

Abstract
Background
Heterozygous human NKX2-5 homeodomain (DNA binding domain) missense mutations are highly penetrant for varied congenital heart defects, including progressive atrioventricular block (AVB) requiring pacemaker implantation. We recently replicated this genetic defect in a murine knock-in model, in which we demonstrated highly penetrant, pleiotropic cardiac anomalies. In this study, we examined postnatal AV conduction in the knock-in mice.
Methods and Results
A murine knock-in model (Arg52Gly, Nkx2-5+/R52G) in a 129/Sv background was analyzed by histopathology, surface and telemetry ECG, and in vivo electrophysiology studies (EPS), comparing with control Nkx2-5+/+ mice at diverse postnatal stages, ranging from postnatal day 1 (P1) to 17 months. PR-prolongation (1st degree AVB) was present at 4 weeks, 7 months, and 17 months of age but not at P1 in the mutant mice. Advanced AVB was also occasionally demonstrated in the mutant mice. EPS showed that AV nodal function, and right ventricular effective refractory period, were impaired in the mutant mice, while sinus nodal function was not affected. AV nodal size was significantly smaller in the mutant mice compared to their controls at 4 weeks of age, corresponding to the presence of PR-prolongation, but not P1, suggesting, at least in part, that the conduction abnormalities are the result of a morphologically atrophic AV node.
Conclusions
The highly penetrant and progressive AVB phenotype seen in human heterozygous missense mutations in NKX2-5 homeodomain was replicated in mice by knocking-in a comparable missense mutation.
Keywords: atrioventricular block, congenital heart disease, genetics, animal models, genetics, human, knock-in
Introduction
To date, nearly 40 mutations in the transcription factor NKX2-5 have been found related to familial congenital heart disease (OMIM, NCBI). Almost half of these mutations have been found in the homeodomain (DNA binding domain)1–6. NKX2-5 homeodomain missense mutations have a high disease penetrance, with pleiotropic cardiac effects when compared to missense mutations outside the homeodomain1–6. In addition, humans harboring a heterozygous homeodomain missense NKX2-5 mutation demonstrate near complete penetrance of progressive atrioventricular block (AVB) during postnatal life1, 7.
In order to understand the potential disease mechanisms underlying heterozygous NKX2-5 mutation, we recently generated and analyzed a heterozygous knock-in mouse model by introducing a disease-causing missense mutation in homeodomain position 52, and Nkx2-5 protein position 188 in Nkx2-5 proteins, Arg52(188)Gly, Nkx2-5+/R52G 8. In contrast to heterozygous knock-out mice (Nkx2-5+/−), we found that the cardiac effects of knock-in mice (Nkx2-5+/R52G), hereafter described as Nkx2-5-KI mice, were similar to those observed in humans. Cardiac malformations were highly penetrant, and the effects on cardiac morphology were pleiotropic. Among them, atrial septal defects characterized by persistent interatrial communication due to deficiency of the flap valve of the oval fossa, are the most prevalent cardiac anomaly found in human patients. In knock-in mice, such interatrial communication was shown to be the consequence of an increased size of the oval fossa and a decreased length of the flap valve that usually closes the fossa, compared to control wild-type mice. In addition, ventricular noncompaction (100%), ventricular septal defects (82%), atrioventricular septal defects (18%) and tricuspid valvar anomalies (47%) were displayed in the knock-in mice. These lesions were not demonstrated in wild-type mice, but some of them were found with lower incidences in heterozygous knock-out mice 8.
In the current study, we analyzed the signature phenotype of human patients having an NKX2-5 mutation, namely progressive AVB observed during postnatal life. We focused on the agreements and disagreements between mice and humans by using the combination of surface and telemetry ECG, in vivo electrophysiology studies (EPS), and histopathology. As in human NKX2-5 mutations, we found a progressive and highly penetrant AVB phenotype in heterozygous Nkx2-5-KI mice.
Materials and Methods
Generation of Nkx2-5+/R52G mice
Nkx2-5+/R52G knock-in mice were generated as reported previously8. In brief, a targeting vector was constructed by introduction of a CGT to GGT point mutation in exon 2, insertion of thymidine kinase (TK) gene for negative selection and floxed-neomycin-resistant gene (NeoR) for positive selection in the intergenic regions. NeoR gene was deleted subsequently by crossing with Cre-deleter mice (ACTB-Cre). Cre-deleter transgenes were eliminated during backcrossing to 129/Sv mice purchased from Charles River (129/SvPasCrl) over 8 generations. All animal experiments were performed with approval from the University of Florida Institutional Animal Care and Use Committee.
Surface 6-lead ECG and telemetry ECG recordings
Surface 6-lead ECG and telemetry ECG recordings were performed as previously described 9, 10. Briefly, mice were anesthetized with isoflurane (1–2%) and placed on a heated pad (using water circulation) at 37°C. ECG recordings were obtained from a 29 gauge subcutaneous electrode. For ambulatory ECG analysis in conscious unrestrained mice, 1.6-g transmitters (ETA-F10. DSI) were implanted. After 72 hours recovery time from surgical instrumentation, ECG recordings were performed in each mouse placed in a separate cage overlying a receiver (DSI) in daytime. Signal-averaged ECGs selected from ~20 stable ECG beats from at least three different time points were analyzed by LabChart with the ECG analysis module (ADInstruments) to confirm the consistency of the data.
Electrophysiology study (EPS)
Simultaneous atrial and ventricular pacing and recording were performed via 1.1F octapolar catheter (EPR-800, Millar) inserted via a jugular vein in mice anesthetized with isoflurane (2–3%). The standard pacing protocols11–13 were programmed in the multichannel stimulator STG 3008 (Multichannel systems), which was analyzed by LabChart (ADInstruments). Sinus nodal function was evaluated by measuring sinus node recovery time (SNRT) at three pacing drive rates (150, 120 and 100 ms) and corrected SNRT (SNRT minus sinus cycle length, SCL). AV conduction properties were assessed with rapid atrial pacing at rates with a minimum pacing cycle length of 50 ms (up to 1,200 bpm). The minimum cycle length maintaining 1:1 AV conduction, the Wenckebach paced cycle length, and the maximum paced cycle length causing 2:1 AV block were determined. AV nodal effective refractory periods were determined by the maximum coupling interval causing AV block at the two pacing drive rates (150 and 120 ms). Right ventricular effective refractory periods were determined by the maximum coupling interval that failed to stimulate ventricles at the two pacing drive rates (150 and 120 ms). Right ventricular burst pacing was performed at rates with a pacing cycle length of 150 to 50 ms (400-1,200 bpm) to assess retrograde ventriculo-atrial conduction.
Histological analysis
Serial paraffin-embedded tissue sectioning of 5 μm thickness was performed to reveal the location and dimensions of the atrioventricular conduction axis as described previously8, 14. For the purpose of this study, we discriminated the conduction axis as having a ventricular component (made up of the ventricular components of the axis and the bundle branches) and a penetrating component (made up of the bundle of His), from a compact atrioventricular nodal component, and its inferior nodal extensions)17,18. We distinguished the penetrating component of the axis on the basis of its insulation from the atrial cardiomyocytes by the fibrous tissues of the central fibrous body. This definition of the node, as opposed to the penetrating bundle, was initially proposed by Tawara15 although subsequent investigators have described so-called “lower nodal cells” as being part of the atrioventricular node. We did not consider these cells as belonging to the node, although it was possible to trace the cells through the basal part of the node, where they became continuous with the inferior nodal extensions.
We then digitized the proximal part of the penetrating bundle immediately it had been insulated from the atrial cardiomyocytes, the compact part of the node, and the inferior nodal extensions so as to measure their areas, nuclear numbers, and cell width using Image J as reported previously.10 The width of the cardiomyocytes within the compact node was measured in the troponin T-positive myocytes at the level of the nuclei as shown previously.10 AV nodal volume was calculated as (r, radius of maximum area size; l, length, 5 μm × number of tissue sections). Paraffin-embedded tissue sectioning or cryosectioning of paraformaldehyde-fixed hearts was utilized for immunostaining. Immunostaining was performed with the following antibodies: actinin (Sigma A7811), connexin 40 (Alpha Diagnostic Inc. cx40-A), connexin 43 (Zymed 71–700), HCN4 (Millipore, AB5808), Nav1.5(α)(Alomone Labs, ASC-005), troponin T (Sigma, T6277) and Nkx2-516. Presence of fibrosis was examined using picrosirius red-stained tissue sections. Briefly, the tissue sections were heated at 60°C for 45 min before deparaffinization and stained in 0.1% direct red 80, 0.1% fast green FCF in 1.2% picric acid for 60 min.
Acetylcholine esterase (AchE) staining using the serial frozen tissue sections (10 μm thickness) was performed as described previously14.
Statistical analysis
Data presented are expressed as mean values plus or minus the standard error (SE) or standard deviation (SD) of the mean. Results were analyzed by SPSS (version 22) using independent T-test. Levene’s test was utilized for equality of variance, and P values were calculated depending on the assurance of equality. P values are 2-sided and those less than 0.05 were considered significant. There are no adjustments for multiple comparison.
Results
Phenotypes associated with Arg52(189)Gly, NKX2-5+/R52,, homeodomain missense mutation
The pedigree of the family having Arg52(189)Gly mutation previously reported5 was recently found to include a new patient in the 5th generation who demonstrated atrioventricular block (AVB), as well as atrial and ventricular septal defects, resulting in six members of the family with a mutation (Figure 1). All 6 genotype-positive individuals demonstrate AVB (first, second or third degree). Atrial septal defect (ASD) was present in 5 patients, ventricular septal defect (VSD) or a small tricuspid valve was present in one patient each.
Figure 1.
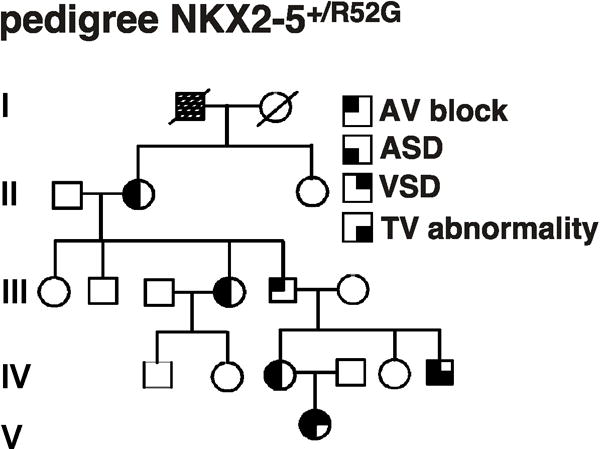
Phenotypes associated with Arg52(189)Gly, NKX2-5+/R52,, homeodomain missense mutation. In the kindred, males are denoted by squares, females by circles. Darkened quadrants indicate AVB, ASD, VSD, or tricuspid valve abnormality. Open symbols denote normal genotype and phenotype. Arg52 is positioned at amino acid 189 in human NKX2-5. ASD, atrial septal defect; VSD, ventricular septal defect; TV, tricuspid valve.
AVB in heterozygous knock-in Nkx2-5 mutant mice, (Arg52(188)Gly, Nkx2-5+/R52G), measured by surface and ambulant ECG recordings
As shown above, the signature phenotype of humans harboring a heterozygous homeodomain missense NKX2-5 mutation is development of progressive AVB during postnatal life. A newly generated mouse model having a single missense mutation by knock-in8, Nkx2-5+/R52G (Nkx2-5-KI), was evaluated by surface ECG recording at postnatal day 1 (P1), 4 weeks, 7 months and 17 months of age (Figures 2A–D). The PR interval was not prolonged in P1 Nkx2-5-KI mice, but was prolonged after 4 weeks of age under low isoflurane anesthesia (1%). The difference was significant; but was subtle, in particular at 4 weeks of age (Figure 2E, Table 1). Exposure to the higher dose of isoflurane (3%) for 5 minutes prolonged the PR interval, even in the control mice (33.2 ± 0.8 vs. 41.5 ± 1.5 ms), but the effect was more profound in Nkx2-5-KI mice (36.5 ± 0.6 vs. 54.2 ± 2.2 ms)(Figure 2F, Supplemental Table 1). These results suggest that the mutant mice demonstrate postnatal progressive AVB with the lower dose of anesthesia, which became more evident under the higher dose of anesthesia.
Figure 2.
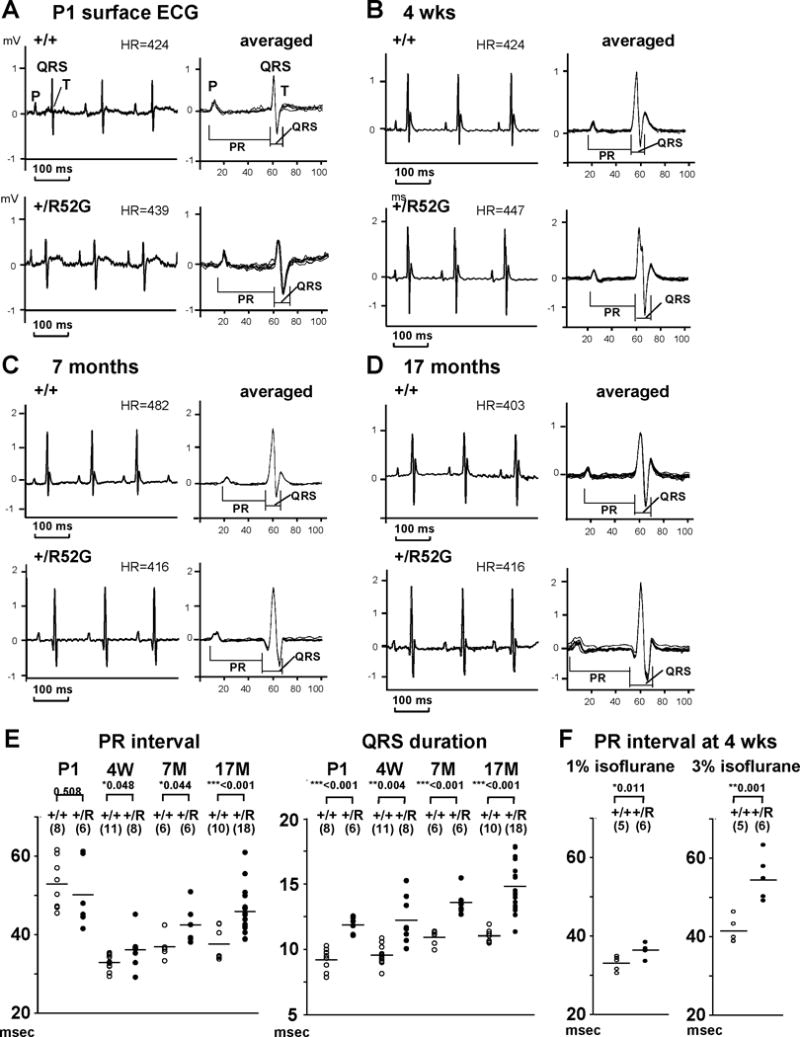
Prolongation of PR interval and QRS duration in Nkx2-5+/R52G mice. Representative surface ECG recordings obtained from P1 (A), 4 weeks (B), 7-month-old (C) and 17 month-old (D) control Nkx2-5+/+ (top) and Nkx2-5+/R52G (bottom) mice. (E) PR-interval and QRS duration over time with individual values plotted from control Nkx2-5+/+ (open circle) and Nkx2-5+/R52G (closed circle) mice. The averages of values, and number of mice examined were indicated. (F) PR interval under two different isoflurane dose (1% and 3%) from control Nkx2-5+/+ (open circle) and Nkx2-5+/R52G (closed circle) mice at 4 weeks of age. Signal-averaged ECG waves were utilized for analysis. Summarized data is shown in Table 1. HR, heart rate. Mean ± SE.
Table 1.
Summary of surface ECG analysis
| genotype | age | SCL (ms) | HR (bpm) | PR (ms) | QRS (ms) | Qtc (ms) | |
|---|---|---|---|---|---|---|---|
| P1 | W/W (n=8) | P1 | 162 ± 16 | 371 ± 33 | 52.9 ± 6.3 | 9.2 ± 0.9 | 23.1 ± 1.8 |
| W/R52G (n=6) | P1 | 172 ± 17 | 351 ± 38 | 50.2 ± 8.6 | 11.9 ± 0.6*** | 25.9 ± 2.6* | |
| 4 wks | W/W (n=11) | 4 weeks | 144 ± 23 | 425 ± 69 | 32.9 ± 1.9 | 9.6 ± 0.8 | 23.1 ± 1.1 |
| W/R52G (n=8) | 4 weeks | 143 ± 11 | 421 ± 38 | 36.1 ± 4.5* | 12.2 ± 1.8** | 26.7 ± 4.7 | |
| adult | W/W (n=6) | 7.4 ± 0.4 M | 141 ± 17 | 429 ± 54 | 36.9 ± 3.0 | 10.9 ± 0.6 | 27.6 ± 3.2 |
| W/R52G (n=6) | 7.4 ± 0.4 M | 145 ± 18 | 419 ± 57 | 42.5 ± 4.9* | 13.6 ± 1.0*** | 29.4 ± 3.9 | |
| aged | W/W (n=10) | 17.4 ± 0.8 M | 146 ± 15 | 413 ± 36 | 37.6 ± 3.4 | 11.2 ± 0.6 | 26.4 ± 2.8 |
| W/R52G (n=18) | 17.5 ± 1.1 M | 145 ± 24 | 422 ± 63 | 45.9 ± 5.6*** | 14.8 ± 1.9*** | 26.5 ± 3.7 |
SCL - sinus cycle length; HR - heart rate; bpm - beats per minute
Mean ± SD. P, P value.
P < 0.05
P < 0.01
P < 0.001.
In order to confirm that the PR prolongation was not a side effect of anesthesia, and to examine the presence of advanced AVB, we utilized ambulatory ECG analysis in conscious unrestrained aged mice at 17 months of age (Figure 3A, Table 2). The PR interval was significantly longer in Nkx2-5-KI mice relative to the controls (38.0 ± 0.9 vs. 33.3 ± 0.9 ms, p< 0.01). One of 16 Nkx2-5-KI mice also demonstrated advanced AVB (complete AVB) (Figure 3B), while none of the control mice showed any degree of AVB.
Figure 3.
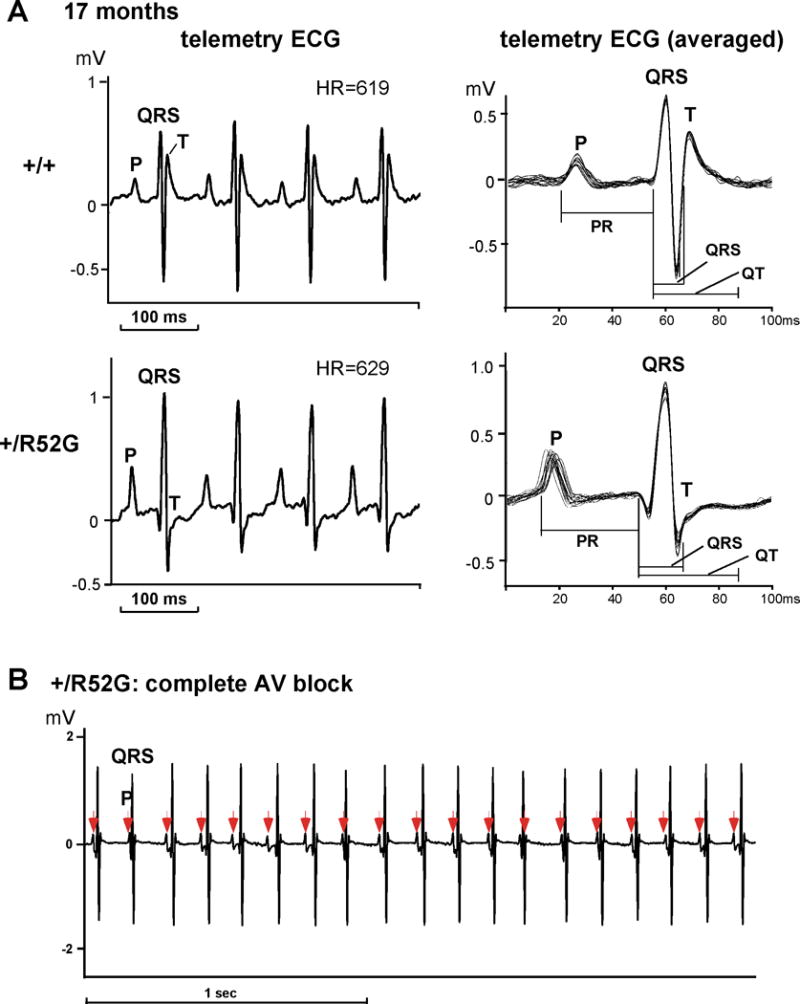
AVB displayed in ambulant telemetry ECG recording from 17 month-old Nkx2-5+/R52G mice. (A) Representative telemetry ECG recordings from control Nkx2-5+/+ (top) and Nkx2-5+/R52G (bottom) mice at 17 months of age. Averaged ECG waves from ~ 20 beats at least three different time points were utilized for analysis shown in Table 2. (B) Representative ECG recording of advanced complete AVB demonstrated in Nkx2-5+/R52G mice. HR, heart rate. Mean ± SE.
Table 2.
Summary of telemetry ECG analysis
| Age (month) | SCL (ms) | HR (bpm) | PR (ms) | QRS (ms) | Qtc (ms) | ||
|---|---|---|---|---|---|---|---|
| aged | W/W (n=10) | 17.4 ± 0.8 | 96 ± 8 | 629 ± 53 | 33.3 ± 2.8 | 11.4 ± 0.7 | 32.8 ± 2.9 |
| W/R52G (n=15) | 17.5 ± 1.2 | 96 ± 9 | 628 ± 55 | 38.0 ± 3.6 (P=0.002)** | 14.6 ± 1.9 (P < 0.001)*** | 35.1 ± 4.3 |
SCL - sinus cycle length; HR - heart rate; bpm - beats per minute
Mean ± SD. P, P value.
P < 0.01
P < 0.001.
Wide QRS, indicative of prolonged ventricular conduction times, was demonstrated in Nkx2-5-KI mice at all ages, with or without anesthesia (Figure 2E, Tables 1, 2). This finding is consistent with the previous studies analyzing conditional Nkx2-5 homozygous, as well as heterozygous, Nkx2-5 knockout mice9, 10, 14, 17, 18. Human patients with NKX2-5 mutations, in contrast, do not demonstrate wide QRS5.
In vivo endocardial electrophysiological studies (EPS)
To determine the electrophysiological functions of conduction systems, in vivo EPS were performed in mice at 7 month of age, when AVB is well-established, and mouse body size is large enough for EPS following the standard pacing protocol with the basal pacing rate of 150 ms. To be paced at 150 ms, endogenous sinus cycle length (CL) was kept longer than 150 ms by a higher dose of isoflurane (2–3%). The sinus CL was maintained between 160–180 ms (heart rate 350–370 bpm) (Table 3), which was slower than those utilized for surface ECG recording at 7 months of age (Figure 2E, Table 1).
Table 3.
Summary of electrophysiology studies (EPS) and simultaneous surface ECG analysis at 7 months of age.
| WW (n=5) | W/R52G (n=6 or 7) | P value | |
|---|---|---|---|
| body weight | 23.1 ± 0.6 | 24.2 ± 1.5 | 0.525 |
| SCL (ms) | 170 ± 14 | 163 ± 5 | 0.261 |
| HR (bpm) | 352 ± 29 | 368 ± 12 | 0.293 |
| PR (ms) | 40.9 ± 1.6 | 53.8 ± 6.2 | 0.003** |
| QRS (ms) | 10.1 ± 1.0 | 14.6 ± 1.4 | <0.001*** |
| QT (ms) | 36.3 ± 3.2 | 36.8 ± 4.7 | 0.845 |
| QTc (ms) | 27.8 ± 3.1 | 31.5 ± 4.2 | 0.136 |
| SNRT 150 (ms) | 212 ± 45 | 204 ± 23 | 0.696 |
| SNRT 120 (ms) | 243 ± 56 | 277 ± 58 | 0.333 |
| SNRT 100 (ms) | 288 ± 53 | 275 ± 66 | 0.742 |
| cSNRT 150 (ms) | 48 ± 42 | 40 ± 21 | 0.666 |
| cSNRT 120 (ms) | 78 ± 53 | 85 ± 29 | 0.762 |
| cSNRT 100 (ms) | 123 ± 51 | 82 ± 44 | 0.165 |
| AVWBCL (ms) | 87 ± 10 | 115 ± 15 | 0.007** |
| AV2:1CL (ms) | 69 ± 12 | 83 ± 9 | 0.047* |
| AVERP 150 (ms) | 46 ± 15 | 70 ± 9 | 0.01* |
| AVERP 120 (ms) | 42 ± 18 | 70 ± 15 | 0.021* |
| VERP 150 (ms) | 37 ± 10 | 60 ± 14 | 0.013* |
| VERP 120 (ms) | 38 ± 11 | 63 ± 18 | 0.021* |
One Nkx2-5+/R52G mouse demonstrating 3:1 AV block could not be assessed for PR and QRS interval from surface ECG, as well as AV nodal function and ventricular conduction due to lack of the 1:1 AV conduction. SCL - sinus cycle length; HR - heart rate; bpm - beats per minute; SNRT - sinus node recovery time; cSNRT – rate corrected SNRT; AVWBCL - atrioventricular Wenckebach block cycle length; AV2:1CL - atrioventricular 2:1 conduction block cycle length; AVERP - atrioventricular effective refractory period; VERP - ventricular effective refractory period.
Mean ± SD. P, P value.
P < 0.05
P < 0.01
P < 0.001.
Under higher isoflurane anesthesia, the PR interval of mice at 7 months of age was prolonged similar to those at 4 weeks of age, even in the control mice (36.9 ± 1.2 vs. 40.9 ± 0.7 ms), compared to those observed in the surface ECG recordings, but the effect was more profound in Nkx2-5-KI mice (42.5 ± 2 vs. 53.8 ± 2.5 ms) (Table 1 vs. 3). EPS in Nkx2-5-KI mice consistently showed impaired AV nodal conduction properties, as assessed by AV nodal effective refractory period, and the longest pacing CL when 1:1 AV conduction failed to Wenckebach type (AVWBCL) and 2:1 AV conduction (AV2:1 CL) (Table 3). In addition, Nkx2-5-KI mice demonstrated a prolonged ventricular effective refractory period, which reflects the wide QRS with an impaired ventricular peripheral conduction system. Sinus nodal conduction properties assessed by sinus node recovery time (SNRT) and rate-corrected SNRT (CSNRT) were unchanged in Nkx2-5-KI mice (Table 3).
One of Nkx2-5-KI mice from a total of 7, demonstrated advanced second degree, 3:1 AVB, shortly after induction of anesthesia (atrial sinus CL, 135 ms), which persisted (Figure 4A, atrial sinus CL 135 ms), while none of the control mice demonstrated advanced AVB. Due to lack of the 1:1 AV conduction, we could not assess PR and QRS interval from surface ECG, as well as AV nodal function and ventricular conduction from this Nkx2-5-KI mouse (see Table 3).
Figure 4.
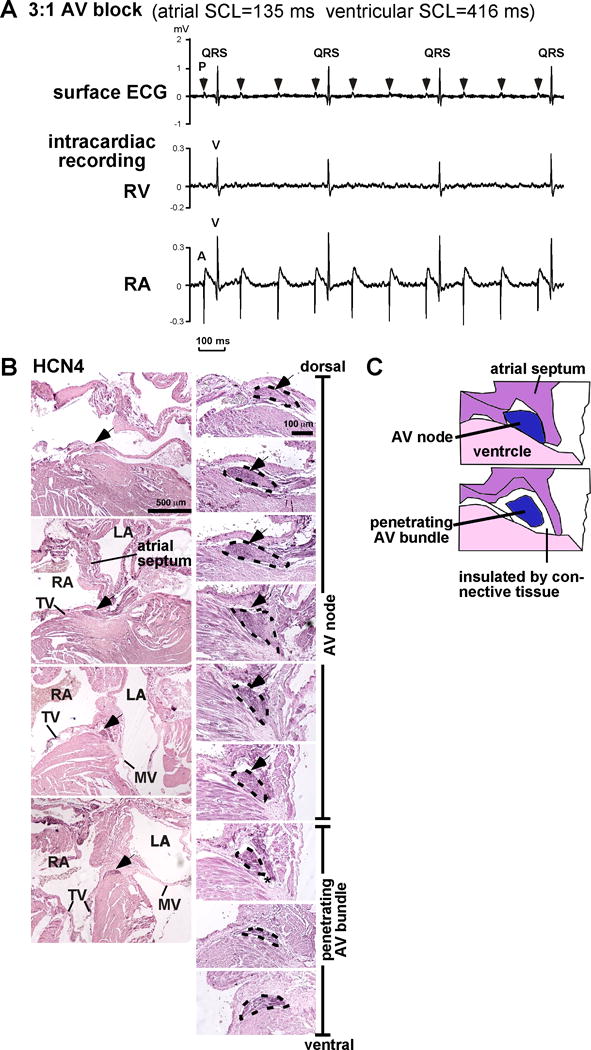
Advanced 3:1 AVB from Nkx2-5+/R52G mouse (7 months of age) detected shortly after anesthesia for EP study and HCN4-positive AV node vs. penetrating AV bundle. (A) Representative recording of surface and intracardiac recordings (RV or RA) demonstrating 3:1 AVB obtained from Nkx2-5+/R52G mice during EP analysis shortly after induction of isoflurane anesthesia with sinus cycle length of 135 ms. (B) Representative HCN4-stained normal heart tissues of Nkx2-5+/+ at 4 weeks of age including AV node and penetrating AV bundles traced by black dots. (C) Simplified illustration to define AV node vs. penetrating AV bundle with or without direct contact to atrial muscle. HCN, hyperpolarization activated cyclic nucleotide-gated potassium channel. A, right atrial electrogram; V, right ventricular electrogram.
Reduced AV nodal size composed of smaller cardiomyocytes in Nkx2-5+/R52G in 4-week-old but not in P1 Nkx2-5+/R52G mice
Using the combination of immunostaining with hyperpolarization activated cyclic nucleotide-gated potassium channel 4 (HCN4) and hematoxylin and eosin staining (H&E), we defined the AV nodal area as being HCN4-positive, containing less-organized sarcomere structure (reduced H&E staining), and having cardiomyocytes in direct contact with atrial muscle19, 20. We excluded from consideration the area of the conduction axis insulated from the atrial muscle by connective tissue, defining this component as the penetrating AV bundle (Figures 4B, C, see Methods, Histological analysis for details).
Atrophy of AV conduction axis has been postulated as an underlying mechanism of the PR-prolongation observed in multiple conditional homozygous Nkx2-5 knockout mice9, 10, 18. In keeping with this notion, we found that, in Nkx2-5-KI mice at 4 weeks of age, when the PR prolongation was first displayed, the area made up of the compact node was markedly reduced, analyzed by AV nodal maximum area size, and estimated AV nodal volume modeled as a 3D cylindrical structure (see Methods)(Figures 5A, B). The areas of the penetrating bundle and nodal extensions were also smaller in Nkx2-5-KI mice. Nuclear density, in contrast, calculated as the number of nuclei per area size in the maximum AV node, was increased. The width of the cardiomyocytes visualized by sarcomeric troponin-staining revealed that the AV nodal cardiomyocytes were smaller in Nkx2-5-KI hearts (Figure 5B). The ratio of the number of Nkx2-5-positive (indicating cardiomyocytes) vs. Nkx2-5-negative (non-myocytes) nuclei was unchanged (Figures 5C, D), suggesting that the cellular component of myocytes and non-myocytes is most likely un-changed between Nkx2-5-KI mice and the controls.
Figure 5.
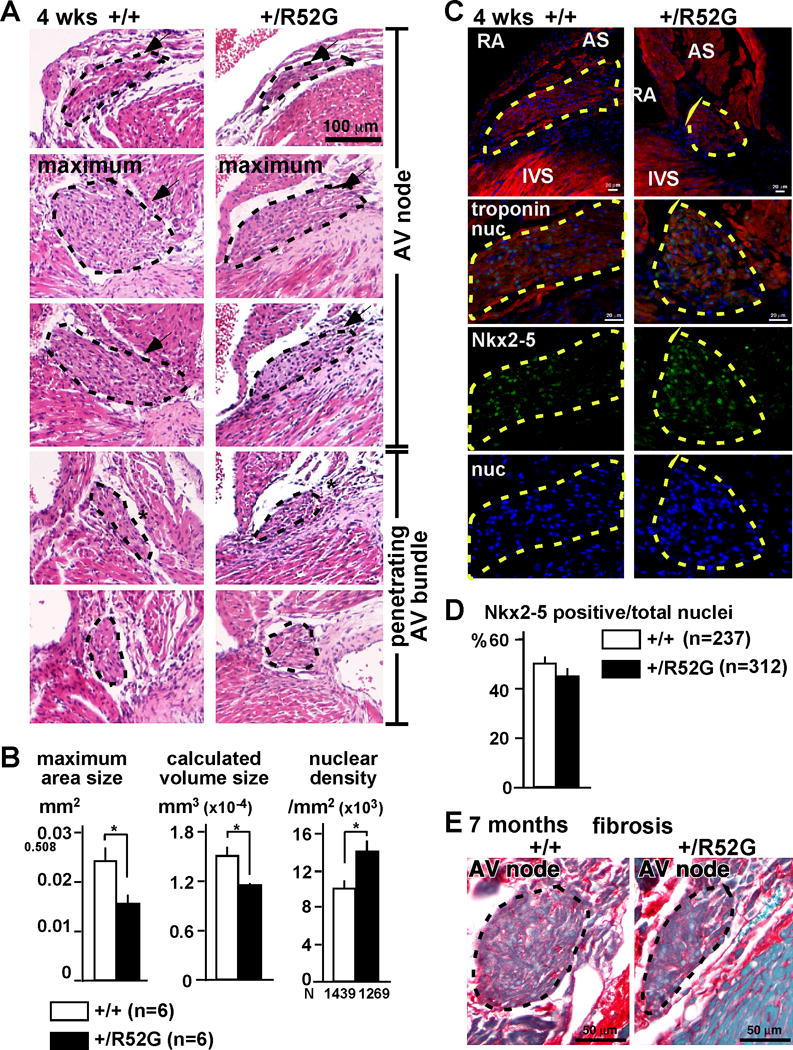
Reduction of AV nodal size composed of smaller cardiomyocytes without apparent increase of fibrosis in 4 weeks or 7 months-old Nkx2-5+/R52G mice. (A) Representative H&E stained heart tissues including AV node and penetrating AV bundles traced in black dots from control Nkx2-5+/+ or Nkx2-5+/R52G mice. (B) Maximum AV nodal area size, calculated AV nodal volume, and nuclear density (number of nuclei × 103/mm2). The numbers of mice and nuclei analyzed were noted. (C) Representative immunofluorescent images of troponin T (red), Nkx2-5 (green) and nuclei (blue) from control Nkx2-5+/+ or Nkx2-5+/R52G mice. AV nodal area is traced by yellow dots. (D) Ratio of Nkx2-5-positive vs. total nuclear number in the AV node. (E) Representative images of picrosirius red-stained tissue sections including AV node from 7-month-old mice. Fibrotic tissues are stained in red and muscle tissues are stained in green. RA, right atrium; AS, atrial septum; IVS, interventricular septum. Mean ± SE.
Massive AV nodal fibrosis has been demonstrated in conditional Nkx2-5 knockout mice using a myosin-light chain 2v-Cre18. As judged by the extent of picrosirius red staining in our material, we did not observe any apparent increase of fibrosis, either at 4 weeks (data not shown) nor 7 months of age (Figure 5E).
In contrast, in P1 mice, when PR-prolongation of Nkx2-5-KI mice was not evident, AV nodal maximum area size and nuclear density were unchanged between Nkx2-5-KI mice and the controls (Figures 6A–D). In addition, immunostaining of positive- or negative-markers for AV node, penetrating AV node and left bundle branch, including HCN4, connexin 40, connexin 43, and Nav1.5 was not different between the two groups neither at P1 (Figure 7A) nor 4 weeks of age, except that the left bundle branch was under-developed in Nkx2-5-KI mice (Supplemental Figure S2). Notably, to detect P1 AV nodes, we needed to utilize picrosirius red staining instead of H&E staining, due to less organized sarcomeric structure in the surrounding atrial myocytes at this age.
Figure 6.
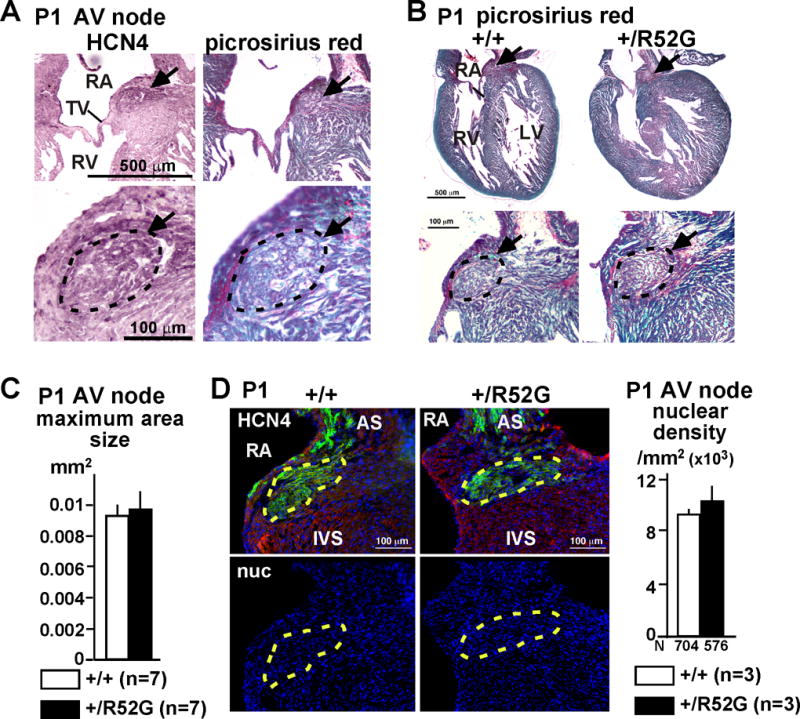
Unchanged AV nodal size in P1 Nkx2-5+/R52G mice. (A) Representative HCN4 and picrosirius red-stained normal heart tissues of P1 Nkx2-5+/+ including AV node traced by black dots. (B) Representative picrosirius red-stained heart tissues including AV node traced in black dots from control Nkx2-5+/+ or Nkx2-5+/R52G mice. (C) Maximum AV nodal area size (n=7 each). (D) Representative immunofluorescent images of HCN4 (green), actinin (red) and nuclei (blue) from control Nkx2-5+/+ or Nkx2-5+/R52G mice. AV nodal area is traced by yellow dots. Nuclear density (number of nuclei × 103/mm2) is measured from the HCN-positive AV nodal area. RA, right atrium; TV, tricuspid valve; RV, right ventricle; LV, left ventricle; AS, atrial septum; IVS, interventricular septum. Mean ± SE.
Figure 7.
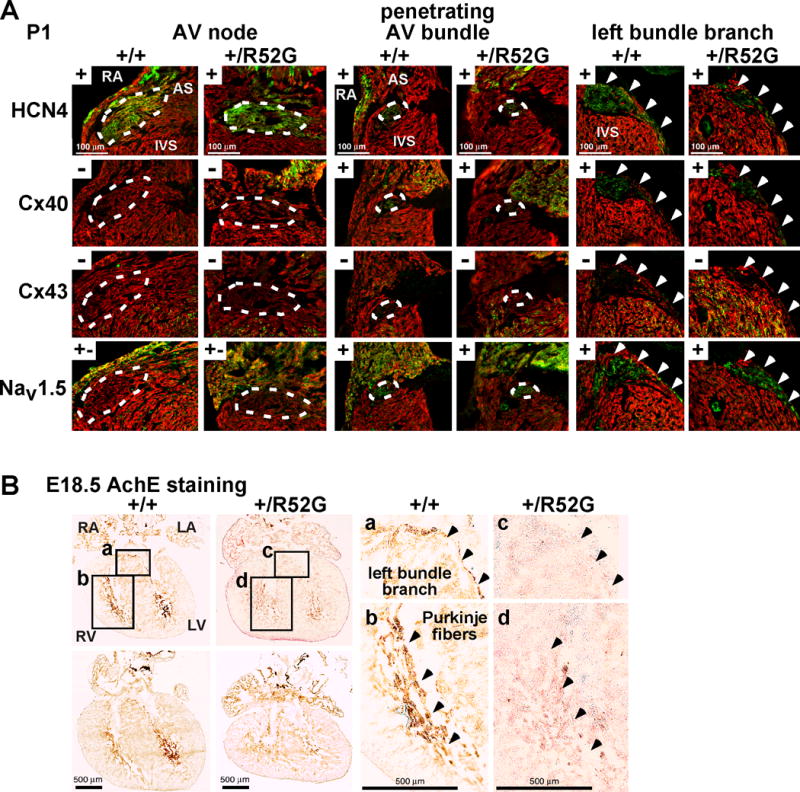
Expression of positive- and negative AV nodal, penetrating AV bundle and left bundle branch markers in P1 hearts, and AchE activities in E18.5 developing hearts. (A) Representative images of serial tissue sections positively (+) or negatively (−) stained with HCN4, connexin 40, connexin 43, and Nav1.5 in AV node, penetrating AV bundle (traced by white dots) and left bundle branch (arrowheads) from P1 mice. (B) Representative images demonstrating AchE activity (brown) in left bundle branch and ventricular trabeculations (Purkinje fibers) from serial tissue sectioning of control Nkx2-5+/+ and Nkx2-5+/R52G (+/R52G) hearts, is weaker in Nkx2-5+/R52G hearts. Areas a–d selected on the left are shown enlarged on the right. RA, right atrium; RV, right ventricle; LA, left atrium; LV, left ventricle; AS, atrial septum; IVS, interventricular septum.
In summary, therefore, Nkx2-5-KI mice were found to have smaller AV nodes at 4 weeks but not at P1. The cardiomyocytes within the compact AV node at 4 weeks were smaller than those of the wild-type animals, but without any noted difference in the ratio of myocyte to non-myocyte nuclei, and without any noted increase in fibrosis.
Reduced acetylcholine esterase (AchE) activity in ventricular trabeculations
Wide QRS, indicative of prolonged ventricular conduction times, was demonstrated in Nkx2-5-KI mice at all ages from the neonatal stage onward. This finding was consistent with P1 hearts demonstrating the under-developed left bundle branch in Nkx2-5-KI compared to control mice (Figure 7A). Further, histopathology of the ventricular conduction system in E18.5 hearts was examined by AchE activity, which is known to coincide with parts of the developing ventricular conduction system and probably controls conduction of electrical impulses during embryonic stages21–23. AchE staining in ventricular trabeculations and left bundle branch was slightly weaker in E18.5 Nkx2-5-KI than in control hearts under the same experimental conditions (Figure 7B, n=4 each). An under-developed ventricular conduction system during the embryonic stage, therefore, could be causative, at least in part, of the wide QRS.
Discussion
Heterozygous missense mutations in the homeodomain of human NKX2-5 lead to near-complete penetrance of AV conduction defects, along with high penetrance of diverse cardiac anomalies compared to mutations outside the homeodomain7. We recently replicated a disease-causing missense mutation in the homeodomain in a knock-in mouse model, Nkx2-5+/R52G, which is currently the most clinically relevant mouse model8. These mice demonstrate a high incidence of diverse cardiac malformations8. Our current study showed that Nkx2-5-KI mice also have developed PR-prolongation sufficient to produce first degree AVB by 4 weeks of age, having by this age smaller AV nodes composed of smaller AV nodal cardiomyocytes. In addition, two Nkx2-5-KI mice demonstrated advanced AVB: 3:1 AVB, and complete AVB, at 7 and 18 months of age respectively.
As yet, the steps involved in the formation of the AV node during development are not fully understood, but it is known that the atrial components of the AV conduction axis, including parts of the AV node, are derived from the embryonic atrioventricular canal, whereas the penetrating AV bundle is developed from the interventricular ring24. It has been postulated that the rate of proliferation of whole AV nodal cells originating from the engaged myocytes from the embryonic stage could be insufficient, and recruitment from adjacent working myocardium is required25. A recent study, in contrast, argued that Tbx3-positive AV nodal cells engaged from the embryonic stage would be sufficient, and would proliferate from embryonic day E14.5 to postnatal day 14, increasing nearly 5-fold24. To our knowledge, however, maintenance of the cellular architecture of the AV node has not been investigated. Additional studies, possibly using laser capture to compare the content of mRNA and proteins in the AV node between control and Nkx2-5-KI mice, could cast further light on this possibility. We are unable to explain, however, why the Nkx2-5(R52G) mutant protein should produce the situation of the compact node being composed of smaller cardiomyocytes.
The population of the Nkx2-5-positive cardiomyocytes as opposed to Nkx2-5-negative non-myocytes was unchanged in the AV nodes of Nkx2-5-KI mice compared to their age-matched controls. Thus, the previously hypothesized mechanisms, including failure of development of the AV nodal cardiomyocytes, or recruitment of cardiomyocytes from the adjacent working myocardium demonstrated in Nkx2-5 knockout mice17, 18, would not be conserved in Nkx2-5-KI mice.
One substantial discordance between the phenotypes observed in Nkx2-5-KI mouse model and human patients with NKX2-5 mutation is the prolongation of QRS, which was uniquely demonstrated in Nkx2-5-KI mice from the neonatal stage onward. AchE-positive ventricular trabeculations, which likely represent components of the developing ventricular conduction system, are morphologically changed and poorly developed in E18.5 Nkx2-5-KI mice shortly before birth. Ventricular noncompaction demonstrated in Nkx2-5-KI mouse could be attributed to abnormal development of the ventricular conduction system. Prolongation of QRS complex has been also demonstrated in multiple heterozygous and conditional homozygous Nkx2-5 knockout mice, and related to poor development of the ventricular conduction system, with reduced expression of selected gap junction protein, such as connexin40, and transcription factor Id29, 10, 14, 17, 18, 26. This suggests that, in mice, Nkx2-5 plays a more substantial role in the formation and function of the ventricular conduction system than it does in humans. Reduction of connexin40, however, was not evident in Nkx2-5-KI mouse when using immunostaining (Figure 7A) nor expression profile from whole hearts (data not shown).
Nkx2-5 is also expressed in the sinus node, and has been implicated in playing a role in nodal formation under the control of Shox2 homeodomain transcription factor27–31. Some heterozygous Nkx2-5 knockout mice, furthermore, demonstrated sinus bradycardia32, while other heterozygous and conditional homozygous Nkx2-5 knockout mice did not9, 10, 17, 18, 33. We also failed to observe any sinus arrhythmias, including sinus bradycardia, nor did we observe sinus nodal dysfunction in our EPS, consistent with the findings in human patients with NKX2-5 mutations5.
In summary, we report the presence of progressive AVB in our recently developed Nkx2-5-KI mouse model having a heterozygous Nkx2-5 missense mutation in the homeodomain. We have focused on the phenotypic similarities and differences between our mice and humans. Our findings suggest that the mouse model is likely to provide important insights into the molecular mechanisms underlying conduction through the AV conduction axis.
Supplementary Material
Acknowledgments
We are grateful to Drs. Jonathan and Christine Seidman, Kevin Fortin, Doug Smith and Miguel Zárate for valuable suggestions and technical support.
Funding Sources: This work was supported by the NIH Recovery Act Grant (HL081577-05S1 to HK), University Florida Undergraduate Research Program (HA, MN), NIH Training Grant (T35 HL007489 to HA) and American Heart Association Training Grant (10PRE4740005 to MM).
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S, Smalls O, Johnson MC, Watson MS, Seidman JG, Seidman CE, Plowden J, Kugler JD. Mutations in the cardiac transcription factor nkx2.5 affect diverse cardiac developmental pathways. J Clin Invest. 1999;104:1567–1573. doi: 10.1172/JCI8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reamon-Buettner SM, Borlak J. Nkx2-5: An update on this hypermutable homeodomain protein and its role in human congenital heart disease (chd) Hum Mutat. 2010;31:1185–1194. doi: 10.1002/humu.21345. [DOI] [PubMed] [Google Scholar]
- 3.Stallmeyer B, Fenge H, Nowak-Gottl U, Schulze-Bahr E. Mutational spectrum in the cardiac transcription factor gene nkx2.5 (csx) associated with congenital heart disease. Clin Genet. 2010;78:533–540. doi: 10.1111/j.1399-0004.2010.01422.x. [DOI] [PubMed] [Google Scholar]
- 4.De Luca A, Sarkozy A, Consoli F, Ferese R, Guida V, Dentici ML, Mingarelli R, Bellacchio E, Tuo G, Limongelli G, Digilio MC, Marino B, Dallapiccola B. Familial transposition of the great arteries caused by multiple mutations in laterality genes. Heart. 2010;96:673–677. doi: 10.1136/hrt.2009.181685. [DOI] [PubMed] [Google Scholar]
- 5.Liu XY, Wang J, Yang YQ, Zhang YY, Chen XZ, Zhang W, Wang XZ, Zheng JH, Chen YH. Novel Nkx2-5 mutations in patients with familial atrial septal defects. Pediatr Cardiol. 2011;32:193–201. doi: 10.1007/s00246-010-9859-6. [DOI] [PubMed] [Google Scholar]
- 6.Wang J, Liu XY, Yang YQ. Novel Nkx2-5 mutations responsible for congenital heart disease. Genet Mol Res. 2011;10:2905–2915. doi: 10.4238/2011.November.29.1. [DOI] [PubMed] [Google Scholar]
- 7.Kasahara H, Benson DW. Biochemical analyses of eight nkx2.5 homeodomain missense mutations causing atrioventricular block and cardiac anomalies. Cardiovasc Res. 2004;64:40–51. doi: 10.1016/j.cardiores.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Ashraf H, Pradhan L, Chang EI, Terada R, Ryan NJ, Briggs LE, Chowdhury R, Zarate MA, Sugi Y, Nam HJ, Benson DW, Anderson RH, Kasahara H. A mouse model of human congenital heart disease: High incidence of diverse cardiac anomalies and ventricular noncompaction produced by heterozygous Nkx2-5 homeodomain missense mutation. Circ Cardiovasc Genet. 2014;7:423–433. doi: 10.1161/CIRCGENETICS.113.000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Briggs LE, Takeda M, Cuadra AE, Wakimoto H, Marks MH, Walker AJ, Seki T, Oh SP, Lu JT, Sumners C, Raizada MK, Horikoshi N, Weinberg EO, Yasui K, Ikeda Y, Chien KR, Kasahara H. Perinatal loss of Nkx2-5 results in rapid conduction and contraction defects. Circ Res. 2008;103:580–590. doi: 10.1161/CIRCRESAHA.108.171835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeda M, Briggs LE, Wakimoto H, Marks MH, Warren SA, Lu JT, Weinberg EO, Robertson KD, Chien KR, Kasahara H. Slow progressive conduction and contraction defects in loss of Nkx2-5 mice after cardiomyocyte terminal differentiation. Lab Invest. 2009;89:983–993. doi: 10.1038/labinvest.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berul CI, Aronovitz MJ, Wang PJ, Mendelsohn ME. In vivo cardiac electrophysiology studies in the mouse. Circulation. 1996;94:2641–2648. doi: 10.1161/01.cir.94.10.2641. [DOI] [PubMed] [Google Scholar]
- 12.Wakimoto H, Kasahara H, Maguire CT, Moskowitz PG, Izumo S, Berul CI. Cardiac electorphysiological phenotypes in postnatal expression of nkx2.5 transgene mice. Genesis. 2003;37:144–150. doi: 10.1002/gene.10236. [DOI] [PubMed] [Google Scholar]
- 13.Wakimoto H, Kasahara H, Maguire CT, Izumo S, Berul CI. Developmentally modulated cardiac conduction failure in transgenic mice with fetal or postnatal overexpression of DNA nonbinding mutant nkx2.5. J Cardiovasc Electrophysiol. 2002;13:682–688. doi: 10.1046/j.1540-8167.2002.00682.x. [DOI] [PubMed] [Google Scholar]
- 14.Terada R, Warren S, Lu JT, Chien KR, Wessels A, Kasahara H. Ablation of Nkx2-5 at mid-embryonic stage results in premature lethality and cardiac malformation. Cardiovasc Res. 2011;91:289–299. doi: 10.1093/cvr/cvr037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tawara S. Das reizleitungssystem des säugetierherzens Eine anatomisch-histologische studie über das atrioventrikularbündel und die purkinjeschen fäden. Jena: Fischer; 1906. [Google Scholar]
- 16.Kasahara H, Bartunkova S, Schinke M, Tanaka M, Izumo S. Cardiac and extracardiac expression of csx/nkx2.5 homeodomain protein. Circ Res. 1998;82:936–946. doi: 10.1161/01.res.82.9.936. [DOI] [PubMed] [Google Scholar]
- 17.Jay PY, Harris BS, Maguire CT, Buerger A, Wakimoto H, Tanaka M, Kupershmidt S, Roden DM, Schultheiss TM, O’Brien TX, Gourdie RG, Berul CI, Izumo S. Nkx2-5 mutation causes anatomic hypoplasia of the cardiac conduction system. J Clin Invest. 2004;113:1130–1137. doi: 10.1172/JCI19846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pashmforoush M, Lu JT, Chen H, Amand TS, Kondo R, Pradervand S, Evans SM, Clark B, Feramisco JR, Giles W, Ho SY, Benson DW, Silberbach M, Shou W, Chien KR. Nkx2-5 pathways and congenital heart disease; loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell. 2004;117:373–386. doi: 10.1016/s0092-8674(04)00405-2. [DOI] [PubMed] [Google Scholar]
- 19.Ho SY, Kilpatrick L, Kanai T, Germroth PG, Thompson RP, Anderson RH. The architecture of the atrioventricular conduction axis in dog compared to man: Its significance to ablation of the atrioventricular nodal approaches. J Cardiovasc Electrophysiol. 1995;6:26–39. doi: 10.1111/j.1540-8167.1995.tb00754.x. [DOI] [PubMed] [Google Scholar]
- 20.Anderson RH, Boyett MR, Dobrzynski H, Moorman AF. The anatomy of the conduction system: Implications for the clinical cardiologist. J Cardiovasc Transl Res. 2013;6:187–196. doi: 10.1007/s12265-012-9433-0. [DOI] [PubMed] [Google Scholar]
- 21.Lamers WH, te Kortschot A, Los JA, Moorman AF. Acetylcholinesterase in prenatal rat heart: A marker for the early development of the cardiac conductive tissue? Anat Rec. 1987;217:361–370. doi: 10.1002/ar.1092170407. [DOI] [PubMed] [Google Scholar]
- 22.Nakamura T, Ikeda T, Shimokawa I, Inoue Y, Suematsu T, Sakai H, Iwasaki K, Matsuo T. Distribution of acetylcholinesterase activity in the rat embryonic heart with reference to hnk-1 immunoreactivity in the conduction tissue. Anat Embryol (Berl) 1994;190:367–373. doi: 10.1007/BF00187294. [DOI] [PubMed] [Google Scholar]
- 23.Franco D, Moorman AF, Lamers WH. Expression of the cholinergic signal-transduction pathway components during embryonic rat heart development. Anat Rec. 1997;248:110–120. doi: 10.1002/(SICI)1097-0185(199705)248:1<110::AID-AR13>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 24.Aanhaanen WT, Mommersteeg MT, Norden J, Wakker V, de Gier-de Vries C, Anderson RH, Kispert A, Moorman AF, Christoffels VM. Developmental origin, growth, and three-dimensional architecture of the atrioventricular conduction axis of the mouse heart. Circ Res. 2010;107:728–736. doi: 10.1161/CIRCRESAHA.110.222992. [DOI] [PubMed] [Google Scholar]
- 25.Cheng G, Litchenberg WH, Cole GJ, Mikawa T, Thompson RP, Gourdie RG. Development of the cardiac conduction system involves recruitment within a multipotent cardiomyogenic lineage. Development. 1999;126:5041–5049. doi: 10.1242/dev.126.22.5041. [DOI] [PubMed] [Google Scholar]
- 26.Moskowitz IP, Kim JB, Moore ML, Wolf CM, Peterson MA, Shendure J, Nobrega MA, Yokota Y, Berul C, Izumo S, Seidman JG, Seidman CE. A molecular pathway including id2, tbx5, and Nkx2-5 required for cardiac conduction system development. Cell. 2007;129:1365–1376. doi: 10.1016/j.cell.2007.04.036. [DOI] [PubMed] [Google Scholar]
- 27.Puskaric S, Schmitteckert S, Mori AD, Glaser A, Schneider KU, Bruneau BG, Blaschke RJ, Steinbeisser H, Rappold G. Shox2 mediates tbx5 activity by regulating bmp4 in the pacemaker region of the developing heart. Hum Mol Genet. 2010;19:4625–4633. doi: 10.1093/hmg/ddq393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Espinoza-Lewis RA, Yu L, He F, Liu H, Tang R, Shi J, Sun X, Martin JF, Wang D, Yang J, Chen Y. Shox2 is essential for the differentiation of cardiac pacemaker cells by repressing Nkx2-5. Dev Biol. 2009;327:376–385. doi: 10.1016/j.ydbio.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blaschke RJ, Hahurij ND, Kuijper S, Just S, Wisse LJ, Deissler K, Maxelon T, Anastassiadis K, Spitzer J, Hardt SE, Scholer H, Feitsma H, Rottbauer W, Blum M, Meijlink F, Rappold G, Gittenberger-de Groot AC. Targeted mutation reveals essential functions of the homeodomain transcription factor shox2 in sinoatrial and pacemaking development. Circulation. 2007;115:1830–1838. doi: 10.1161/CIRCULATIONAHA.106.637819. [DOI] [PubMed] [Google Scholar]
- 30.Espinoza-Lewis RA, Liu H, Sun C, Chen C, Jiao K, Chen Y. Ectopic expression of nkx2.5 suppresses the formation of the sinoatrial node in mice. Dev Biol. 2011;356:359–369. doi: 10.1016/j.ydbio.2011.05.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Munshi NV. Gene regulatory networks in cardiac conduction system development. Circ Res. 2012;110:1525–1537. doi: 10.1161/CIRCRESAHA.111.260026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Risebro CA, Petchey LK, Smart N, Gomes J, Clark J, Vieira JM, Yanni J, Dobrzynski H, Davidson S, Zuberi Z, Tinker A, Shui B, Tallini YI, Kotlikoff MI, Miquerol L, Schwartz RJ, Riley PR. Epistatic rescue of nkx2.5 adult cardiac conduction disease phenotypes by prospero-related homeobox protein 1 and hdac3. Circ Res. 2012;111:e19–31. doi: 10.1161/CIRCRESAHA.111.260695. [DOI] [PubMed] [Google Scholar]
- 33.Biben C, Weber R, Kesteven S, Stanley E, McDonald L, Elliott DA, Barnett L, Koentgen F, Robb L, Feneley M, Harvey RP. Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2-5. Circ Res. 2000;87:888–895. doi: 10.1161/01.res.87.10.888. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.