

Abstract
Varkud Satellite (VS) ribozyme mediates rolling circle replication of a plasmid found in the Neurospora mitochondria. We report crystal structures of this ribozyme at 3.1Å resolution, revealing an intertwined dimer formed by an exchange of substrate helices. Within each protomer, an arrangement of three-way helical junctions organizes seven helices into a global fold that creates a docking site for the substrate helix of the other protomer, resulting in the formation of two active sites in trans. This mode of RNA-RNA association resembles the process of domain swapping in proteins and has implications for RNA regulation and evolution. Within each active site, adenine and guanine nucleobases abut the scissile phosphate, poised to serve direct roles in catalysis. Similarities to the active sites of the hairpin and hammerhead ribozymes highlight the functional significance of active site features, underscore the ability of RNA to access functional architectures from distant regions of sequence space, and suggest convergent evolution.
Introduction
Ribozymes comprise an important class of non-coding RNAs that play prominent roles in splicing, tRNA maturation, replication of sub-genomic elements, translation and genetic control, and may have played a critical role at an early stage in the evolution of life1. Six unique nucleolytic ribozyme classes are known, several of which occur in many eukaryotic species. All catalyze site-specific RNA cleavage and some catalyze ligation reactions as well. In each case the cleavage reaction involves nucleophilic attack of a ribose 2′-O on the adjacent phosphorus atom, with the departure of the 5′-O to yield cyclic 2′-3′ phosphate and 5′-hydroxyl groups. In protein ribonucleases such as ribonuclease A, imidazole side chains of two histidine residues facilitate the cleavage reaction, acting as general acid/base catalysts that activate the 2′O nucleophile and 5′-O leaving group2. In the nucleolytic ribozymes, nucleobases play key roles in mediating catalytic interactions1,3.
Within the mitochondria of a natural isolate of Neurospora, transcription of the 881 bp VS plasmid gives rise to a multimeric RNA containing a self-cleaving motif involved in the processing of intermediates during rolling circle replication4,5. The motif, known as the VS ribozyme, contains seven helical segments (1 through 7) separated by three-way helical junctions6,7 (Fig. 1 and Supplementary Fig. 1). Cleavage and ligation reactions occur in helix 1, historically referred to as the substrate-helix, although it also contains one of the proposed catalytic nucleobases G638. These reactions are thought to be general acid-base catalyzed with the nucleobases G638 and A756 acting as general base and general acid, respectively, in the cleavage reaction and general acid and general base in the ligation reaction. Many biochemical investigations have focused on a trans acting form of the ribozyme, with the substrate helix physically disconnected from the remaining RNA8. However, the intact enzyme is active as a dimer in which the substrate-helix of one ribozyme docks into and is cleaved by helices 2–7 of the other9 (Supplementary Fig. 2).
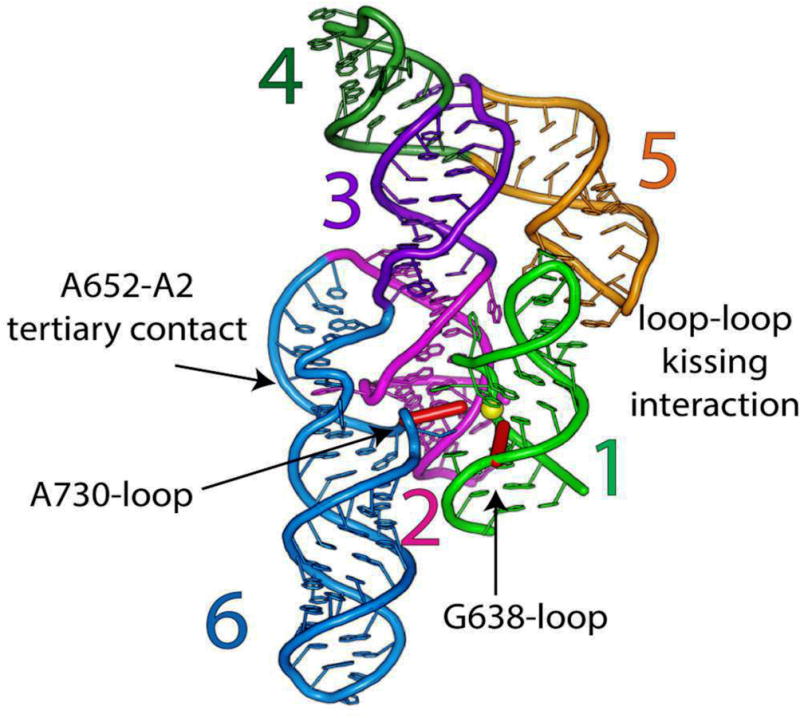
Figure 1. Global architecture of the crystallized dimeric VS ribozyme.

a, Revised secondary structure of the VS ribozyme reflecting the coaxial stacking and relative domain organization in the dimer as evident from the crystal structure. Coloring of the top ribozyme according to numbered helical elements corresponds to the canonical representation depicted in Supplementary Fig. 1. The bottom ribozyme is in yellow. Yellow circles represent scissile phosphates, and red diamonds represent catalytic nucleotides. b, Overall structure of the dimer in cartoon representation. Color scheme as in a. Yellow sphere correspond to scissile phosphates, and red sticks represent catalytic nucleotides.. c, Same as b, rotated 90° about the y-axis with helices 7 removed for clarity. Cartoons correspond to the structural model derived from the VSx_G638A ribozyme; the VSx_A756G ribozyme adopts essentially the same structure.
Despite being completely unrelated in sequence, secondary or tertiary structure, the VS ribozyme shares several important features with the smaller hairpin ribozyme. Mechanistic analysis of both ribozymes has linked key guanine and adenine nucleobases to nucleophile activation and leaving group stabilization, respectively (G8 and A38 in the hairpin10 and G63811 and A75612,13 in the VS) in the cleavage reaction. The roles of these catalytic nucleobases are reversed in the ligation reaction, according to the principle of microscopic reversibility. In both ribozymes, the two key residues occur in the same order relative to the scissile phosphate, and the active sites are assembled by interactions between internal loops found within two separate helices (historically termed the G638 and A730 loops in the VS ribozyme)14. These analogies led to the suggestion that the two ribozymes bear mechanistic and active site structural similarities, possibly representing a case of convergent evolution14. A crystal structure of the hairpin ribozyme in complex with a transition state analogue revealed the guanine and adenine juxtaposed with the reaction nucleophile and leaving group respectively, poised to participate directly in catalysis15. Although a rich literature describing VS ribozyme structural and mechanistic features has accumulated over the past two decades, the RNA has eluded high-resolution structure determination and the precise architecture and active site configuration have remained unknown. We now report the first crystal structure of the VS ribozyme at 3.1Å resolution.
Results
Crystallization construct and overall structure
Our crystallization construct closely resembles the full length wild-type ribozyme (Supplementary Fig. 1). We found that the following modifications enhanced the conformational homogeneity and decreased aggregation of the sample (Supplementary Fig. 1b): First, we installed the C634G mutation, which constitutively shifts the secondary structure of helix 1b and thus precludes the need for substrate helix rearrangement upon active site docking. This type of construct has been used for numerous biochemical studies16,17,18. Stem 4 was shortened by 3 bp and the sequence of its loop replaced with one (AAACA) predicted to be more flexible19, and stems 7a and 6c were mutated to enhance stability. The three altered stems are remote from the active site. This construct (VS_G638) populates only the monomeric and dimeric states, and is active in vitro (See Online Methods). To facilitate a homogeneous population of dimeric, uncleaved ribozyme, we made two more constructs, VSx_G638A and VSx_A756G, in which the active site catalytic nucleobases were mutated separately (Supplementary Fig. 1c, d). The use of mutations to trap the precursor state rather than deactivation of the 2′-OH nucleophile by 2′-deoxy- or 2′-methoxynucleotide substitution reflects the need to carry out native purification of the RNA from the transcription reaction. Phases were determined by SAD using an iridium hexamine derivative construct VSx_G638A_tGU (for the VSx_G638A structure) and MR (for the VSx_A756G structure) (Supplementary Table 1) and the structures of the VSx_G638A and VSx_A756G variants were refined at 3.1Å resolution in each case to Rwork/Rfree values of 0.17/0.21 and 0.23/0.27, respectively. Crystal contacts involved interactions of the AAACA loop with two other dimers in the lattice via helix 6 and 7, respectively (Supplementary Fig. 3). Both ribozyme constructs fold into essentially identical overall structures with the only differences localized near the scissile phosphate.
The crystal structures reveal that the VS ribozyme forms a symmetric dimer (Fig. 1), with an intricate interdigitation of helical segments from the two subunits (Fig. 1b and Supplementary Fig. 4a, b) that is unprecedented among known ribozymes. Dimerization creates two hybrid active sites, in which each protomer donates its substrate-helix to the catalytic domain of the other (Fig. 2). This structural exchange resembles the process of domain swapping observed in proteins where protein segments exchange part of their structure to form an intertwined dimer or higher-order oligomer20.
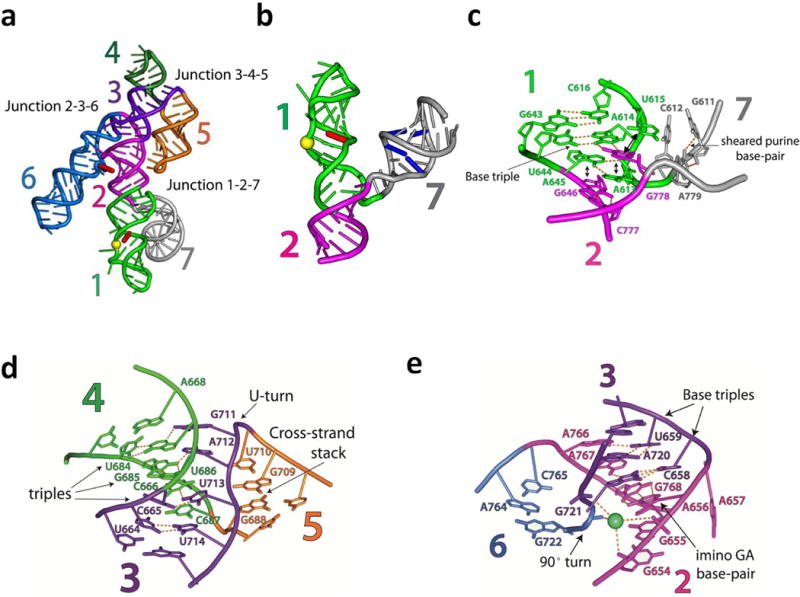
Figure 2. Trans-form of the VS ribozyme including docked substrate.
a, Catalytic domain (helices 2–6) of one protomer and the substrate-helix (helix 1) donated by the other protomer (shown in green for consistency in coloring scheme; see Supplementary Fig. 2c). The three-way helical junctions 2-3-6 and 3-4-5 organize the overall fold of the catalytic domain. The yellow sphere depicts the scissile phosphate. Red sticks correspond to the catalytic nucleobases. Junction 1-2-7 and accompanying helices 1 and 7 have been omitted for clarity..
The secondary structure of the VS ribozyme in the crystal conforms to the model derived from biochemical footprinting and mutagenesis6, with the exception of junction 1-2-7 whose secondary structure had not been defined (Fig. 1 and Supplementary Fig. 4) Electrophoretic/FRET/SAXS data on the monomeric species generally agreed with our structure21,22. Two adjacent multi-helix stacks run along the central axis of the dimer. Each stack consists of helices 1–4 of each protomer, extended further to include helix 5 of the other protomer via an intermolecular “kissing” interaction with stem-loop 1. Helices 5, 6, and 7 emerge from each stack at their respective three-way junctions, organizing the global architecture of the RNA21,22 (Fig. 1 and Supplementary Fig. 4). As helix 5 runs roughly parallel to the coaxial stack from which it protrudes, the kissing interactions lead to a close lateral association of the two multi-helix stacks. Helix 7, which pairs the 3′ and 5′ ends of the RNA chain, protrudes from the otherwise-globular structure. An angle of 55° between helix 2 and helix 6 (Supplementary Fig. 4c) creates a cleft for docking of the substrate helix (helix 1) of the other protomer (Fig. 2). SAXS analysis of the VS dimer construct VSx_G638A confirmed the overall model derived from crystallography. (Supplementary Fig. 5)
Trans docking of the substrate helix
The structure can be thought of as two enzyme-substrate complexes, with each ribozyme core consisting of helices 2–6 plus the substrate helix (helix 1) of the opposite subunit (Fig. 2). Helix 1 contains the putative general base (G638) as well as the scissile phosphate, while helix 6 contains the putative general acid (A756). The active site is formed by close melding of helix 2 with the internal loops of helices 1 and 6 (the G638 and A730 loops, respectively). As described below, tertiary structure formation alters the conformation of both of these internal loops.
The loop-loop kissing interactions with helix 5 further stabilize docking of helix 1. A U-turn motif within each stem loop organizes the nucleotides into an A-form conformation to promote formation of the inter-loop base pairs (Supplementary Fig. 6a), consistent with mutational analysis, 2′-deoxyribose and phosphorothioate interference and NMR data6,23–25. The loop-loop interaction contains three Watson-Crick base pairs and one non-canonical base pair, C629:A701 (Supplementary Fig. 6a, b)24,25. The latter is reinforced by extrusion of the U700 nucleobase from the stack (Supplementary Fig. 6a), which enables hydrogen bonding between its 5′ phosphate group and the base 3′ to it, A701 (Supplementary Fig. 6b). A kissing complex formed by just the isolated stem-loops retains these features25. Disruption of the kissing interaction decreases both binding of the substrate-helix18 and dimerization26. The kissing loop-loop interaction induces a secondary structure rearrangement in helix 1b (referred to as ‘Shifting’)16,17, which has significance for substrate recognition18.
Junction 1-2-7 projects the substrate helix outward
Nucleotides in junction 1-2-7 form an intricate network of non-canonical interactions (Fig. 3a–c and Supplementary Figs. 1 and 7). These include two purine/purine base-pairs and a base triple, as well as cross-strand purine stacks that provide base-stacking continuity between helices 1 and 2 (Fig. 3c and Supplementary Fig. 7). The junction orients helix 7 outward from the dimerization interface and directs the substrate-helix (helix 1) away from its own catalytic domain and toward that of the partner protomer7 (Fig. 1b, c and Fig. 3a, b). This conformation likely accounts for the in vivo preponderance of the monomeric, circular (ligated) form of the VS RNA and explains the observation that in concatameric VS RNA, a given ribozyme core strongly prefers to cleave the downstream-substrate helix over its own substrate helix27.
Figure 3. Three way junctions in the VS ribozyme monomer.
a, VS monomer, rendered as a cartoon, showing the position and orientation of the three-way junctions 1-2-7, 3-4-5 and 2-3-6. b, Isolated junction 1-2-7. Cartoon expanded and reoriented relative to a for clarity. The U782:G608 and C785:G606 base-pairs, known to favor ligation, are depicted in blue7. The yellow sphere and the red stick correspond to the scissile phosphate and the catalytic nucleobase, G638, respectively.c, Local interactions in junction 1-2-7 (see Supplementary Fig. 7 for further details). Orientation corresponds to that shown in ‘b’.Double-arrowheads indicate stacking interactions. d, Local interactions in Junction 3-4-5 (see Supplementary Fig. 8a–d for further details). e, Local interactions in junction 2-3-6 (see Supplementary Fig. 8e–g for further details). In this and subsequent figures in order to match the nucleotide numbering in the PDB file, add 5 to any nucleotide whose index is greater than 675 (See Supplementary Table 2).
The intricate architecture of junction 1-2-7 immediately raises the question of whether a given VS ribozyme core could achieve cleavage of its own substrate helix (reaction in cis), as the junction would need to undergo substantial rearrangement necessary to allow the substrate-helix to pivot toward its own catalytic domain. As proposed previously but not yet tested7, junction 1-2-7 might exist as an equilibrium between two conformations, one of which could enable formation of the active site in cis. Alternatively, formation of the active site in cis may have significance during ligation to give circular RNA7,27. Following the cleavage of the concatameric transcript to unit length RNAs, helices 1a and 7 must undergo strand exchange and reform intramolecularly with the 3′-end of transcript. The severed connection between helix 1a and 1b in the substrate helix could free stem loop 1b from the constraints imposed by the structure of junction 1-2-7 and allow formation of the loop-loop kissing interaction in cis, resulting in positioning of the 5′-OH at the active site for ligation. Ligation could occur following formation of the new helix 1a and helix 7, before junction 1-2-7 becomes fully structured. Following ligation, restructuring of the junction would draw the substrate helix away from its own active site, favoring RNA circles.
Two three-way junctions organize the catalytic domain
The global architecture of the core is dictated by two three-way helical junctions (3-4-5 and 2-3-6) connected through their common helix 3 (Fig. 3a, d, e). Junction 3-4-5 resembles the NMR structure of the isolated junction28 and is stabilized mainly by base triple interactions (Fig. 3d and Supplementary Fig. 8a–d). This junction orients helix 5 for formation of the loop-loop kissing interaction (Supplementary Fig. 6a).
Junction 2-3-6 (Fig. 3e and Supplementary Fig. 8e–g) orients helices 2 and 6 in a conformation that facilitates docking of the substrate helix and formation of the active site. It juxtaposes the minor groove of helix 2 with the 3′-strand of the A730-loop (containing A756) within helix 6. Docking of the substrate-helix via the minor groove of its G638-loop positions the scissile phosphate for catalysis (Fig. 2). Junction 2-3-6 includes several non-canonical base interactions and a divalent metal ion (Fig. 3e). Helices 2 and 3 are coaxial (θII-III = 151°), although one nucleotide (A657) at their junction is extruded from the stack (it packs against a A622 extruded from helix 1, as described in more detail below). The last base pair of helix 2 is an imino G:A pair as predicted by biochemical data. (Fig. 3e and Supplementary Fig. 8g)29. Base stacking is continued by two purine nucleobases in junction J6–2 that form base-triples with the minor groove face of helix 3 (Supplementary Fig. 8e, f). Junction J3–6 contains no unpaired nucleotides and its phosphate backbone makes a sharp turn, likely stabilized by the metal ion coordinated to its own phosphodiester backbone and that of helix 2 (Fig. 3e).
The orientation of helices 2 and 6 established by junction 2-3-6 is reinforced by an additional, tertiary interaction between bulges within each of these helices (Fig. 2 and Supplementary Fig. 6c). A652 flips out of helix 2 and inserts into helix 6, stacking between paired bases and forming a cis WC-sugar edge base-pair with A725 of the “A2 bulge” motif of helix 6. The backbone of the A2 bulge forms an S-turn motif, in which the direction of ribose sugars in the backbone invert twice, extruding A726 into the solvent and positioning A725 for pairing with A652. The A652-A2 tertiary interaction is consistent with a large body of solution data6,12
Active site architecture and catalysis
Association of the internal loops of helices 1 and 6 (the G638- and A730-loops, respectively) and the minor groove of helix 2b buries 1247Å2 of solvent accessible surface area creating the catalytic core (Figs. 2 and 4a). Helix 1- helix 2b interactions are mediated largely by two nucleobases that extrude from helix 1: A622 docks into the minor groove of helix 2b and also stacks against an extrahelical nucleobase from that helix (Fig. 4a and Supplementary Fig. 6d), while A621, the nucleotide immediately downstream of the scissile phosphate, forms an A-minor interaction with helix 2b (Fig. 4a and Supplementary Fig.6e). Helix 1 - helix 6 interactions are mediated by substantial inter-helix stacking: C755 and A756 (the general acid) flip out from helix 6 and are sandwiched between A621 and A639 of helix 1 (Fig. 4a and Supplementary Fig. 9a). Consistent with this stacking interaction, ultraviolet light induces a crosslink between A756 and a 4-thiouridine at position 62130.
Figure 4. Local environment of the VS active site.

a, The active site of the VS ribozyme. The proposed general acid A756 and general base A638 (G638 in WT) in the cleavage reaction are in orange. The scissile phosphate is shown as a yellow sphere. b, Schematic of biochemically inferred mechanism of catalysis by the VS ribozyme involving general acid-base catalysis by G638 and A756. Arrows indicate bonds that are formed or broken during the transition state. c, Active site nucleotides in VSx_G638A. Red spheres depict the 2′OH nucleophile, scissile phosphate and leaving group. Arrows depict movement of electrons in the cleavage reaction. N1 of A756 sits 4.2 Å from the 5′O leaving group (dashed yellow line). N1 of A638 (G638 in WT) sits within 4.9 Å of the nucleophilic 2′OH (dashed yellow line). The angle (τ) subtended by the 2′O, scissile P and 5′O is 97°. d, Active site in VSx_A756G with color coding as in c. Arrows depict movement of electrons in the cleavage reaction. N1 of G638 sits within 5.7 Å of the nucleophilic 2′OH (dashed yellow line), and the N1 of G756 (A756 in WT) sits 4.0 Å from the 5′O leaving group (dashed yellow line). N1 and N2 of G638 reside within H-bonding distance of non-bridging oxygens of the scissile phosphate (dashed green lines). These interactions could favor an in-line conformation and stabilize the transition state. Consistent with this hypothesis, the angle (τ) subtended by the 2′O, scissile P and 5′O is 129°, much closer to the ideal in-line angle than in the G638A mutant. (See Supplementary Fig. 10b, c for 2|Fo|-|Fc| simulated annealing, composite maps for the active site).
At the active site, two separate stacks of nucleotide bases create a ‘V’ shaped arrangement that organizes the catalytic nucleobases around the scissile phosphate and splays the cleavage site nucleotides G620 and A621 (Fig. 4a and Supplementary Fig. 9a). One edge of this ‘V’ contains the proposed general base, G638 (A638 in VSx_G638A) and consists exclusively of nucleotides from helix 1. Stacking on the 5′ side of G638 is intra-strand, whereas on the 3′ side, G638 forms a cross-strand stack with G620, which harbours the 2′O nucleophile. The other edge of the V contains the proposed general acid, A756 (G756 in VSx_A756G), and consists of nucleotides from both loops, including the nucleotide 3′ of the scissile phosphate, A621. Hydrogen bonding interactions with the cleavage site nucleotides further facilitate the splayed conformation and include the sheared base pair between G620 and A639 near the apex of the V and the A-minor interaction between A621 and helix 2b. (Fig. 4a and Supplementary Figs. 9b and 6e, respectively). The catalytic nucleobases G638 and A756 are both unpaired and extend toward the scissile phosphate that lies between them.
The splayed conformation, a feature observed in the active sites of the five other nucleolytic ribozymes4,31–33, allows acquisition of the in-line geometry required for the reaction: ideally a 180° angle (τ) defined by the nucleophile, the phosphorus reaction center, and the leaving group (this angle is on average 70° for an A-form RNA helix1). Our structures are consistent with the wealth of biochemical data implicating nucleobase catalysis by G638 and A756 in the cleavage mechanism of the VS ribozyme,11,12–14,34(Fig. 4b). Nevertheless, in our pre-catalytic structure of the G638A mutant, the reacting groups deviate significantly from in-line geometry (τ = 97°), and the catalytic nucleobases reside slightly beyond hydrogen bonding distance of the scissile phosphate (Fig. 4c). Clearly adjustments must occur to bring the ribozyme active site into a fully-activated state. Inspection of the A756G structure suggests that such adjustments could be restricted to local atomic movements. The A756G ribozyme, which adopts the same overall fold and active site architecture as the G638A ribozyme, configures the reacting groups closer to the in-line geometry (τ = 129°). In addition, the imino group of G638 resides within hydrogen bonding distance of the scissile phosphate (Fig. 4d). These results suggest that G638 may stabilize the in-line conformation and facilitate catalysis directly via hydrogen bonding or proton transfer, analogous to that proposed for the hairpin ribozyme10,35,36. Nevertheless, the molecular strategy by which the active site G mediates catalysis in this and other ribozymes requires further functional analysis. Our structures will provide a powerful framework for evaluating mechanistic proposals. Regarding residue 756, in both structures N1 of the purine nucleobase sits close to the 5′-oxygen leaving group, poised to mediate proton transfer (Fig. 4c, d).
Considering the use of inactivating mutations to trap the precursor, the deviations from in-line geometry observed in VSx_G638A and VSx_A756G could reflect misfolding of these mutant RNAs. However, substrate helices bearing mutations at G638, while severely affecting the cleavage reaction rate, retain in-line probing profiles and binding affinities similar to those of the corresponding helix lacking the mutation11. In addition, the A756G ribozyme retains full activity against a substrate bearing a 5′-suflur leaving group34. These data and the strong agreement between the structural and biochemical data support the functional relevance of our structures. Modeling the natural catalytic nucleobases into each structure by isosteric replacement suggests that the active site configuration of VSx_A756G forms potential hydrogen bonds to the scissile phosphate from the amino and guanidino groups of A756 and G638, respectively. More speculatively, if our structures reflect progressive stages along the reaction coordinate, then formation of new hydrogen bonds might accompany acquisition of the in-line geometry. Structural analysis of the hairpin ribozyme has also implicated a catalytic strategy involving increased hydrogen bonding to the scissile phosphate as the reaction progresses37.
Docking of the substrate remodels A730- and G638-loops
Comparison of the NMR structures of stem loops 1 and 6 in isolation with the crystal structure of the intact ribozyme shows that critical portions of the active site are remodeled by the formation of tertiary structure38 (Supplementary Fig. 9c–e). The overall features of the A730 loop within helix 6 are similar in both structures: an A730:G757 cis WC-WC base-pair widens helix 6 (Supplementary Figs. 1a and 9c, d, f, g) allowing formation of an S-turn motif that extrudes C755 and the catalytic nucleobase A756 from the helical stack (Supplementary Fig. 9g). However, the crystallographic model exhibits a more pronounced widening of the minor groove and different overall curvature of the backbone near the S-turn, with the extruded nucleobases projecting further outward from the helical stack to make interactions with nucleotides in the active site region of helix 1 (Supplementary Fig. 9c, d). In the NMR structure of stem-loop I in isolation39–41, A621 and A622 remain inside the loop whereas in the crystal structure these nucleotides protrude from the helical stack to make specific interactions with the ribozyme core (Supplementary Fig. 9e, h). Remodeling also occurs in loops A and B of the hairpin ribozyme, suggested by comparison of the crystal structure of the complete ribozyme with the NMR structure of the isolated loops42,43.
Discussion
We have crystallized and solved structures of the VS ribozyme, revealing a domain swapped dimer with unique implications for RNA catalysis, evolution, and regulation. Two unconventional strategies enabled us to obtain VS ribozyme crystals with good diffraction quality. A common approach in RNA construct design entails replacing flexible, nonessential loops with structured GNRA tetraloops. These loops impart stability and local rigidity and provide an opportunity to create lattice contacts through interactions with the minor groove of RNA helices. In this work, we found that a nonessential tetraloop was contributing to RNA aggregation, and that, unexpectedly, replacing the tetraloop with a more flexible pentaloop eliminated the aggregation problem and allowed isolation of the ribozyme as a dimer. Moreover, the pentaloop, which was intended to provide an epitope for Fab binding19, adjusted its conformation (relative to the one observed previously in complex with the Fab) to mediate lattice contacts with two other VS ribozymes (Supplementary Fig. 3). In light of these observations, it seems that in some cases of RNA crystallization it may prove advantageous to screen constructs in which nonessential loops, including tetraloops, are replaced with flexible pentaloops19.
On the basis of biochemical data noted above and now structural data, the VS and hairpin ribozymes both appear to facilitate self-cleavage via direct participation of adenine and guanine nucleobases, serving analogous roles in catalysis by each ribozyme (Fig. 5a, b). Possibly, the mechanistic similarity simply reflects the limited collection of side-chains that RNA has available for catalysis. However, the similarities between the VS and hairpin ribozymes extend beyond identity of catalytic nucleobases and into architectural features of the active sites. First, in addition to the splayed conformation of the nucleotides flanking the scissile phosphate, in both ribozymes the catalytic G stacks below the N-1 nucleotide and the catalytic A stacks above the N+1 nucleotide (Fig. 5c). Second, in both ribozymes an S-turn motif15,38 directs the catalytic A towards the leaving group and positions an upstream nucleotide coaxially to complete a stacking sandwich. Third, the two ribozymes use analogous motifs, referred to here as the L-platform (Fig. 5c), to position the catalytic guanine nucleobase toward the nucleophilic 2′OH (Fig. 5c) and complete a stacking sandwich (Fig. 5c) with the base of the L formed by a non-canonical pair between the N-1 nucleotide and the nucleotide immediately 3′ of the general base (Supplementary Fig. 10a). The conservation of these features in both ribozymes highlights their functional significance.
Figure 5. Active site similarities in the VS, hairpin, and hammerhead ribozymes.
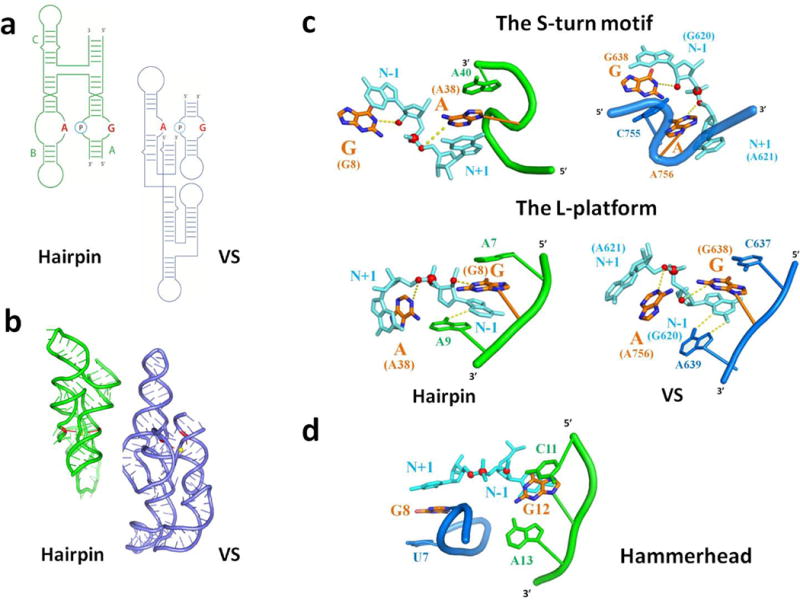
a, Secondary structures of the hairpin (green) and VS (blue) ribozymes. Identical color scheme is followed in Figs 5a–c. b, Tertiary structures of the hairpin (green) (PDB-ID: 1M5O) and VS (blue) ribozymes. The yellow sphere and red sticks represent the scissile phosphate and catalytic nucleobases, respectively. c, Similarity in the local active site structure of the hairpin and VS ribozymes. The proposed catalytic nucleobases form stacking interactions with nucleotides flanking the scissile phosphate (cyan). Red spheres correspond to oxygen atoms of the 2′OH nucleophile, scissile phosphate and leaving group. The S-turn motif projects the adenine general-acid toward the leaving group and provides an auxiliary nucleobase that stacks against the opposite side of the general acid. The L-platform positions the catalytic G nucleobase. The base of the L corresponds to a purine-purine base-pair between the N-1 nucleotide and the nucleotide 3′ of the catalytic G that rotates the N-1 nucleobase toward the major groove. This facilitates formation of the stem of the L: a three-base stack between the N-1 nucleobase, the catalytic G and the nucleotide 5′ of the catalytic G. (see Supplementary Fig. 10a for further illustration of the L-platform). VS structures were derived from the VSx_G638A ribozyme. A guanosine nucleobase is modeled in place of A638, preserving the position of the purine base itself. VSx_A756G has the same motifs with the only differences indicated in Figures 4c and d. d, The hammerhead ribozyme contains an approximate S-turn and L-platform for positioning the catalytic groups and splaying the nucleotides that flank the cleavage site.
The similar active site motifs employed by the VS and hairpin ribozymes, despite having low sequence and structural homology (Fig. 5 a, b), upholds the possibility that these ribozymes represent an example of convergent evolution14. In the classic case of convergent evolution among protein enzymes that catalyze analogous reactions, the proteases chymotrypsin and subtilisin form a common Asp/His/Ser catalytic triad organized by distinct tertiary scaffolds44. In the context of nucleases, a similar His(Tyr)/His/Lys catalytic triad at the active sites of RNase A and the unrelated tRNA endoribonuclease EndA may reflect convergent evolution45. In contrast to proteins, RNA probably has fewer catalytic motifs available for building a nuclease active site. Nevertheless, the VS and hairpin ribozymes accessed the same catalytic motif from distinct sequences and structures. Evolutionary history notwithstanding, two dissimilar ribozymes sharing mechanistic and active site features implicate RNA as a robust scaffold that can access functional architectures from distant regions of sequence space.
Analysis of the remaining nucleolytic ribozymes unexpectedly revealed the presence of these architectural features in the hammerhead (HH) ribozyme as well (Fig. 5d). The HH ribozyme forms its active site through interactions within a three-way helical junction rather than through interactions between internal loops and facilitates self-cleavage in a distinct manner. Like the VS and hairpin ribozymes, nucleophile activation involves a guanine nucleobase, but a 2′-OH from another guanosine residue (rather than an adenine nucleobase) appears poised to stabilize the leaving group46. In addition, a divalent metal ion facilitates catalysis via interactions with the scissile phosphate47. Despite these mechanistic differences, the HH ribozyme organizes its active site using motifs similar to the hairpin and VS ribozymes. The catalytic G stacks between the N-1 nucleotide and the nucleotide 5′ of it as part of an L-platform. The guanine nucleobase harboring the catalytic 2′-OH stacks with the N+1 nucleotide and emerges from an approximate S-turn motif that positions the upstream nucleobase coaxially46 (Fig. 5d). Thus the L-platform/S-turn, associated coaxial alignments, and mutualistic positioning of the cleavage site and catalytic groups through stacking appear to be a general architectural feature that can facilitate nucleolytic cleavage using different catalytic strategies. The presence of these architectural features in the context of what appears to be a distinct mechanism of catalysis underscores their functional significance and bolsters the case for convergent evolution.
Our structure also reveals a mode of RNA association, in which one domain from a monomeric RNA replaces the same domain from an identical RNA chain resulting in an intertwined dimer. This process, referred to as domain swapping, occurs commonly among natural proteins and provides a mechanism to form higher order oligomers and regulate folding and function20. Together with gene duplication events, domain swapping may have provided a mechanism for proteins to evolve larger, complex folds from smaller, simpler ones20. Perhaps in the RNA world this mode of association served analogous roles in regulation of RNA function and the emergence of complex folds. The peptidyl transferase center (PTC) of the ribosome contains an A- and P-regions that form symmetrical pocket-like structure with RNA backbone folds related by internal twofold symmetry, prompting speculation that the PTC evolved from a dimeric RNA48. Our findings reveal domain swapping as a dimerization mechanism through which a symmetrical structure could emerge in RNA.
Online Methods
Cloning of crystallization constructs
The crystals producing the native datasets were of constructs VSx_G638A and VSx_A756G (solved by molecular replacement of VSx_G638A) (Supplementary Fig.1c, d). DNA representing VSx_G638A and VSx_A756G ribozyme constructs were synthesized by PCR with overlapping primers (IDT). The resulting PCR fragment was subcloned into EcoRI and XbaI sites of pUC19 (New England Biolabs) under a T7 promoter to make the plasmids pVSx_G638A and pVSx_A756G. The relevant sequence of the VSx_G638A and VSx_A756G inserts are (See Supplementary Table 3):
VSx_G638A (See Supplementary Fig.1c):
taatacgactcacttataGGCGCTGTGTCGCAATCTGCGAAGGGCGTCGTCGGCCCAAGCGGTAGTAAGCAGGGAACTCACCTCCAATGAAACACATTGTCGTAGCAGTTGACTACTGTTATGTGATTGGTAGAGGCTAAGTGACGGTATTGGCGTAAGCCAATACCGCAGCACAGCACAAGCCCGCTTGCGAGATTACAGCGC
VSx_A756G (See Supplementary Fig.1d):
taatacgactcactataGGCGCTGTGTCGCAATCTGCGAAGGGCGTCGTCGGCCCGAGCGGTAGTAAGCAGGGAACTCACCTCCAATGAAACACATTGTCGTAGCAGTTGACTACTGTTATGTGATTGGTAGAGGCTAAGTGACGGTATTGGCGTAAGCCAATACCGCGGCACAGCACAAGCCCGCTTGCGAGATTACAGCGC
The lowercase letters indicate the T7 promoter, the bold and underlined nucleotides illustrate the only differences between the two constructs and bold nucleotides denote the 5′ and the 3′ ends of the ribozyme. G600 was added as the 5′ overhang to accommodate efficient transcription initiation with T7 RNA polymerase. Mutations A782→U, U785→C, U786→A, and U787→G (shown as blue sticks in Fig. 3b) were chosen because they were shown to increase VS ligation efficiency7. G604→C, A605→U mutations were made to restore base-pairing with G787 and A786 in the 5′ strand of helix 7. Two UA base-pairs in helix 7 were converted to GC base-pairs (U602→C:A789→G and A603→G:U788→C) to increase the stability of the helix. C634→G mutation forces the substrate-helix to constitutively adopt the shifted conformation16,17. G638→A (in VSx_G638A) and A756→G (in VSx_A756G) mutations were chosen to inhibit the catalytic activity of VS ribozyme and trap the ribozyme in the pre-cleaved state34 (See Supplementary Fig. 1c, d). Helix 4 was shortened by three base-pairs. We replaced the SL-4 with a pentaloop graft from the in vitro selected class-I ligase ribozyme to create a binding site for the crystallization chaperone Fab BL3–619. The AAACA hairpin was closed with 5′-G:C-3′ base-pair to increase affinity of BL3-6 for the hairpin19. Neither the AAACA hairpin graft onto loop 4 nor the 3bp shortening of helix 4, alone or in combination (Supplementary Fig. 1b), affect significantly the cleavage/ligation equilibrium of the complete cis-form of VS ribozyme (ratio of cleaved to ligated RNA was approximately 50: 50) (Supplementary Fig. 11a). This construct populates only the monomeric and dimeric states (Supplementary Fig. 11a, b). Mutations in helix 6c (U746→C, G749→A, U752→C and U753→C) were made to stabilize the helix.
The construct VSx_G638A_tGU, was designed to include two tandem GU wobble base-pairs in helix 6 of the ribozyme construct VSx_G638A in order to create a cation binding motif49 (Supplementary Fig. 1c). Plasmid pVSx_G638A_tGU was prepared by the same method as pVSx_G638A and pVSx_A756G. The relevant sequence of the insert was:
taatacgactcacttataGGCGCTGTGTCGCAATCTGCGAAGGGCGTCGTCGGCCCAAGCGGTAGTAAGCAGGGAACTCACCTCCAATGAAACACATTGTCGTAGCAGTTGACTACTGTTATGTGATTGGTAGAGGCTAAGTGACGGGTGTGGCGTAAGCCACGTCCGCAGCACAGCACAAGCCCGCTTGCGAGATTACAGCGC
The lowercase letters indicate the T7 promoter; bold nucleotides denote the 5′ and the 3′ ends of the ribozyme and the underlined residues reflect the tandem GU pair (Supplementary Table 3).
RNA synthesis and purification
Templates for transcription reactions were prepared by PCR amplification of pVSx_G638A, pVSx_A756G and pVSx_G638A_tGU. The primer sequences were: 5′-CAG TGA ATT CCG TAA TAC GAC TCA CTA TAG-3′ and 5′-mGmCG CTG TAA TCT CGC AAG C-3′ for the forward and the reverse primer, respectively. The first two nucleotides of the reverse primer were designed to contain the 2′OMe modification (those preceded by lowercase “m”) to reduce transcriptional heterogeneity at the 3′ end.
RNA was prepared by in vitro transcription for 2 hrs at 37°C in buffer containing 40 mM Tris-HCl pH 7.9, 2 mM spermidine, 10 mM NaCl, 25 mM MgCl2, 10mM DTT, 30 U/mL RNase Inhibitor (NEB), 2.5 U/mL TIPPase (NEB), 4 mM of each NTPs, DNA template 30 pmol/mL, T7 RNA polymerase 40 μg/mL. Transcription reactions were quenched by the addition of 10 U/mL DNase I (Promega) and incubation at 37°C for 30 min. RNA was P/C/I extracted (pH 4.3) three times and loaded onto NAP-10 column pre-equilibrated with gel filtration (GF) buffer (10 mM Tris-HCl pH 7.0, 25 mM KCl, 5 mM MgCl2). RNA was eluted with 1.5 mL of GF buffer and loaded onto HiLoad 16/60 superdex 200pg gel filtration column (GE). All gel filtration runs were carried out at 4°C. Elution peaks containing the VS ribozyme dimer were collected and concentrated to 10 mg/mL using an Amicon Ultra-15 column (30 kDa molecular weight cut-off). RNA was aliquoted into small fractions, flash frozen in liquid nitrogen and stored at −80°C.
Test of ribozyme activity
Ribozyme activity was evaluated by approximately quantifying the ratio of the precursor and product species in the cleavage reaction during transcription. Transcription reactions were quenched after 10 min in gel loading dye (90% (v/v) formamide, 0.05% (w/v) bromophenol blue, 0.05% (w/v) xylene cyanol) in 1:1 ratio and heated to 95°C for 2 min. Chemical equilibrium between cleavage and ligation was assessed by electrophoresis on a 10% denaturing polyacrylamide gel in the presence of 8 M urea. The gel was stained with EtBr and visualized by fluorescence (UVP) (Supplementary Fig. 11a).
Control sequences are (See Supplementary Table 3):
VS_WT: construct contains WT helix 4 (See Supplementary Fig. 1a). GGCGCUGUGUCGCAAUCUGCGAAGGGCGUCGUCGGCCCGAGCGGUAGUAAGCAGGGAACUCACCUCCAAUUUCAGUACUGAAAUUGUCGUAGCAGUUGACUACUGUUAUGUGAUUGGUAGAGGCUAAGUGACGGUAUUGGCGUAAGCCAAUACCGCAGCACAGCACAAGCCCGCUUGCGAGAUUACAGCGC
VS_G638A: construct contains WT helix 4 and mutation of the putative general base guanine (G638) to adenine. GGCGCUGUGUCGCAAUCUGCGAAGGGCGUCGUCGGCCCAAGCGGUAGUAAGCAGGGAACUCACCUCCAAUUUCAGUACUGAAAUUGUCGUAGCAGUUGACUACUGUUAUGUGAUUGGUAGAGGCUAAGUGACGGUAUUGGCGUAAGCCAAUACCGCAGCACAGCACAAGCCCGCUUGCGAGAUUACAGCGC
VS_G638 (Supplementary Fig.1b): same as crystallization construct VSx_G638A but with WT putative general base guanine. GGCGCUGUGUCGCAAUCUGCGAAGGGCGUCGUCGGCCCGAGCGGUAGUAAGCAGGGAACUCACCUCCAAUGAAACACAUUGUCGUAGCAGUUGACUACUGUUAUGUGAUUGGUAGAGGCUAAGUGACGGUAUUGGCGUAAGCCAAUACCGCAGCACAGCACAAGCCCGCUUGCGAGAUUACAGCGC
Light Scattering
VS ribozyme monomer and dimer fractions were passed through 0.2 μm cutoff centrifugal filter units (Millipore) and equilibrated to room temperature. Light scattering measurements were performed with DynaPro spectrometer (Wyatt). Data analysis was performed using native DynaPro software package (Supplementary Fig. 11c, d).
Crystallization
We found that the crystallization constructs dimerize during in vitro transcription when RNA concentration is ≥ 0.5 μM (Supplementary Fig. 11c, d), consistent with the activity of the dimeric form of the ribozyme9. We isolated the dimer directly from the transcription reaction without the use of any denaturation steps to avoid contamination from non-native states induced by renaturation26. RNA was rapidly thawed and diluted to 7.5 mg/mL in GF buffer. To decrease the number of nucleation events, RNA was passed over 0.2 μm centrifugal filter units (Millipore). Mosquito liquid handling robot (TTP Labtech) was used to set up HT hanging drop vapor diffusion crystallization screens using Crystal Screen I/II (Hampton), Index Screen I/II (Hampton), Natrix Screen I/II (Hampton) and RNA crystallization screen (Sigma). HT-crystallization trays were stored at RT (20–25°C) and 4°C. High diffracting crystals were observed in a condition from Index I Screen (Hampton Research): 100 mM bis-Tris pH 6.5, 2.0 M ammonium sulfate. Crystals appeared and grew to full size within 2–3 days. For cryoprotection, drops bearing suitable crystals were brought to 4.4 M ammonium acetate, keeping all other buffer compositions isotonic. Crystals were picked off directly from 96-well plate.
To better accommodate metal soaking experiments, crystals were obtained by scaling up from the HT-crystallization plate to standard VDX 24-well-plate (Hampton Research) using 500 μL of mother liquor in the reservoir. The buffer in mother liquor (100 mM bis-tris pH 6.5) was switched to sodium cacodylate pH 6.8. Drops were set up on siliconized glass slides (Hampton Research) and mixed with RNA/mother-liquor in 1:1 ratio. Crystals grew to their full size of 100×100×500 μm3. Derivative crystals were prepared by soaking native VSx_G638A_tGU crystals in a stabilizing solution supplemented with ~ 80 mM iridium hexamine49. The VSx_A756G crystals were grown in 100mM sodium citrate pH 5.8, 4% 1, 3-butanediol and 2.0 M ammonium sulfate.
Data Collection and Processing
Screening of hits from high throughput crystallization screens was performed at the Advanced Photon Source (APS) GM/CA-CAT, SBC-CAT and APS NE-CAT. The derivative data sets were collected at APS NE-CAT.
Single wavelength anomalous diffraction experiment was carried out on the VSx_G638A_tGU crystals soaked in a stabilizing solution supplemented with ~ 80 mM iridium hexamine49. Clear diffraction was observed to at least 4.5 Å resolution. We indexed, integrated and scaled the data using HKL2000/300050, identified heavy atom sites and calculated phases with SHELXD/AutoSol51 and carried out refinement with Phenix/ERRASER52,53. Fifteen iridium sites were found using SHELXD51, aided by the hkl2map GUI54_ENREF_32. SAD phasing was performed using AutoSol with the four highest occupancy heteroatom sites from SHELXD as input parameters. Initial density modification and model building was done by AutoBuild52. The Mean Figure of Merit (FOM) before and after density modification are 0.47 and 0.76, respectively. Ellipsoidal truncation and anisotropic scaling were applied to the native dataset prior to refinement using the Diffraction Anisotropy Server (http://services.mbi.ucla.edu/anisoscale/).The final model had an Rwork of 0.17 and Rfree of 0.21 (Supplementary Table 1a). Coordinate and Phase errors were 0.36Å and 25.2°, respectively. Electron density was observed for 185 of 186 phosphates and 183 of the 186 nucleobases (Supplementary Fig. 11e).
VSx_A756G datasets were integrated and scaled by RAPD (http://necat.chem.cornell.edu/). Phases were obtained by molecular replacement using the VSx_G638A as search model using Phaser52. Model building for both structures was completed with COOT55 with aid of RCrane56. Refinement was carried out with the Phenix/ERRASER pipeline52,53. The final model had an Rwork of 0.23 and Rfree of 0.27. Coordinate and Phase errors were 0.55Å and 40.3°, respectively.
Structure analysis
Helical axes for all stems were computed using Curves+57. The interhelical angles were computed by vector methods in MatLab (MathWorks). Solvent accessible surface area was calculated using POPs58. Tertiary. Torsion angles were calculated using Amigos II59. All figures were made in Pymol (Schrodinger) and edited in Illustrator (Adobe).
SAXS
SAXS experiments were conducted on the SIBYLS beamline at the Advanced Light Source synchrotron as previously described60. The samples were purified and prepared as described above. For each experiment, concentrated samples (1.5 mg/ml, 2.0 mg/ml (two wells), 2.5 mg/ml, 3.0 mg/ml, 3.5 mg/ml, 4.0 mg/ml, 4.5 mg/ml in 25 μl were placed in a 96-well plate. SAXS data were collected continuously with Q ranging from 0.012 – 0.324 with exposures of 0.5, 1.6 seconds (Supplementary Fig. 5a, Supplementary Table 4). Buffer (10 mM Tris (pH 7.5), 25 mM KCl and 5 mM MgCl2) blanks were performed both before and after each sample exposure and subtracted from the sample signal. Within each concentration, each buffer-subtracted exposure was checked for radiation damage and any oversaturated points were removed before being averaged together (Supplementary Fig. 5a). The final experimental scattering curve was calculated by scaling the averaged datasets for each concentration to the highest concentration (4.5 mg/ml) dataset and merging with ALMERGE61, extrapolating to infinite dilution (Supplementary Fig. 5a). SAXS curves were calculated from the crystal structure atomic coordinates and fit to the experimental data using the FOXS62. SAXS data collection, scattering-derived parameters and programs used for analysis are presented in Supplementary Table 4. Data analysis and super-imposition of SAXS envelope with the crystal structure is presented in Supplementary Figure 5b–d.
The coordinates and structure factors have been deposited to RCSB Protein Data Bank. SAXS data have been deposited to SASDB under SASDB ID code SASDAC9 (VSx_G638A).
Supplementary Material
Acknowledgments
We thank Jeffrey Kieft for the generous gift of iridium hexamine. Special gratitude goes to Xiaojing Yang, Sherwin Montaño, Kay Perry, Balasundaresan Dhakshnamoorthy and Kyle Dolan for advice with crystallographic data processing and Raymond Hulse, Irina Dementieva, Agnieszka Mateja, Francis Qufei, Darya Urusova and Gosia Dobosz for recommendations on growing crystals. We thank Joe Olvera for expression and purification the T7 RNA polymerase. We thank staff of the Advanced Photon Source at Argonne National Laboratory for providing technical advice on X-Ray data-collection: Kay Perry, Craig Ogata, Nagarajan Venugopalan, Bogislav Nocek, and Surajit Banerjee, the staff of SIBYLS beam line at Lawrence Berkely National Laboratory for SAXS data collection. We thank Shohei Koide, Tobin Sosnick, Sean Crosson for their valuable feedback and discussion on the project. We express gratitude to Anthony Kossiakoff and Keith Moffat for use of their data processing stations. We also thank Fang-Chieh Chao and Rhiju Das for their aid with ERRASER software. We thank Sandip Shelke for help with figures, Yaming Shao for help with Amigos II and Nicole Tuttle, Qing Dai, Nan-sheng Li, Kostek Ceslinski, Sebastian Fica, Raghuvir Sengupta, Tim Wilson, Tom Cech and Anna Pyle for helpful discussion and comments on the manuscript. This work was supported by grants from the National Institutes of Health (R01AI081987 and R01GM102489) to J.A.P. This work is based on research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamline which are supported by a grant from the National Institute of General Medical Sciences (P41 GM103403) from the National Institutes of Health (NIH) and Advanced Light Source beamline SIBYLS, all supported by USA Department of Energy (DOE). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357 and Advanced Light Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Lawrence Berkeley National Laboratory under Integrated Diffraction Analysis (IDAT) grant contract number DE-AC02-05CH11231.
Footnotes
Supplementary Notes and Online Methods are linked to the online version of the paper.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
Accession codes
The coordinates and structure factors have been deposited to RCSB Protein Data Bank under PDB ID code 4R4V and RCSB ID code rcsb086901 (VSx_G638A) and 4R4P and RCSB ID code rcsb086895 (VSx_A756G).
SAXS data have been deposited to SASBDB under SASBDB ID code SASDAC9 (VSx_G638A).
Author Contributions
N.B.S., S. D. and J.A.P. designed the study; N.B.S., S. D. and H.H. set up high-throughput crystallization experiments; N.B.S. and S. D. screened crystals and optimized crystallization conditions; N.B.S., S. D. and P.A.R. collected the data; N.B.S. and S. D. phased and solved the structures; N. B. S. collected SAXS data, J. R. F., S. D and J. A. P. analyzed the SAXS data. N.B.S., S. D. D.M. J. L., P.A.R. and J.A.P. analyzed the overall data and wrote the manuscript.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Lilley DMJ, Eckstein F. In: Ribozymes and RNA Catalysis. Lilley DMJ, Eckstein F, editors. RSC Publishing; Cambridge: 2008. pp. 1–7. [Google Scholar]
- 2.Thompson JE, Raines RT. Value of general acid-base catalysis to Ribonuclease A. J Am Chem Soc. 1994;116:5467–5468. doi: 10.1021/ja00091a060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cochrane JC, Strobel SA. Catalytic strategies of self-cleaving ribozymes. Accounts Chem Res. 2008;41:1027–1035. doi: 10.1021/ar800050c. [DOI] [PubMed] [Google Scholar]
- 4.Kennell JC, et al. The VS catalytic RNA replicates by reverse transcription as a satellite of a retroplasmid. Genes Dev. 1995;9:294–303. doi: 10.1101/gad.9.3.294. [DOI] [PubMed] [Google Scholar]
- 5.Saville BJ, Collins RA. A site-specific self-cleavage reaction performed by a novel RNA in Neurospora mitochondria. Cell. 1990;61:685–696. doi: 10.1016/0092-8674(90)90480-3. [DOI] [PubMed] [Google Scholar]
- 6.Beattie TL, Olive JE, Collins RA. A secondary-structure model for the self- cleaving region of Neurospora VS RNA. Proc Natl Acad Sci USA. 1995;92:4686–4690. doi: 10.1073/pnas.92.10.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones FD, Ryder SP, Strobel SA. An efficient ligation reaction promoted by a Varkud Satellite ribozyme with extended 5′- and 3′- termini. Nucleic Acids Res. 2001;29:5115–5120. doi: 10.1093/nar/29.24.5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo HC, Collins RA. Efficient trans-cleavage of a stem-loop RNA substrate by a ribozyme derived from neurospora VS RNA. EMBO J. 1995;14:368–376. doi: 10.1002/j.1460-2075.1995.tb07011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ouellet J, Byrne M, Lilley DM. Formation of an active site in trans by interaction of two complete Varkud Satellite ribozymes. RNA. 2009;15:1822–1826. doi: 10.1261/rna.1759009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kath-Schorr S, Wilson TJ, Li NS, Lu J, Piccirilli JA, Lilley DMJ. General Acid–Base Catalysis Mediated by Nucleobases in the Hairpin Ribozyme. J Am Chem Soc. 2012;134:16717–24. doi: 10.1021/ja3067429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilson TJ, McLeod AC, Lilley DM. A guanine nucleobase important for catalysis by the VS ribozyme. EMBO J. 2007;26:2489–2500. doi: 10.1038/sj.emboj.7601698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sood VD, Collins RA. Identification of the catalytic subdomain of the VS ribozyme and evidence for remarkable sequence tolerance in the active site loop. J Mol Biol. 2002;320:443–454. doi: 10.1016/s0022-2836(02)00521-1. [DOI] [PubMed] [Google Scholar]
- 13.Lafontaine DA, Wilson TJ, Zhao ZY, Lilley DM. Functional group requirements in the probable active site of the VS ribozyme. J Mol Biol. 2002;323:23–34. doi: 10.1016/s0022-2836(02)00910-5. [DOI] [PubMed] [Google Scholar]
- 14.Wilson TJ, Lilley DMJ. Do the hairpin and VS ribozymes share a common catalytic mechanism based on general acid-base catalysis? A critical assessment of available experimental data. RNA. 2011;17:213–221. doi: 10.1261/rna.2473711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rupert PB, Ferré-D’Amaré AR. Crystal structure of a hairpin ribozyme-inhibitor complex with implications for catalysis. Nature. 2001;410:780–786. doi: 10.1038/35071009. [DOI] [PubMed] [Google Scholar]
- 16.Andersen AA, Collins RA. Intramolecular secondary structure rearrangement by the kissing interaction of the Neurospora VS ribozyme. Proc Natil Acad Sci USA. 2001;98:7730–7735. doi: 10.1073/pnas.141039198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersen AA, Collins RA. Rearrangement of a stable RNA secondary structure during VS ribozyme catalysis. Mol Cell. 2000;5:469–478. doi: 10.1016/s1097-2765(00)80441-4. [DOI] [PubMed] [Google Scholar]
- 18.Zamel R, Collins RA. Rearrangement of substrate secondary structure facilitates binding to the Neurospora VS ribozyme. J Mol Biol. 2002;324:903–915. doi: 10.1016/s0022-2836(02)01151-8. [DOI] [PubMed] [Google Scholar]
- 19.Koldobskaya Y, Duguid EM, Shechner DM, Suslov NB, Ye JD, Sidhu SS, Bartel DP, Koide S, Kossiakoff AA, Piccirilli JA. A portable RNA sequence, whose recognition by a synthetic antibody facilitates structural determination. Nat Struct Mol Biol. 2011;18:100–106. doi: 10.1038/nsmb.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rousseau F, Schymkowitz J, Itzhaki LS. Implications of 3D domain swapping for protein folding, misfolding and function. Adv Exp Med Biol. 2012;747:137–52. doi: 10.1007/978-1-4614-3229-6_9. [DOI] [PubMed] [Google Scholar]
- 21.Lafontaine DA, Norman DG, Lilley DM. The global structure of the VS ribozyme. EMBO J. 2002;21:2461–2471. doi: 10.1093/emboj/21.10.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lipfert J, Ouellet J, Norman DG, Doniach S, Lilley DM. The complete VS ribozyme in solution studied by small-angle X-ray scattering. Structure. 2008;16:1357–1367. doi: 10.1016/j.str.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rastogi T, Beattie TL, Olive JE, Collins RA. A long range pseudoknot is required for activity of the Neurospora VS ribozyme. EMBO J. 1996;15:2820–2825. [PMC free article] [PubMed] [Google Scholar]
- 24.Bouchard P, et al. Role of SLV in SLI substrate recognition by the Neurospora VS ribozyme. RNA. 2008;14:736–748. doi: 10.1261/rna.824308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bouchard P, Legault P. A remarkably stable kissing-loop interaction defines substrate recognition by the Neurospora Varkud Satellite ribozyme. RNA. 2014;20:1451–64. doi: 10.1261/rna.046144.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suslov NB. Crystal structure of the VS ribozyme: implications for RNA catalysis, biology and evolution. Thesis (University of Chicago) 2012 [Google Scholar]
- 27.Poon AHL, Olive JE, McLaren M, Collins RA. Identification of separate structural features that affect rate and cation concentration dependence of self-cleavage by the Neurospora VS ribozyme. Biochemistry. 2006;45:13394–13400. doi: 10.1021/bi060769+. [DOI] [PubMed] [Google Scholar]
- 28.Bonneau E, Legault P. Nuclear magnetic resonance structure of the III-IV-V three-way junction from the Varkud satellite ribozyme and identification of magnesium-binding sites using paramagnetic relaxation enhancement. Biochemistry. 2014;53:6262–6275. doi: 10.1021/bi500826n. [DOI] [PubMed] [Google Scholar]
- 29.Lafontaine DA, Norman DG, Lilley DM. Structure, folding and activity of the VS ribozyme: importance of the 2–3–6 helical junction. EMBO J. 2001;20:1415–1424. doi: 10.1093/emboj/20.6.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hiley SL, Sood VD, Fan J, Collins RA. 4-thio-U cross-linking identifies the active site of the VS ribozyme. EMBO J. 2002;21:4691–4698. doi: 10.1093/emboj/cdf462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Wilson TJ, McPhee S, Lilley DMJ. Crystal structure and mechanistic investigation of the Twister ribozyme. Nat Chem Biol. 2014;10:739–744. doi: 10.1038/nchembio.1587. [DOI] [PubMed] [Google Scholar]
- 32.Eiler D, Wang J, Steitz TA. Structural basis for the fast self-cleavage reaction catalyzed by the twister ribozyme. Proc Natl Acad Sci USA. 2014;111:13028–13033. doi: 10.1073/pnas.1414571111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ren A, et al. In-line alignment and Mg2+ coordination at the cleavage site of the env22 twister ribozyme. Nat Commun. 2014;5:5534. doi: 10.1038/ncomms6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson TJ, et al. Nucleobase-mediated general acid-base catalysis in the Varkud satellite ribozyme. Proc Natl Acad Sci USA. 2010;107:11751–11756. doi: 10.1073/pnas.1004255107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pinard R, et al. Functional involvement of G8 in the hairpin ribozyme cleavage mechanism. EMBO J. 2001;20:6343–6442. doi: 10.1093/emboj/20.22.6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heldenbrand H, et al. Evidence for the role of active site residues in the Hairpin ribozyme from molecular simulations along the reaction path. J Am Chem Soc. 2014;136:7789–7792. doi: 10.1021/ja500180q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rupert PB, Massey AP, Sigurdsson STh, Ferré-D’Amaré A. Transition State Stabilization by a Catalytic RNA. Science. 2002;298:1421–1424. doi: 10.1126/science.1076093. [DOI] [PubMed] [Google Scholar]
- 38.Desjardins G, Bonneau E, Girard N, Boisbouvier J, Legault P. NMR structure of the A730 loop of the Neurospora VS ribozyme: insights into the formation of the active site. Nucleic Acids Res. 2011;39:4427–4437. doi: 10.1093/nar/gkq1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoffmann B, et al. NMR structure of the active conformation of the Varkud satellite ribozyme cleavage site. Proc Natl Acad Sci USA. 2003;100:7003–7008. doi: 10.1073/pnas.0832440100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flinders J, Dieckmann T. A pH controlled conformational switch in the cleavage site of the VS ribozyme substrate RNA. J Mol Biol. 2001;308:665–679. doi: 10.1006/jmbi.2001.4627. [DOI] [PubMed] [Google Scholar]
- 41.Michiels PJA, Schouten CHJ, Hilbers CW, Heus HA. Structure of ribozyme substrate hairpin of Neurospora VS RNA: A close look at the cleavage site. RNA. 2000;6:1821–1832. doi: 10.1017/s1355838200001394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cai Z, Tinoco IJ. Solution structure of loop A from the hairpin ribozyme from tobacco ringspot virus satellite. Biochemistry. 1996;35:6026–6036. doi: 10.1021/bi952985g. [DOI] [PubMed] [Google Scholar]
- 43.Butcher SE, Allain FH, Feigon J. Solution structure of the loop B domain from the hairpin ribozyme. Nat Str Biol. 1999;6:212–216. doi: 10.1038/6651. [DOI] [PubMed] [Google Scholar]
- 44.Kraut J. Serine proteases: structure and mechanism of catalysis. Annu Rev Biochem. 1977;46:331–358. doi: 10.1146/annurev.bi.46.070177.001555. [DOI] [PubMed] [Google Scholar]
- 45.Bujnicki JM, Rychlewski L. Unusual evolutionary history of the tRNA splicing endonuclease EndA: relationship to the LAGLIDADG and PD-(D/E)XK deoxyribonucleases. Protein Sci. 2001;10:656–660. doi: 10.1110/ps.37101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martick M, Scott WG. Tertiary contacts distant from the active site prime a ribozyme for catalysis. Cell. 2006;126:309–320. doi: 10.1016/j.cell.2006.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang S, Karbstein K, Perecchi A, Beigelman L, Herschlag D. Identification of the hammerhead ribozyme metal ion binding site responsible for rescue of the deletrious effect of a cleavage site phosphorothioate. Biochemistry. 1999;38:14363–14378. doi: 10.1021/bi9913202. [DOI] [PubMed] [Google Scholar]
- 48.Belousoff MJ, et al. Ancient machinery embedded in the contemporary ribosome. Biochem Soc Trans. 2010;38:422–427. doi: 10.1042/BST0380422. [DOI] [PubMed] [Google Scholar]
- 49.Keel AY, Rambo RP, Batey RT, Kieft JS. A general strategy to solve the phase problem in RNA crystallography. Structure. 2007;15:761–772. doi: 10.1016/j.str.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 51.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 52.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chou F-C, Sripakdeevong P, Das R. Correcting pervasive errors in RNA Crystallography with Rosetta. Nature Methods. 2013;10:74–76. doi: 10.1038/nmeth.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pape T, Schneider TR. HKL2MAP: a graphical user interface for phasing with SHELX programs. J Appl Cryst. 2004;37:843–844. [Google Scholar]
- 55.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 56.Keating KS, Pyle AM. Semiautomated model building for RNA crystallography using a directed rotameric approach. Proc Natl Acad Sci USA. 2010;107:8177–8182. doi: 10.1073/pnas.0911888107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lavery R, Moakher M, Maddocks JH, Petkeviciute D, Zakrzewska K. Conformational analysis of nucleic acids revisited: Curves+ Nucleic Acids Res. 2009;37:5917–5929. doi: 10.1093/nar/gkp608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cavallo L, Kleinjung J, Fraternali F. POPS: A fast algorithm for solvent accessible surface areas at atomic and residue level. Nucleic Acids Res. 2003;31:3364–3366. doi: 10.1093/nar/gkg601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wadley LM, Pyle AM. The identification of novel RNA structural motifs using COMPADRES: an automated approach to structural discovery. Nucleic Acids Res. 2004;32:6650–6659. doi: 10.1093/nar/gkh1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hura GL, et al. Robust, high-throughput solution structural analyses by small angle X-ray Scattering (SAXS) Nat Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Franke D, Kikhney AG, Svergun DI. Automated Acquisition and Analysis of Small Angle X-ray Scattering Data. Nuc Inst Meth A. 2012;689:52–59. [Google Scholar]
- 62.Schneidman-Duhovny D, Hammel M, Tainer JA, Sali A. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys J. 2013;105:962–974. doi: 10.1016/j.bpj.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.