evidence at any stage of plexus resolution that would suggest that remodeling is accomplished through breakage and regression of epithelial segments, as reported for vascular plexus remodeling (Franco et al. 2015). Rather, we observed an apparent progressive consolidation of epithelial plexus segments that effectively reduces the complexity of, and eventually eliminates, the plexus web to generate the final ductal tree. Our analysis of Ngn3−/− pancreata allows us to rule out a model in which plexus remodeling is primarily driven by epithelial cell departure (delamination) of Ngn3+ endocrine precursors. While Ngn3-dependent processes are clearly important for regulating and propagating a “normal” plexus state, the observation of the late stage corrective remodeling demonstrates that the epithelial remodeling program, albeit with substantial delay, eventually enforces a relatively normal morphogenesis even in the absence of Ngn3. Therefore, in normal tissue, we propose that the function of Ngn3 in maintaining and propagating the progenitor-rich plexus is balanced with an opposing and apparently self-organizing epithelial morphogenesis process that eliminates the plexus intermediate in favor of the typical ductal structures of the mature epithelium. It remains to be determined how other known regulators of pancreatic epithelial morphogenesis and growth, especially those derived from epithelium-extrinsic sources such as the mesenchyme (Hart et al. 2003; Kobberup et al. 2010; Landsman et al. 2011) and vasculature (Magenheim et al. 2011b; Villasenor and Cleaver 2012), contribute to localized epithelial remodeling patterns during plexus growth, plexus-to-duct transformation, and epithelial branch specification and outgrowth. We propose that the detailed analysis of the dual-mode morphogenesis program reported here should enable future connection of such extrinsic cues to specific epithelial remodeling outcomes.
Endocrine progenitor dynamics during the secondary transition
Our EdU pulse-chase and endocrine yield analyses quantified in vivo the balance between endocrine differentiation and maintenance/growth of the epithelial progenitor pool. Our population-based measurements leverage a high temporal resolution enabled by a short bioavailability of EdU, a known injection time, and a set of directly detected and marker-defined cell states that arise during progenitor replication and endocrine commitment (Figs. 3, 4). We estimate ∼12 h for both the cell cycle period and Ngn3 protein-positive period in vivo. Consistent with recent reporter-based single-cell live-imaging analyses in explants (Kim et al. 2015), the rapid EdU acquisition in Ngn3+ populations indicates that a large portion of endocrine-committing cells acquire Ngn3 immunoreactivity shortly after a division event (Fig. 4). Our measurements suggest that, as a population, upward of a one-third fraction of progenitors in the plexus is allocated to the Ngn3+ state approximately every 12 h (Fig. 5). These data are consistent with a highly processive plexus state from which endocrine cells are continuously born. The increased understanding of endocrine progenitor dynamics in the context of plexus remodeling should provide a foundation for deeper analyses of the distribution, timing, potency, and molecular regulation of individual differentiation events.
Notch and endocrine differentiation-mediated feedback effects regulate distinct endocrine progenitor behaviors
Our finding that Hes1 production is maintained locally within the plexus even at late gestational stages (Fig. 6) leads to interesting questions regarding how duct versus endocrine cell fate allocation might be dynamically influenced by Notch activity. Our data showing that Hes1, similar to Ngn3, is enriched in the plexus and that Notch inhibition by DBZ increases the portion of the plexus cell population activating Ngn3 expression fit a model in which Notch-based signals derived locally from Ngn3+ cells maintain epithelial cells in an undifferentiated and Notch-responsive state. Ultimately, Sox9+ cells of the duct state epithelium (that is, in the core ducts and peripheral DB) down-regulate Hes1 and Sox9 and become a poor source of Ngn3+ cells both normally and under Notch-inhibited conditions, consistent with the idea that nonplexus state cells have truly entered a ductal differentiation program. Our observations thus begin to outline spatial variations in the level of Notch pathway activity and the differentiation response to perturbing Notch, which are correlated with distinct states of epithelial morphology arising during organogenesis. Identifying markers that distinguish states of pro-duct specification versus undifferentiated bipotent progenitors will likely aid in defining the non-cell-autonomous cues that instruct duct specification or maintain the bipotent pool, perhaps as obligatory features of developmental coordination in the plexus.
We observed profound reductions in progenitor replication under conditions where Ngn3 is inactivated but not when Notch signal processing is inhibited. While spatiotemporally more precise Notch perturbation may uncover roles in regulating the replicative amplification of endocrine progenitors in mice, our results raise the possibility that a delamination function for Ngn3 may determine replication rates in the epithelial population. Indeed, studies in Drosophila demonstrate that cell delamination limits tissue overcrowding and promotes epithelial growth (Marinari et al. 2012), suggesting that biomechanical mechanisms and/or pathways regulating cell shape-dependent or density-dependent growth (Huang and Ingber 1999) could be critical determinants of pancreatic progenitor growth and replication.
Intrinsic versus feedback-based regulation of endocrine lineage allocation, progenitor maintenance, and plexus morphogenesis during the secondary transition
In contrast to growth compensation programs in the liver, the pancreatic growth program is regulated intrinsically by the number of MPCs initially allocated to the pancreatic anlagen (Stanger et al. 2007). We speculate that an internally regulated, feedback-rich process of plexus morphogenesis from its initial establishment to maturation as a ductal arbor could explain how endocrine lineage output is “predetermined” by founder MPC number. Endocrine yield is robust within the plexus at all stages but is rapidly reduced upon its transformation into nonplexus states. The initial number of MPCs allocated to the initial plexus state could influence how long the plexus is maintained and thereby prolong or shorten the period of endocrine lineage allocation. Of relevance is the effect of ectopic Nkx6.1 expression in pancreatic MPCs. During stages concomitant with plexus formation, Nkx6.1 antagonizes the protip gene Ptf1a, diverting MPCs toward bipotent trunk progenitors (Schaffer et al. 2010). This transcriptional antagonism effect on tissue compartment allocation is Notch-regulated, providing a potential link between the timing and/or levels of Notch in the Nkx6.1+ MPCs during initial plexus formation and the final endocrine cell output (Afelik et al. 2012, Qu et al. 2013). Current translational objectives regarding the differentiation of pluripotent cells toward the endocrine cell fate in vitro could benefit from investigating how the size of the “initiating plexus” and the duration of its maintenance impact the final size and composition of the endocrine pancreas and whether these parameters can be controlled by modulation of Notch, Nkx6.1, and Ptf1a.
Our data put forward the possibility that endocrine differentiation dynamics, in terms of the rate of endocrine flux and/or the specific type of endocrine cells produced, could be guided by epithelium-intrinsic morphogenetic influences. Of particular interest to endocrine subtype specification, a previous study (Johansson et al. 2007) showed that competence windows for the differentiation of endocrine cell subtypes are regulated by factors intrinsic to the pancreatic epithelium. Such windows of competence could be connected to specific tissue-level or cell-biological alterations associated with plexus formation, expansion, and remodeling phases. (The α-cell competence window could correlate with initial plexus formation [E9.5–E12.5]; β-cell birth, more with the mid-gestation plexus expansion phase [E12.5–E15.5]; and the later production of δ/ε/PP cells with plexus-to-duct transformation [E16.5–E18.5].) We envisage the future testing of the idea that specific and perhaps relatively subtle perturbations to the plexus morphogenesis program might alter the epithelial environment in ways that influence the number and type of endocrine cells born.
Linked programs of endocrine progenitor maintenance, differentiation, and morphogenesis
Emerging reports suggest that some aspects of endocrine progenitor differentiation and proliferative expansion are dependent on molecular determinants of cell and tissue morphology. For instance, a requirement for Cdc42-mediated apicobasal polarization has already been linked to the generation of an endocrine differentiation-competent progenitor pool (Kesavan et al. 2009) and could thus be considered as a central contributor to the generation of the plexus. Additionally, ablation of Strad13, a negative regulator of RhoA–ROCK–nmMyoII, reduces the size and replicative activity of pancreatic MPCs (Petzold et al. 2013), and defects in planar cell polarity are associated with reduced Ngn3+ cell numbers (Cortijo et al. 2012). While the detailed mechanistic underpinnings causing defective endocrine lineage development in these cases remain unclear, the picture emerging is that insights into how endocrine progenitors and committed endocrine precursors are efficiently maintained and generated, respectively, may be gleaned from characterizing, with increased spatiotemporal resolution, how cytoskeletal dynamics, cell polarity, actomyosin contractility, and/or the trafficking and distribution of receptors such as Notch functionally link cell and tissue morphogenesis processes with cell fate determination.
Materials and methods
Mice
Ngn3EGFP knock-in mice from Guoqiang Gu (Vanderbilt University) were genotyped as described (Lee et al. 2001). Ngn3EGFP/+ and Ngn3EGFP/EGFP embryos were phenotyped by EGFP fluorescence and lack of endocrine cells. Wild-type mice were of mixed genotypes. All protocols were approved by the Vanderbilt University Institutional Animal Care and Use Committee. See Supplemental Table 3 for a list of genotyping primers.
Immunodetection
Embryonic pancreata were paraformaldehyde-fixed (4%) for 4–6 h at 4°C. For cryosectioning, samples were sucrose-equilibrated (30%) overnight at 4°C and OCT-embedded (Tissue-Tek). Thick cryosections (30–45 μm) were obtained on a Leica CM3050 S. For whole mounts, pancreata were manually divided into core and peripheral regions and processed as described (Ahnfelt-Ronne et al. 2007). See Supplemental Table 2 for a list of the antibodies used.
qRT–PCR
Total RNA was TRIzol-isolated from dorsal pancreata, and cDNA was synthesized with iScript cDNA synthesis kit (Bio-Rad) for PCR in a Bio-Rad CFX96 with SsoFast EvaGreen Supermix (Bio-Rad). Expression level was normalized to GAPDH by the ΔΔCt method. See Supplemental Table 3 for a list primer sequences.
DBZ and EdU administration and quantification
DBZ (Cayman Chemical) was resuspended finely in ME4M slow-delivery vehicle (Methocell) using a motorized pestle and was injected intraperitoneally. EdU (10 mg/kg) (Life Technologies) was given intraperitoneally and was detected by the Click-iT Plus EdU imaging kit (Molecular Probes). S-phase indices and percent EdU incorporation were counted manually.
Image acquisition and 3D reconstructions
Confocal images were from a Zeiss LSM 510 META (details are in the figure legends), and 3D reconstructions were rendered using Imaris software.
Quantification and statistical analyses
Muc1+ plexus and duct areas were traced manually as indicated in the figure legends. Pixel areas were obtained using ImageJ. All sectional analyses covered ∼30% of the dorsal pancreas. For endocrine yield measurements within the plexus, duct, and DB states, the morphology types were based on the Muc1 lumen structure. Sox9+ and Ngn3+ cells were counted manually. Muc1+ lumen that was not unambiguously scorable as plexus, duct, or DB was not analyzed. For Hes1 immunofluorescence, mean pixel intensity was determined using ImageJ. For Sox9 immunofluorescence, mean pixel intensity was determined using Imaris image analysis software (dots function, 4-μm nuclear sphere).
Supplementary Material
Acknowledgments
We thank Tetsuo Sudo (Toray Industries) for the gift of Hes1 antibody, and the Vanderbilt Cell Imaging Shared Resource for training and access to equipment and image processing software. We thank Anna Means, Guoqiang Gu, and members of the Wright/Gu laboratories for discussions. Imaging scholarships were from the Vanderbilt University Medical Center Digestive Disease Research Center and Diabetes Research and Training Center and the Vanderbilt-Ingram Cancer Center (supported by National Institutes of Health grants CA68485, DK20593, DK58404, and DK59637). This study was supported by the National Institutes of Health (1UO1DK089570-01).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.267914.115.
References
- Afelik S, Qu X, Hasrouni E, Bukys M, Deering T, Nieuwoudt S, Rogers W, MacDonald R, Jensen J. 2012. Notch-mediate patterning and cell fate allocation of pancreatic progenitor cells. Development 139: 1744–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahnfelt-Ronne J, Jorgensen MC, Hald J, Madsen OD, Serup P, Hecksher-Sorensen J. 2007. An improved method for three-dimensional reconstruction of protein expression patterns in intact mouse and chicken embryos and organs. J Histochem Cytochem 55: 925–930. [DOI] [PubMed] [Google Scholar]
- Apelqvist A, Hao H, Sommer L, Beatus P, Anderson DJ, Honjo T, Hrabe de Angelis M, Lendahl U, Edlund H. 1999. Notch signaling controls pancreatic cell differentiation. Nature 400: 877–881. [DOI] [PubMed] [Google Scholar]
- Cheraghali A, Knaus E, Wiebe L. 1994. Bioavailability and pharmacokinetic parameter for 5-ethyl-2′-deoxyuridine. Antiviral Res 25: 259–267. [DOI] [PubMed] [Google Scholar]
- Cortijo C, Gouzi M, Tissir F, Grapin-Botton A. 2012. Planar cell polarity controls pancreatic beta cell differentiation and glucose homeostasis. Cell Rep 2: 1593–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desgraz R, Herrera PL. 2009. Pancreatic neurogenin 3-expressing cells are unipotent islet precursors. Development 136: 3567–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vas MG, Kopp J, Heliot C, Sander M, Cereghini S, Haumaitre C. 2015. Hnf1b controls pancreas morphogenesis and the generation of Ngn3+ endocrine progenitors. Development 142: 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox P, Vought V, Hanazawa MH, Lee M, Maine E, Schedl T. 2011. Cyclin E and CDK-2 regulate proliferative cell fate and cell cycle progression in the C. elegans germline. Development 138: 2223–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco CA, Jones ML, Bernabeu MO, Geudens I, Mathivet T, Rosa A, Lopes FM, Lima AP, Ragab A, Collins RT, et al. 2015. Dynamic endothelial cell rearrangements drive developmental vessel regression. PLoS Biol 13: 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs E, Tumbar T, Guasch G. 2004. Socializing with the neighbors: stem cells and their niche. Cell 116: 769–778. [DOI] [PubMed] [Google Scholar]
- Gouzhi M, Kim Y, Katsumoto K, Johansson K, Grapin-Botton A. 2011. Neurogenin3 initiates stepwise delamination of differentiating endocrine cells during pancreas development. Dev Dyn 240: 589–604. [DOI] [PubMed] [Google Scholar]
- Gradwohl G, Dierech A, LeMeur M, Gillemot F. 2000. Neurogenin3 is required for the development of the four endocrine cell lineages. Proc Natl Acad Sci 97: 1607–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grapin-Botton A. 2005. Ductal cells of the pancreas. Int J Biochem Cell Biol 37: 504–510. [DOI] [PubMed] [Google Scholar]
- Gu G, Dubauskiate J, Melton D. 2002. Direct evidence for the pancreatic lineage: Ngn3+ cells are islet progenitors and are distinct from duct progenitors. Development 129: 2447–2457. [DOI] [PubMed] [Google Scholar]
- Hart A, Papadopoulou S, Edlund H. 2003. Fgf10 maintains Notch activation, stimulates proliferation, and blocks differentiation of pancreatic epithelial cells. Dev Dyn 228: 185–193. [DOI] [PubMed] [Google Scholar]
- Hick AC, van Eyll JM, Sabine C, Forez C, Passante L, Kohara H, Nagasawa T, Vanderhaeghen P, Courtoy P, Rousseau G, et al. 2009. Mechanism of primitive duct formation in the pancreas and submandibular glands: a role for SDF-1. BMC Dev Biol 9: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan BL. 1999. Morphogenesis. Cell 96: 225–233. [DOI] [PubMed] [Google Scholar]
- Huang S, Ingber DE. 1999. The structural and mechanical complexity of cell-growth control. Nat Cell Biol 1: 131–138. [DOI] [PubMed] [Google Scholar]
- Johansson KA, Dursun U, Jordan N, Gu G, Beermann F, Gradwohl G, Grapin-Botton A. 2007. Temporal control of neurogenin3 activity in pancreas progenitors reveals competence windows for the generation of different endocrine cell types. Dev Cell 12: 457–465. [DOI] [PubMed] [Google Scholar]
- Kageyama R, Ohtsuka T, Shimojo H, Imayoshi I. 2008. Dynamic Notch signaling in neural progenitors and a revised view of lateral inhibition. Nat Neurosci 11: 1247–1251. [DOI] [PubMed] [Google Scholar]
- Kesavan G, Sand FW, Greiner TU, Johansson JK, Kobberup S, Xunwei W, Brakebusch C, Semb H. 2009. Cdc42-mediated tubulogenesis controls cell specification. Cell 139: 791–801. [DOI] [PubMed] [Google Scholar]
- Kim YH, Larsen HL, Rué P, Lemaire LA, Ferrer J, Grapin-Botton A. 2015. Cell cycle-dependent differentiation balances growth and endocrine differentiation in the pancreas. PLoS Biol 13: 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobberup S, Schmerr M, Dang ML, Nyeng P, Jensen JN, MacDonald RJ, Jensen J. 2010. Conditional control of the differentiation competence of pancreatic endocrine and ductal cells by Fgf10. Mech Dev 127: 220–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopinke D, Brailsford M, Shea JE, Leavitt R, Scaife CL, Murtaugh LC. 2011. Lineage tracing reveals the dynamic contribution of Hes1+ cells to the developing and adult pancreas. Development 138: 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp JL, Dubois C, Schaffer AE, Hao E, Shih HP, Seymour P, Ma J, Sander M. 2011. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development 138: 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammert E, Brown J, Melton D. 2000. Notch gene expression during pancreatic organogenesis. Mech Dev 94: 199–203. [DOI] [PubMed] [Google Scholar]
- Landsman L, Nijagal A, Whitchurch TJ, VanderLaan RL, Zimmer WE, MacKenzie TC, Hebrok M. 2011. Pancreatic mesenchyme regulates epithelial organogenesis throughout development. PLoS Biol 9: e1001143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Smith SB, Watada H, Lin J, Scheel D, Wang J, Mirmira RG, German MS. 2001. Regulation of the pancreatic pro-endocrine gene neurogenin3. Diabetes 50: 928–936. [DOI] [PubMed] [Google Scholar]
- Li L, Xie T. 2005. Stem cell niche: structure and function. Annu Rev Cell Dev Biol 21: 605–631. [DOI] [PubMed] [Google Scholar]
- Magenhiem J, Klein AM, Stanger BZ, Ashery-Padan R, Sosa-Pineda B, Gu G, Dor Y. 2011a. Ngn3+ endocrine progenitor cells control the fate and morphogenesis of pancreatic ductal epithelium. Dev Biol 359: 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magenheim J, Ilovich O, Lazarus A, Klochendler A, Ziv O, Werman R, Hija A, Cleaver O, Mishani E, Keshet E, et al. 2011b. Blood vessels restrain pancreas branching, differentiation and growth. Development 138: 4743–4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinari E, Mehonic A, Curran S, Gale J, Duke T Baum B. 2012. Live-cell delamination counterbalances epithelial growth to limit tissue overcrowding. Nature 484: 542–545. [DOI] [PubMed] [Google Scholar]
- Metzger RJ, Klein OD, Martin GR, Krasnow MA. 2008. The branching programme of mouse lung development. Nature 453: 745–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger DE, Gasperowicz M, Otto F, Cross JC, Gradwohl G, Zaret KS. 2012. The transcriptional co-repressor Grg3/Tle3 promotes pancreatic endocrine progenitor delamination and β-cell differentiation. Development 139: 1447–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milano J, McKay J, Dagenais C, Foster-Brown L, Pognan F, Gradient R, Jacobs R, Zacco A, Greenberg B, Ciacco P. 2004. Modulation of Notch processing by γ-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol Sci 82: 341–358. [DOI] [PubMed] [Google Scholar]
- Miyatsuka T, Kosaka Y, Kim H, German MS. 2011. Neurogenin3 inhibits proliferation in endocrine progenitors by inducing Cdkn1a. Proc Natl Acad Sci 108: 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. 2003. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci 100: 14920–14925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninov N, Borius M, Stainier DY. 2012. Different levels of Notch signaling regulate quiescence, renewal, and differentiation in pancreatic endocrine progenitors. Development 139: 1557–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan FC, Wright C. 2011. Pancreas organogenesis: from bud to plexus to gland. Dev Dyn 240: 530–565. [DOI] [PubMed] [Google Scholar]
- Petzold KM, Naumann H, Spagnoli FM. 2013. Rho signalling restriction by the RhoGAP Stard13 integrates growth and morphogenesis in the pancreas. Development 140: 126–135. [DOI] [PubMed] [Google Scholar]
- Pierfelice T, Alberi L, Gaiano N. 2011. Notch in the vertebrate nervous system: an old dog with new tricks. Neuron 69: 840–855. [DOI] [PubMed] [Google Scholar]
- Pierreux CE, Poll AV, Kemp CR, Clotman F, Maestro MA, Cordi S, Ferrer J, Leyns L, Rousseau GG, Lemaigre FP. 2006. The transcription factor hepatocyte nuclear factor-6 controls the development of pancreatic ducts in the mouse. Gastroenterology 130: 532–541. [DOI] [PubMed] [Google Scholar]
- Qu X, Afelik S, Jensen JN, Bukys MA, Kobberup S, Schmerr M, Xiao F, Nyeng P, Veronica Albertoni M, et al. 2013. Notch-mediated post-translational control of Ngn3 protein stability regulates pancreatic patterning and cell fate commitment. Dev Biol 376: 1–12. [DOI] [PubMed] [Google Scholar]
- Reichert M, Rustgi AK. 2011. Pancreatic ductal cells in development, regeneration, and neoplasia. J Clin Invest 121: 4572–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieck S, Bankaitis ED, Wright C. 2012. Lineage determinants in early endocrine development. Semin Cell Dev Biol 23: 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salic A, Mitchison TJ. 2008. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci 105: 2415–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer AE, Freude K, Nelson S, Sander M. 2010. Nkx6 transcription factors and Ptf1a function as antagonistic lineage determinants in multipotent pancreatic progenitors. Dev Cell 18: 1022–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwitzgebel VM, Scheel DW, Conners JR, Kalamaras J, Lee JE, Anderson DJ, Sussel L, Johnson JD, German MS. 2000. Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development 127: 3533–3542. [DOI] [PubMed] [Google Scholar]
- Serup P. 2012. Signaling pathways regulating murine pancreas development. Semin Cell Dev Biol 23: 663–672. [DOI] [PubMed] [Google Scholar]
- Seymour P, Freude K, Dubois C, Shih HP, Patel N, Sander M. 2008. A dosage-dependent requirement for Sox9 in pancreatic endocrine cell formation. Dev Biol 323: 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih HP, Kopp J, Sandhu M, Dubois C, Seymour P, Grapin-Botton A, Sander M. 2012. A Notch-dependent molecular circuitry initiates pancreatic endocrine and ductal cell differentiation. Development 139: 2488–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SB, Watada H, German MS. 2004. Neurogenin3 activates the islet differentiation program while repressing its own expression. Mol Endocrinol 18: 142–149. [DOI] [PubMed] [Google Scholar]
- Solar M, Cardala C, Houbracken I, Martin M, Maestro MA, De Medts N, Xu X, Grau V, Heimberg H, Bouwens L, Ferrer J. 2009. Pancreatic exocrine duct cells give rise to insulin-producing β cells during embryogenesis but not after birth. Dev Cell 17: 849–860. [DOI] [PubMed] [Google Scholar]
- Stanger BZ, Akemi J, Melton D. 2007. Organ size is limited by the number of embryonic progenitor cells in the pancreas but not the liver. Nature 445: 886–891. [DOI] [PubMed] [Google Scholar]
- van Es J, van Gijn M, Riccio O, van den Born M, Vooijs M, Begthel H, Cozijnsen M, Robine S, Winton D, Radtke F, et al. 2005. Notch/γ-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 435: 959–963. [DOI] [PubMed] [Google Scholar]
- Villasenor A, Cleaver O. 2012. Crosstalk between the developing pancreas and its blood vessels: an evolving dialog. Semin Cell Dev Biol 23: 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villasenor A, Chong DC, Cleaver O. 2008. Biphasic Ngn3 expression in the developing pancreas. Dev Dyn 237: 3270–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villasenor A, Chong DC, Henkemeyer M, Cleaver O. 2010. Epithelial dynamics of pancreatic branching morphogenesis. Development 137: 4295–4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Kilic G, Aydin M, Burke Z, Oliver G, Sosa-Pineda B. 2005. Prox1 activity controls pancreas morphogenesis and participates in the production of ‘secondary transition’ pancreatic endocrine cells. Dev Biol 286: 182–194. [DOI] [PubMed] [Google Scholar]
- Wang S, Yan J, Anderson D, Yanwen X, Maneesh K, Zheng C, Wright C, Gu G. 2010. Neurog3 gene dosage regulates allocation of endocrine and exocrine cell fates in the developing mouse pancreas. Dev Biol 339: 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmoreland JJ, Kilic G, Sartain C, Sirma S, Blain J, Rehg J, Harvey N, Sosa-Pineda B. 2012. Pancreas-specific deletion of Prox1 affects development and disrupts homeostasis of the exocrine pancreas. Gastroenterology 142: 999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Law AC, Rajagopal J, Anderson WJ, Gray PA, Melton DA. 2007. A multipotent progenitor domain guides pancreatic organogenesis. Dev Cell 13: 103–114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.